
Sulfatide Deficiency, an Early Alzheimer’s Lipidomic Signature, Causes Brain Ventricular Enlargement in the Absence of Classical Neuropathological Hallmarks

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Mild Sulfatide Depletion Causes Brain Ventricular Enlargement
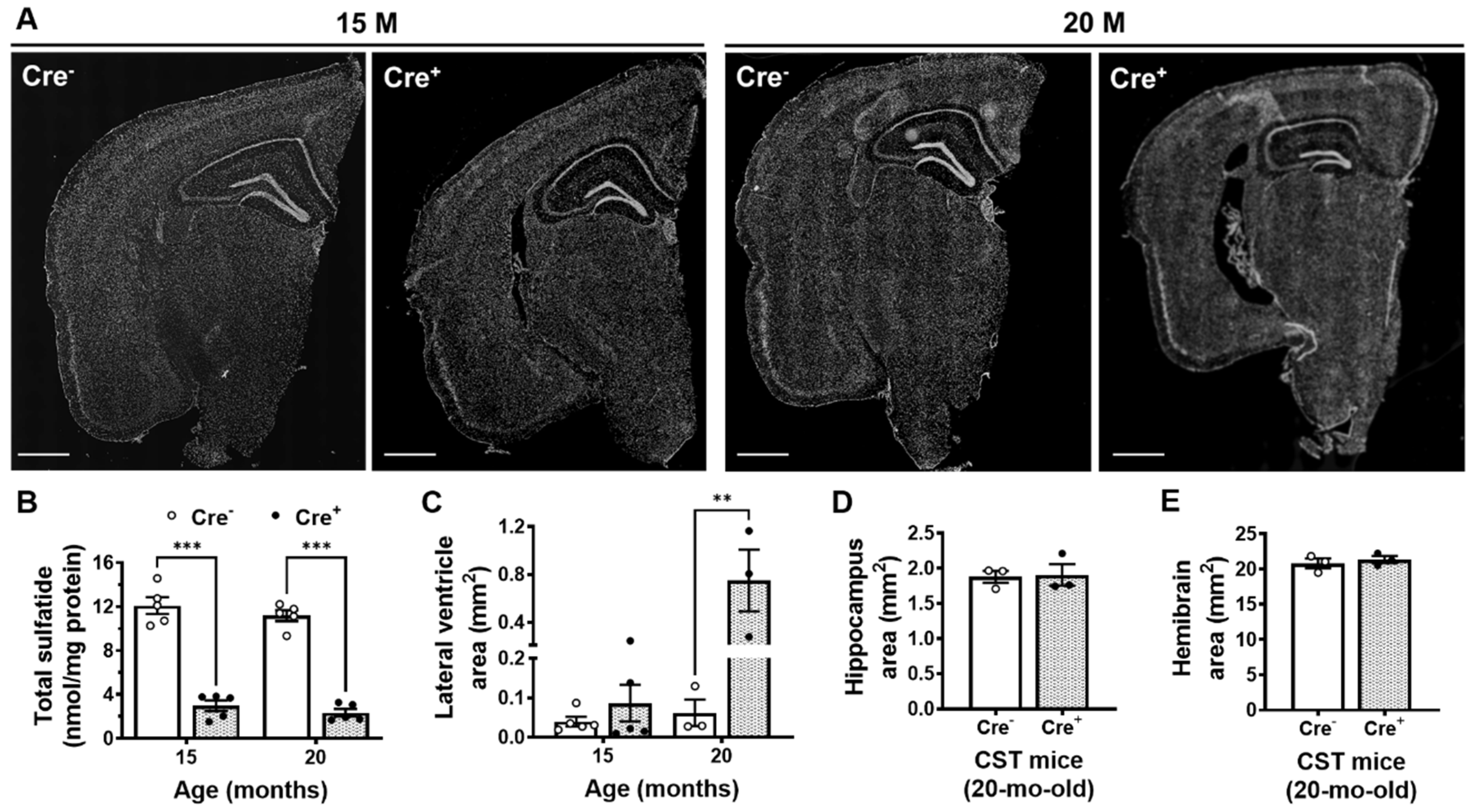
2.2. Adult-Onset CNS-Specific Sulfatide Deficiency Leads to Brain Ventricular Enlargement
2.3. Mild Sulfatide Deficiency and Ventricular Enlargement Observed in APOE4 Compared to APOE2 Knock-in Adult Mice
2.4. Sulfatide Deficiency Induces Ventricular Enlargement by Increasing AQP4 Expression
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Histology
4.3. Lipidomics
4.4. Brain MRI
4.5. Western Blot
4.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Villemagne, V.L.; Burnham, S.; Bourgeat, P.; Brown, B.; Ellis, K.A.; Salvado, O.; Szoeke, C.; Macaulay, S.L.; Martins, R.; Maruff, P.; et al. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: A prospective cohort study. Lancet Neurol. 2013, 12, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, O.T.; Kuller, L.H.; Lopez, O.L.; Thompson, P.M.; Dutton, R.A.; Lu, A.; Lee, S.E.; Lee, J.Y.; Aizenstein, H.J.; Meltzer, C.C.; et al. Cerebral ventricular changes associated with transitions between normal cognitive function, mild cognitive impairment, and dementia. Alzheimer Dis. Assoc. Disord. 2007, 21, 14–24. [Google Scholar] [CrossRef]
- Luxenberg, J.S.; Haxby, J.V.; Creasey, H.; Sundaram, M.; Rapoport, S.I. Rate of ventricular enlargement in dementia of the Alzheimer type correlates with rate of neuropsychological deterioration. Neurology 1987, 37, 1135–1140. [Google Scholar] [CrossRef]
- Nestor, S.M.; Rupsingh, R.; Borrie, M.; Smith, M.; Accomazzi, V.; Wells, J.L.; Fogarty, J.; Bartha, R. Alzheimer’s Disease Neuroimaging, I. Ventricular enlargement as a possible measure of Alzheimer’s disease progression validated using the Alzheimer’s disease neuroimaging initiative database. Brain 2008, 131, 2443–2454. [Google Scholar] [CrossRef]
- Frisoni, G.B.; Fox, N.C.; Jack, C.R., Jr.; Scheltens, P.; Thompson, P.M. The clinical use of structural MRI in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 67–77. [Google Scholar] [CrossRef]
- Apostolova, L.G.; Green, A.E.; Babakchanian, S.; Hwang, K.S.; Chou, Y.Y.; Toga, A.W.; Thompson, P.M. Hippocampal atrophy and ventricular enlargement in normal aging, mild cognitive impairment (MCI), and Alzheimer Disease. Alzheimer Dis. Assoc. Disord. 2012, 26, 17–27. [Google Scholar] [CrossRef]
- Breteler, M.M.; van Amerongen, N.M.; van Swieten, J.C.; Claus, J.J.; Grobbee, D.E.; van Gijn, J.; Hofman, A.; van Harskamp, F. Cognitive correlates of ventricular enlargement and cerebral white matter lesions on magnetic resonance imaging. The Rotterdam Study. Stroke 1994, 25, 1109–1115. [Google Scholar] [CrossRef]
