


Metabolomics and Lipidomics Screening Reveal Reprogrammed Signaling Pathways toward Cancer Development in Non-Alcoholic Steatohepatitis
 , , ,
, , ,  and
and
Abstract
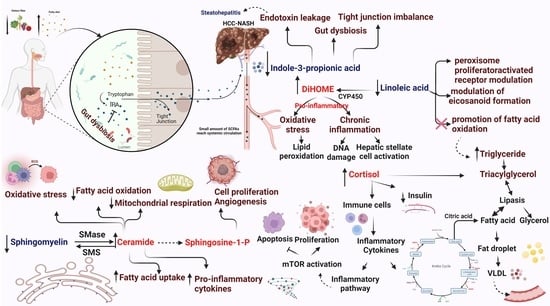
1. Introduction
2. Results
2.1. Clinical Characteristics of NASH and NASH-HCC Patients
2.2. Hematological and Biochemical Analysis
2.3. Tumor Markers and Diabetic Profile
2.4. Metabolomic Profiling of NASH and NASH-HCC Patients
2.5. Targeted Analysis of Lipids
2.6. Targeted Analysis of Vitamins and Amino Acids
2.7. Targeted Analysis of Adenosine, ATP, and 5′-AMP
2.8. Metabolomics and Clinical Data Clustering between NASH and NASH-HCC Patients
2.9. Pathway Enrichment Analysis
2.10. An Integrative Metabolite–Protein Interactions Network
3. Discussion
4. Materials and Methods
4.1. Chemical and Reagents
4.2. Study Population and Sample Collection
4.3. Blood Analysis
4.3.1. Hematological and Biochemical Test
4.3.2. Diabetic Profile
4.3.3. Tumor Markers
4.4. Samples and Standards Preparation
4.4.1. Sample Preparation for Metabolomics Analysis
4.4.2. Standards Preparation
4.5. LC-MS/MS Analysis Using the SWATH Acquisition Method
4.5.1. LC Method
4.5.2. Mass Spectrometry Method
4.5.3. Sample Processing
4.6. Metabolomics Data Analysis
4.6.1. Non-Targeted Analysis
4.6.2. Targeted Analysis
4.7. Pre-Processing and Statistical Analysis
4.7.1. Data Pre-Processing
4.7.2. Data Normalization and Transformation
4.7.3. Statistical Analysis
4.8. Pathway Enrichment Analysis and Metabolite–Protein Knowledge-Based Integration
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dufour, J.-F.; Scherer, R.; Balp, M.-M.; McKenna, S.J.; Janssens, N.; Lopez, P.; Pedrosa, M. The global epidemiology of nonalcoholic steatohepatitis (NASH) and associated risk factors–A targeted literature review. Endocr. Metab. Sci. 2021, 3, 100089. [Google Scholar] [CrossRef]
- Kim, G.-A.; Lee, H.C.; Choe, J.; Kim, M.-J.; Lee, M.J.; Chang, H.-S.; Bae, I.Y.; Kim, H.-K.; An, J.; Shim, J.H. Association between non-alcoholic fatty liver disease and cancer incidence rate. J. Hepatol. 2018, 68, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Cholankeril, G.; Patel, R.; Khurana, S.; Satapathy, S.K. Hepatocellular carcinoma in non-alcoholic steatohepatitis: Current knowledge and implications for management. World J. Hepatol. 2017, 9, 533. [Google Scholar] [CrossRef] [PubMed]
- Raza, S.; Rajak, S.; Anjum, B.; Sinha, R.A. Molecular links between non-alcoholic fatty liver disease and hepatocellular carcinoma. Hepatoma Res. 2019, 5, 42. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, F.; Kramer, J.R.; Mapakshi, S.; Natarajan, Y.; Chayanupatkul, M.; Richardson, P.A.; Li, L.; Desiderio, R.; Thrift, A.P.; Asch, S.M. Risk of hepatocellular cancer in patients with non-alcoholic fatty liver disease. Gastroenterology 2018, 155, 1828–1837.e1822. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; El-Serag, H.B.; Sada, Y.H.; Kanwal, F.; Duan, Z.; Temple, S.; May, S.B.; Kramer, J.R.; Richardson, P.A.; Davila, J.A. Hepatocellular carcinoma in the absence of cirrhosis in United States veterans is associated with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2016, 14, 124–131.e1. [Google Scholar] [CrossRef] [PubMed]
- Paradis, V.; Zalinski, S.; Chelbi, E.; Guedj, N.; Degos, F.; Vilgrain, V.; Bedossa, P.; Belghiti, J. Hepatocellular carcinomas in patients with metabolic syndrome often develop without significant liver fibrosis: A pathological analysis. Hepatology 2009, 49, 851–859. [Google Scholar] [CrossRef]