- Barron, S.A.; Jacobs, L.; Kinkel, W.R. Changes in size of normal lateral ventricles during aging determined by computerized tomography. Neurology 1976, 26, 1011–1013. [Google Scholar] [CrossRef]
- Chou, Y.Y.; Lepore, N.; Avedissian, C.; Madsen, S.K.; Parikshak, N.; Hua, X.; Shaw, L.M.; Trojanowski, J.Q.; Weiner, M.W.; Toga, A.W.; et al. Mapping correlations between ventricular expansion and CSF amyloid and tau biomarkers in 240 subjects with Alzheimer’s disease, mild cognitive impairment and elderly controls. Neuroimage 2009, 46, 394–410. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.M.; Hayashi, K.M.; De Zubicaray, G.I.; Janke, A.L.; Rose, S.E.; Semple, J.; Hong, M.S.; Herman, D.H.; Gravano, D.; Doddrell, D.M.; et al. Mapping hippocampal and ventricular change in Alzheimer disease. Neuroimage 2004, 22, 1754–1766. [Google Scholar] [CrossRef] [PubMed]
- Chou, Y.Y.; Lepore, N.; Saharan, P.; Madsen, S.K.; Hua, X.; Jack, C.R.; Shaw, L.M.; Trojanowski, J.Q.; Weiner, M.W.; Toga, A.W.; et al. Ventricular maps in 804 ADNI subjects: Correlations with CSF biomarkers and clinical decline. Neurobiol. Aging 2010, 31, 1386–1400. [Google Scholar] [CrossRef] [PubMed]
- Josephs, K.A.; Whitwell, J.L.; Ahmed, Z.; Shiung, M.M.; Weigand, S.D.; Knopman, D.S.; Boeve, B.F.; Parisi, J.E.; Petersen, R.C.; Dickson, D.W.; et al. Beta-amyloid burden is not associated with rates of brain atrophy. Ann. Neurol. 2008, 63, 204–212. [Google Scholar] [CrossRef]
- Eckhardt, M. The role and metabolism of sulfatide in the nervous system. Mol. Neurobiol. 2008, 37, 93–103. [Google Scholar] [CrossRef]
- Grassi, S.; Prioni, S.; Cabitta, L.; Aureli, M.; Sonnino, S.; Prinetti, A. The Role of 3-O-Sulfogalactosylceramide, Sulfatide, in the Lateral Organization of Myelin Membrane. Neurochem. Res. 2016, 41, 130–143. [Google Scholar] [CrossRef]
- Marcus, J.; Honigbaum, S.; Shroff, S.; Honke, K.; Rosenbluth, J.; Dupree, J.L. Sulfatide is essential for the maintenance of CNS myelin and axon structure. Glia 2006, 53, 372–381. [Google Scholar] [CrossRef]
- Palavicini, J.P.; Wang, C.; Chen, L.; Ahmar, S.; Higuera, J.D.; Dupree, J.L.; Han, X. Novel molecular insights into the critical role of sulfatide in myelin maintenance/function. J. Neurochem. 2016, 139, 40–54. [Google Scholar] [CrossRef]
- Hirahara, Y.; Bansal, R.; Honke, K.; Ikenaka, K.; Wada, Y. Sulfatide is a negative regulator of oligodendrocyte differentiation: Development in sulfatide-null mice. Glia 2004, 45, 269–277. [Google Scholar] [CrossRef]
- Shroff, S.M.; Pomicter, A.D.; Chow, W.N.; Fox, M.A.; Colello, R.J.; Henderson, S.C.; Dupree, J.L. Adult CST-null mice maintain an increased number of oligodendrocytes. J. Neurosci. Res. 2009, 87, 3403–3414. [Google Scholar] [CrossRef]
- Baba, H.; Ishibashi, T. The Role of Sulfatides in Axon-Glia Interactions. Adv. Exp. Med. Biol. 2019, 1190, 165–179. [Google Scholar] [CrossRef] [PubMed]
- McGonigal, R.; Barrie, J.A.; Yao, D.; McLaughlin, M.; Cunningham, M.E.; Rowan, E.G.; Willison, H.J. Glial Sulfatides and Neuronal Complex Gangliosides Are Functionally Interdependent in Maintaining Myelinating Axon Integrity. J. Neurosci. 2019, 39, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, T.; Dupree, J.L.; Ikenaka, K.; Hirahara, Y.; Honke, K.; Peles, E.; Popko, B.; Suzuki, K.; Nishino, H.; Baba, H. A myelin galactolipid, sulfatide, is essential for maintenance of ion channels on myelinated axon but not essential for initial cluster formation. J. Neurosci. 2002, 22, 6507–6514. [Google Scholar] [CrossRef] [PubMed]
- Honke, K.; Hirahara, Y.; Dupree, J.; Suzuki, K.; Popko, B.; Fukushima, K.; Fukushima, J.; Nagasawa, T.; Yoshida, N.; Wada, Y.; et al. Paranodal junction formation and spermatogenesis require sulfoglycolipids. Proc. Natl. Acad. Sci. USA 2002, 99, 4227–4232. [Google Scholar] [CrossRef]
- Takahashi, T.; Suzuki, T. Role of sulfatide in normal and pathological cells and tissues. J. Lipid Res. 2012, 53, 1437–1450. [Google Scholar] [CrossRef]
- Han, X. Lipid alterations in the earliest clinically recognizable stage of Alzheimer’s disease: Implication of the role of lipids in the pathogenesis of Alzheimer’s disease. Curr. Alzheimer Res. 2005, 2, 65–77. [Google Scholar] [CrossRef]
- Cheng, H.; Wang, M.; Li, J.L.; Cairns, N.J.; Han, X. Specific changes of sulfatide levels in individuals with pre-clinical Alzheimer’s disease: An early event in disease pathogenesis. J. Neurochem. 2013, 127, 733–738. [Google Scholar] [CrossRef]
- Han, X.; Holtzman, D.M.; McKeel, D.W., Jr.; Kelley, J.; Morris, J.C. Substantial sulfatide deficiency and ceramide elevation in very early Alzheimer’s disease: Potential role in disease pathogenesis. J. Neurochem. 2002, 82, 809–818. [Google Scholar] [CrossRef]
- Han, X.; Fagan, A.M.; Cheng, H.; Morris, J.C.; Xiong, C.; Holtzman, D.M. Cerebrospinal fluid sulfatide is decreased in subjects with incipient dementia. Ann. Neurol. 2003, 54, 115–119. [Google Scholar] [CrossRef]
- Han, X.; Cheng, H.; Fryer, J.D.; Fagan, A.M.; Holtzman, D.M. Novel role for apolipoprotein E in the central nervous system. Modulation of sulfatide content. J. Biol. Chem. 2003, 278, 8043–8051. [Google Scholar] [CrossRef]