- Lewinska, M.; Santos-Laso, A.; Arretxe, E.; Alonso, C.; Zhuravleva, E.; Jimenez-Agüero, R.; Eizaguirre, E.; Pareja, M.J.; Romero-Gómez, M.; Arrese, M. The altered serum lipidome and its diagnostic potential for Non-Alcoholic Fatty Liver (NAFL)-associated hepatocellular carcinoma. EBioMedicine 2021, 73, 103661. [Google Scholar] [CrossRef]
- Khalil, A.; Elfert, A.; Ghanem, S.; Helal, M.; Abdelsattar, S.; Elgedawy, G.; Obada, M.; Abdel-Samiee, M.A.; El-Said, H. The role of metabolomics in hepatocellular carcinomas. Egypt. Liver J. 2021, 11, 1–8. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Negro, F. Natural history of NASH and HCC. Liver Int. 2020, 40 (Suppl. S1), 72–76. [Google Scholar] [CrossRef] [PubMed]
- De Matteis, S.; Ragusa, A.; Marisi, G.; De Domenico, S.; Casadei Gardini, A.; Bonafe, M.; Giudetti, A.M. Aberrant Metabolism in Hepatocellular Carcinoma Provides Diagnostic and Therapeutic Opportunities. Oxidative Med. Cell. Longev. 2018, 2018, 7512159. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.-X.; Zheng, C.-Y.; Deng, Y.-J.; Tang, K.-R.; Nie, H.; Xie, J.-Q.; Liu, D.-D.; Tu, G.-F.; Yang, Q.-H.; Zhang, Y.-P. Hepatic protective effects of Shenling Baizhu powder, a herbal compound, against inflammatory damage via TLR4/NLRP3 signalling pathway in rats with nonalcoholic fatty liver disease. J. Integr. Med. 2021, 19, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Han, J.; Xing, H.; Zhang, H.; Li, Z.; Liang, L.; Li, C.; Dai, S.; Wu, M.; Shen, F.; et al. Dysregulated fatty acid metabolism in hepatocellular carcinoma. Hepatic Oncol. 2016, 3, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Todisco, S.; Convertini, P.; Iacobazzi, V.; Infantino, V. TCA Cycle Rewiring as Emerging Metabolic Signature of Hepatocellular Carcinoma. Cancers 2019, 12, 68. [Google Scholar] [CrossRef] [PubMed]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Tenen, D.G.; Chai, L.; Tan, J.L. Metabolic alterations and vulnerabilities in hepatocellular carcinoma. Gastroenterol. Rep. 2020, 9, 1–13. [Google Scholar] [CrossRef]
- Léveillé, M.; Estall, J.L. Mitochondrial Dysfunction in the Transition from NASH to HCC. Metabolites 2019, 9, 233. [Google Scholar] [CrossRef]
- Raza, S.; Tewari, A.; Rajak, S.; Sinha, R.A. Vitamins and non-alcoholic fatty liver disease: A molecular insight. Liver Res. 2021, 5, 62–71. [Google Scholar] [CrossRef]
- Sunshine, H.; Iruela-Arispe, M.L. Membrane lipids and cell signaling. Curr. Opin. Lipidol. 2017, 28, 408. [Google Scholar] [CrossRef]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Clish, C.B. Metabolomics: An emerging but powerful tool for precision medicine. Cold Spring Harb. Mol. Case Stud. 2015, 1, a000588. [Google Scholar] [CrossRef] [PubMed]
- Dhamija, E.; Paul, S.B.; Kedia, S. Non-alcoholic fatty liver disease associated with hepatocellular carcinoma: An increasing concern. Indian J. Med. Res. 2019, 149, 9. [Google Scholar] [PubMed]
- Gawlik, A.; Shmoish, M.; Hartmann, M.F.; Wudy, S.A.; Olczak, Z.; Gruszczynska, K.; Hochberg, Z. Steroid metabolomic signature of liver disease in nonsyndromic childhood obesity. Endocr. Connect. 2019, 8, 764–771. [Google Scholar] [CrossRef]
- Wang, K.K.; Czaja, A.J. Hepatocellular carcinoma in corticosteroid-treated severe autoimmune chronic active hepatitis. Hepatology 1988, 8, 1679–1683. [Google Scholar] [CrossRef]
- Tian, Y.; Li, Y.; Wang, W.-X.; Jiang, W.-L.; Fei, J.; Li, C.-Y. Novel strategy for validating the existence and mechanism of the “gut–liver axis” in vivo by a hypoxia-sensitive NIR fluorescent probe. Anal. Chem. 2020, 92, 4244–4250. [Google Scholar] [CrossRef]
- Albhaisi, S.A.; Bajaj, J.S.; Sanyal, A.J. Role of gut microbiota in liver disease. Am. J. Physiol.-Gastrointest. Liver Physiol. 2020, 318, G84–G98. [Google Scholar] [CrossRef]
- Zhao, Z.-H.; Xin, F.-Z.; Xue, Y.; Hu, Z.; Han, Y.; Ma, F.; Zhou, D.; Liu, X.-L.; Cui, A.; Liu, Z. Indole-3-propionic acid inhibits gut dysbiosis and endotoxin leakage to attenuate steatohepatitis in rats. Exp. Mol. Med. 2019, 51, 1–14. [Google Scholar] [CrossRef]
- Wong, R.J.; Cheung, R.; Ahmed, A. Nonalcoholic steatohepatitis is the most rapidly growing indication for liver transplantation in patients with hepatocellular carcinoma in the US. Hepatology 2014, 59, 2188–2195. [Google Scholar] [CrossRef]
- Liu, F.; Sun, C.; Chen, Y.; Du, F.; Yang, Y.; Wu, G. Indole-3-Propionic Acid-Aggravated CCl4-Induced Liver Fibrosis via the TGF-β1/Smads Signaling Pathway. J. Clin. Transl. Hepatol. 2021, 9, 917. [Google Scholar] [CrossRef]
- Han, J.; Dzierlenga, A.L.; Lu, Z.; Billheimer, D.D.; Torabzadeh, E.; Lake, A.D.; Li, H.; Novak, P.; Shipkova, P.; Aranibar, N.; et al. Metabolomic profiling distinction of human nonalcoholic fatty liver disease progression from a common rat model. Obesity (Silver Spring Md.) 2017, 25, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Jiang, M.; Zhao, J.; Song, Y.; Du, W.; Shi, J. The Mechanism Underlying the Influence of Indole-3-Propionic Acid: A Relevance to Metabolic Disorders. Front. Endocrinol. 2022, 13, 841703. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Coker, O.O.; Chu, E.S.; Fu, K.; Lau, H.C.; Wang, Y.-X.; Chan, A.W.; Wei, H.; Yang, X.; Sung, J.J. Dietary cholesterol drives fatty liver-associated liver cancer by modulating gut microbiota and metabolites. Gut 2021, 70, 761–774. [Google Scholar] [CrossRef]
- Li, Y.; Xu, W.; Zhang, F.; Zhong, S.; Sun, Y.; Huo, J.; Zhu, J.; Wu, C. The gut microbiota-produced indole-3-propionic acid confers the antihyperlipidemic effect of mulberry-derived 1-deoxynojirimycin. Msystems 2020, 5, e00313-20. [Google Scholar] [CrossRef] [PubMed]
- Tajiri, K.; Shimizu, Y. Branched-chain amino acids in liver diseases. World J. Gastroenterol. 2013, 19, 7620. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, K.; Yu, L.; Nagasue, N. Direct effect of branched-chain amino acids on the growth and metabolism of cultured human hepatocellular carcinoma cells. Nutr. Cancer 1998, 31, 62–68. [Google Scholar] [CrossRef]
- Miuma, S.; Ichikawa, T.; Arima, K.; Takeshita, S.; Muraoka, T.; Matsuzaki, T.; Ootani, M.; Shibata, H.; Akiyama, M.; Ozawa, E. Branched-chain amino acid deficiency stabilizes insulin-induced vascular endothelial growth factor mRNA in hepatocellular carcinoma cells. J. Cell. Biochem. 2012, 113, 3113–3121. [Google Scholar] [CrossRef]
- Ninomiya, S.; Shimizu, M.; Imai, K.; Takai, K.; Shiraki, M.; Hara, T.; Tsurumi, H.; Ishizaki, S.; Moriwaki, H. Possible role of visfatin in hepatoma progression and the effects of branched-chain amino acids on visfatin-induced proliferation in human hepatoma cells. Cancer Prev. Res. 2011, 4, 2092–2100. [Google Scholar] [CrossRef]
- Hagiwara, A.; Nishiyama, M.; Ishizaki, S. Branched-chain amino acids prevent insulin-induced hepatic tumor cell proliferation by inducing apoptosis through mTORC1 and mTORC2-dependent mechanisms. J. Cell. Physiol. 2012, 227, 2097–2105. [Google Scholar] [CrossRef]