- Cheng, H.; Zhou, Y.; Holtzman, D.M.; Han, X. Apolipoprotein E mediates sulfatide depletion in animal models of Alzheimer’s disease. Neurobiol. Aging 2010, 31, 1188–1196. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Palavicini, J.P.; Han, X. Myelin lipid deficiency: A new key driver of Alzheimer’s disease. Neural. Regen. Res. 2023, 18, 121–122. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Palavicini, J.P.; Wang, J.; Gonzalez, N.S.; He, S.; Dustin, E.; Zou, C.; Ding, L.; Bhattacharjee, A.; Van Skike, C.E.; et al. Adult-onset CNS myelin sulfatide deficiency is sufficient to cause Alzheimer’s disease-like neuroinflammation and cognitive impairment. Mol. Neurodegener. 2021, 16, 64. [Google Scholar] [CrossRef] [PubMed]
- Agre, P. The aquaporin water channels. Proc. Am. Thorac. Soc. 2006, 3, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Filippidis, A.S.; Carozza, R.B.; Rekate, H.L. Aquaporins in Brain Edema and Neuropathological Conditions. Int. J. Mol. Sci. 2016, 18, 55. [Google Scholar] [CrossRef]
- Amiry-Moghaddam, M.; Ottersen, O.P. The molecular basis of water transport in the brain. Nat. Rev. Neurosci. 2003, 4, 991–1001. [Google Scholar] [CrossRef]
- Papadopoulos, M.C.; Verkman, A.S. Aquaporin water channels in the nervous system. Nat. Rev. Neurosci. 2013, 14, 265–277. [Google Scholar] [CrossRef]
- Wang, C.; Wang, M.; Zhou, Y.; Dupree, J.L.; Han, X. Alterations in mouse brain lipidome after disruption of CST gene: A lipidomics study. Mol. Neurobiol. 2014, 50, 88–96. [Google Scholar] [CrossRef][Green Version]
- DeCarli, C.; Haxby, J.V.; Gillette, J.A.; Teichberg, D.; Rapoport, S.I.; Schapiro, M.B. Longitudinal changes in lateral ventricular volume in patients with dementia of the Alzheimer type. Neurology 1992, 42, 2029–2036. [Google Scholar] [CrossRef]
- Friedland, R.P.; Koss, E.; Haxby, J.V.; Grady, C.L.; Luxenberg, J.; Schapiro, M.B.; Kaye, J. NIH conference. Alzheimer disease: Clinical and biological heterogeneity. Ann. Intern. Med. 1988, 109, 298–311. [Google Scholar] [CrossRef]
- Guptha, S.H.; Holroyd, E.; Campbell, G. Progressive lateral ventricular enlargement as a clue to Alzheimer’s disease. Lancet 2002, 359, 2040. [Google Scholar] [CrossRef] [PubMed]
- Sturrock, R.R. Myelination of the mouse corpus callosum. Neuropathol. Appl. Neurobiol. 1980, 6, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Williamson, J.M.; Lyons, D.A. Myelin Dynamics Throughout Life: An Ever-Changing Landscape? Front. Cell Neurosci. 2018, 12, 424. [Google Scholar] [CrossRef] [PubMed]
- Torello, L.B.; Yates, A.J.; Hart, R.; Leon, K.S. A comparative-evolutionary study of lipids in the aging brain of mice. Neurobiol. Aging 1986, 7, 337–346. [Google Scholar] [CrossRef]
- Couttas, T.A.; Kain, N.; Tran, C.; Chatterton, Z.; Kwok, J.B.; Don, A.S. Age-Dependent Changes to Sphingolipid Balance in the Human Hippocampus Are Gender-Specific and May Sensitize to Neurodegeneration. J. Alzheimers Dis. 2018, 63, 503–514. [Google Scholar] [CrossRef]
- Svennerholm, L.; Bostrom, K.; Jungbjer, B.; Olsson, L. Membrane lipids of adult human brain: Lipid composition of frontal and temporal lobe in subjects of age 20 to 100 years. J. Neurochem. 1994, 63, 1802–1811. [Google Scholar] [CrossRef]
- Svennerholm, L.; Bostrom, K.; Jungbjer, B. Changes in weight and compositions of major membrane components of human brain during the span of adult human life of Swedes. Acta Neuropathol. 1997, 94, 345–352. [Google Scholar] [CrossRef]
- Marmarou, A.; Foda, M.A.; Bandoh, K.; Yoshihara, M.; Yamamoto, T.; Tsuji, O.; Zasler, N.; Ward, J.D.; Young, H.F. Posttraumatic ventriculomegaly: Hydrocephalus or atrophy? A new approach for diagnosis using CSF dynamics. J. Neurosurg. 1996, 85, 1026–1035. [Google Scholar] [CrossRef]
- Saunders, A.; Macosko, E.Z.; Wysoker, A.; Goldman, M.; Krienen, F.M.; de Rivera, H.; Bien, E.; Baum, M.; Bortolin, L.; Wang, S.; et al. Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell 2018, 174, e1016. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Ximerakis, M.; Lipnick, S.L.; Innes, B.T.; Simmons, S.K.; Adiconis, X.; Dionne, D.; Mayweather, B.A.; Nguyen, L.; Niziolek, Z.; Ozek, C.; et al. Single-cell transcriptomic profiling of the aging mouse brain. Nat. Neurosci. 2019, 22, 1696–1708. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, J.A.; Hsu, M.S.; Seldin, M.M.; Binder, D.K. Expression of the Astrocyte Water Channel Aquaporin-4 in the Mouse Brain. ASN Neuro 2015, 7, 1759091415605486. [Google Scholar] [CrossRef] [PubMed]
- Skjolding, A.D.; Rowland, I.J.; Sogaard, L.V.; Praetorius, J.; Penkowa, M.; Juhler, M. Hydrocephalus induces dynamic spatiotemporal regulation of aquaporin-4 expression in the rat brain. Cereb. Fluid Res. 2010, 7, 20. [Google Scholar] [CrossRef]
- Bloch, O.; Auguste, K.I.; Manley, G.T.; Verkman, A.S. Accelerated progression of kaolin-induced hydrocephalus in aquaporin-4-deficient mice. J. Cereb. Blood Flow Metab. 2006, 26, 1527–1537. [Google Scholar] [CrossRef]
- Bradley, W.G., Jr. CSF Flow in the Brain in the Context of Normal Pressure Hydrocephalus. AJNR Am. J. Neuroradiol. 2015, 36, 831–838. [Google Scholar] [CrossRef]
- Cheng, H.; Guan, S.; Han, X. Abundance of triacylglycerols in ganglia and their depletion in diabetic mice: Implications for the role of altered triacylglycerols in diabetic neuropathy. J. Neurochem. 2006, 97, 1288–1300. [Google Scholar] [CrossRef]
- Cheng, H.; Jiang, X.; Han, X. Alterations in lipid homeostasis of mouse dorsal root ganglia induced by apolipoprotein E deficiency: A shotgun lipidomics study. J. Neurochem. 2007, 101, 57–76. [Google Scholar] [CrossRef]
- Wang, C.; Palavicini, J.P.; Han, X. Lipidomics Profiling of Myelin. Methods Mol. Biol. 2018, 1791, 37–50. [Google Scholar] [CrossRef]