- Muto, Y.; Sato, S.; Watanabe, A.; Moriwaki, H.; Suzuki, K.; Kato, A.; Kato, M.; Nakamura, T.; Higuchi, K.; Nishiguchi, S. Overweight and obesity increase the risk for liver cancer in patients with liver cirrhosis and long-term oral supplementation with branched-chain amino acid granules inhibits liver carcinogenesis in heavier patients with liver cirrhosis. Hepatol. Res. 2006, 35, 204–214. [Google Scholar] [CrossRef]
- Kobayashi, M.; Ikeda, K.; Arase, Y.; Suzuki, Y.; Suzuki, F.; Akuta, N.; Hosaka, T.; Murashima, N.; Saitoh, S.; Someya, T. Inhibitory effect of branched-chain amino acid granules on progression of compensated liver cirrhosis due to hepatitis C virus. J. Gastroenterol. 2008, 43, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, K.; Okabayashi, T.; Maeda, H.; Namikawa, T.; Iiyama, T.; Sugimoto, T.; Kobayashi, M.; Mimura, T.; Hanazaki, K. Oral supplementation of branched-chain amino acids reduces early recurrence after hepatic resection in patients with hepatocellular carcinoma: A prospective study. Surg. Today 2013, 43, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Behiry, E.G.; Mahmoud, S.K.; Swelim, M.A.; El-dougdoug, K.A.; Attia, A.; Hussein, A.M. Changes in tryptophan and phenylalanine in chronic HCV patients treated with direct acting antiviral (sofosbuvir). Bull. Natl. Res. Cent. 2018, 42, 1–7. [Google Scholar] [CrossRef]
- de Mello, V.D.; Sehgal, R.; Männistö, V.; Klåvus, A.; Nilsson, E.; Perfilyev, A.; Kaminska, D.; Miao, Z.; Pajukanta, P.; Ling, C. Serum aromatic and branched-chain amino acids associated with NASH demonstrate divergent associations with serum lipids. Liver Int. 2021, 41, 754–763. [Google Scholar] [CrossRef]
- Jain, M.; Nilsson, R.; Sharma, S.; Madhusudhan, N.; Kitami, T.; Souza, A.L.; Kafri, R.; Kirschner, M.W.; Clish, C.B.; Mootha, V.K. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 2012, 336, 1040–1044. [Google Scholar] [CrossRef]
- Gomes, R.N.; Felipe da Costa, S.; Colquhoun, A. Eicosanoids and cancer. Clinics 2018, 73, e530s. [Google Scholar] [CrossRef]
- Jee, S.H.; Kim, M.; Kim, M.; Yoo, H.J.; Kim, H.; Jung, K.J.; Hong, S.; Lee, J.H. Metabolomics profiles of hepatocellular carcinoma in a Korean prospective cohort: The Korean Cancer Prevention Study-II. Cancer Prev. Res. 2018, 11, 303–312. [Google Scholar] [CrossRef]
- Lu, X.; Yu, H.; Ma, Q.; Shen, S.; Das, U.N. Linoleic acid suppresses colorectal cancer cell growth by inducing oxidant stress and mitochondrial dysfunction. Lipids Health Dis. 2010, 9, 1–11. [Google Scholar] [CrossRef]
- Ma, C.; Kesarwala, A.H.; Eggert, T.; Medina-Echeverz, J.; Kleiner, D.E.; Jin, P.; Stroncek, D.F.; Terabe, M.; Kapoor, V.; ElGindi, M. NAFLD causes selective CD4+ T lymphocyte loss and promotes hepatocarcinogenesis. Nature 2016, 531, 253–257. [Google Scholar] [CrossRef]
- Paul, B.; Lewinska, M.; Andersen, J.B. Lipid alterations in chronic liver disease and liver cancer. JHEP Rep. 2022, 4, 100479. [Google Scholar] [CrossRef]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.-S.; Xu, A.; Huang, T.; Yu, X.; Li, D. Low docosahexaenoic acid content in plasma phospholipids is associated with increased non-alcoholic fatty liver disease in China. Lipids 2012, 47, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Tiwari-Heckler, S.; Gan-Schreier, H.; Stremmel, W.; Chamulitrat, W.; Pathil, A. Circulating phospholipid patterns in NAFLD patients associated with a combination of metabolic risk factors. Nutrients 2018, 10, 649. [Google Scholar] [CrossRef] [PubMed]