- Han, X.; Yang, K.; Gross, R.W. Microfluidics-based electrospray ionization enhances the intrasource separation of lipid classes and extends identification of individual molecular species through multi-dimensional mass spectrometry: Development of an automated high-throughput platform for shotgun lipidomics. Rapid Commun. Mass Spectrom. 2008, 22, 2115–2124. [Google Scholar] [CrossRef]
- Yang, K.; Cheng, H.; Gross, R.W.; Han, X. Automated lipid identification and quantification by multidimensional mass spectrometry-based shotgun lipidomics. Anal. Chem. 2009, 81, 4356–4368. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, C.; Han, X. Selection of internal standards for accurate quantification of complex lipid species in biological extracts by electrospray ionization mass spectrometry—What, how and why? Mass Spectrom. Rev. 2017, 36, 693–714. [Google Scholar] [CrossRef] [PubMed]
- Strupp, J.P. Stimulate: A GUI Based fMRI Analysis Software Package. Neuroimage 1996, 3, S607. [Google Scholar] [CrossRef]
- Wang, L.; Li, C.; Sun, Q.; Xia, D.; Kao, C.Y. Active contours driven by local and global intensity fitting energy with application to brain MR image segmentation. Comput. Med. Imaging Graph. 2009, 33, 520–531. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palavicini, J.P.; Ding, L.; Pan, M.; Qiu, S.; Wang, H.; Shen, Q.; Dupree, J.L.; Han, X. Sulfatide Deficiency, an Early Alzheimer’s Lipidomic Signature, Causes Brain Ventricular Enlargement in the Absence of Classical Neuropathological Hallmarks. Int. J. Mol. Sci. 2023, 24, 233. https://doi.org/10.3390/ijms24010233
Palavicini JP, Ding L, Pan M, Qiu S, Wang H, Shen Q, Dupree JL, Han X. Sulfatide Deficiency, an Early Alzheimer’s Lipidomic Signature, Causes Brain Ventricular Enlargement in the Absence of Classical Neuropathological Hallmarks. International Journal of Molecular Sciences. 2023; 24(1):233. https://doi.org/10.3390/ijms24010233
Chicago/Turabian StylePalavicini, Juan Pablo, Lin Ding, Meixia Pan, Shulan Qiu, Hu Wang, Qiang Shen, Jeffrey L. Dupree, and Xianlin Han. 2023. "Sulfatide Deficiency, an Early Alzheimer’s Lipidomic Signature, Causes Brain Ventricular Enlargement in the Absence of Classical Neuropathological Hallmarks" International Journal of Molecular Sciences 24, no. 1: 233. https://doi.org/10.3390/ijms24010233
APA StylePalavicini, J. P., Ding, L., Pan, M., Qiu, S., Wang, H., Shen, Q., Dupree, J. L., & Han, X. (2023). Sulfatide Deficiency, an Early Alzheimer’s Lipidomic Signature, Causes Brain Ventricular Enlargement in the Absence of Classical Neuropathological Hallmarks. International Journal of Molecular Sciences, 24(1), 233. https://doi.org/10.3390/ijms24010233