- Gorden, D.L.; Myers, D.S.; Ivanova, P.T.; Fahy, E.; Maurya, M.R.; Gupta, S.; Min, J.; Spann, N.J.; McDonald, J.G.; Kelly, S.L. Biomarkers of NAFLD progression: A lipidomics approach to an epidemic1 [S]. J. Lipid Res. 2015, 56, 722–736. [Google Scholar] [CrossRef]
- Skill, N.J.; Scott, R.E.; Wu, J.; Maluccio, M.A. Hepatocellular carcinoma associated lipid metabolism reprogramming. J. Surg. Res. 2011, 169, 51–56. [Google Scholar] [CrossRef]
- Puri, P.; Baillie, R.A.; Wiest, M.M.; Mirshahi, F.; Choudhury, J.; Cheung, O.; Sargeant, C.; Contos, M.J.; Sanyal, A.J. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 2007, 46, 1081–1090. [Google Scholar] [CrossRef]
- Rein-Fischboeck, L.; Haberl, E.M.; Pohl, R.; Feder, S.; Liebisch, G.; Krautbauer, S.; Buechler, C. Variations in hepatic lipid species of age-matched male mice fed a methionine-choline-deficient diet and housed in different animal facilities. Lipids Health Dis. 2019, 18, 1–12. [Google Scholar] [CrossRef]
- Musso, G.; Cassader, M.; Paschetta, E.; Gambino, R. Bioactive lipid species and metabolic pathways in progression and resolution of nonalcoholic steatohepatitis. Gastroenterology 2018, 155, 282–302.e288. [Google Scholar] [CrossRef]
- Li, Y.; Lin, N.; Xu, J.; Lu, Y.; Chen, S.; Pan, C.; Wang, C.; Xu, M.; Zhou, B.; Xu, R. Measurement of serum and hepatic eicosanoids by liquid chromatography tandem-mass spectrometry (LC-MS/MS) in a mouse model of hepatocellular carcinoma (HCC) with delivery of c-Met and activated β-catenin by hepatocyte hydrodynamic injection. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2018, 24, 1670. [Google Scholar] [CrossRef]
- Hardwick, J.P.; Osei-Hyiaman, D.; Wiland, H.; Abdelmegeed, M.A.; Song, B.J. PPAR/RXR Regulation of Fatty Acid Metabolism and Fatty Acid omega-Hydroxylase (CYP4) Isozymes: Implications for Prevention of Lipotoxicity in Fatty Liver Disease. PPAR Res. 2009, 2009, 952734. [Google Scholar] [CrossRef]
- de la Cruz-Ojeda, P.; Flores-Campos, R.; Dios-Barbeito, S.; Navarro-Villarán, E.; Muntané, J. Role of Nitric Oxide in Gene Expression Regulation during Cancer: Epigenetic Modifications and Non-Coding RNAs. Int. J. Mol. Sci. 2021, 22, 6264. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, Y.; Hu, X.; Qin, Y.; Zhong, W.; Meng, J.; Xiao, T.; Zhang, C.; Li, M.; Chen, S.; et al. Thymidine phosphorylase promotes metastasis and serves as a marker of poor prognosis in hepatocellular carcinoma. Lab. Investig. 2017, 97, 903–912. [Google Scholar] [CrossRef] [PubMed]
- Da Li, F.-F.B.; Chen, N.-N.; Cao, J.-M.; Sun, W.-P.; Zhou, Y.-M.; Cao, C.; Li, C.-Y.; Yang, Q. Epigenetic repression of phosphatidylethanolamine N-methyltransferase (PEMT) in BRCA1-mutated breast cancer. Oncotarget 2014, 5, 1315. [Google Scholar]
- European Association For The Study Of The Liver. EASL clinical practice guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.-C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Kleiner, D.; Makhlouf, H.; Program, D.; Investigation, P.; Branch, R.; Shams, A. Histology of NAFLD and NASH in adults and children. Clin. Liver Dis. 2017, 20, 293–312. [Google Scholar] [CrossRef]
- Group, F.M.C.S.; Bedossa, P. Intraobserver and interobserver variations in liver biopsy interpretation in patients with chronic hepatitis C. Hepatology 1994, 20, 15–20. [Google Scholar]
- Altman, D.G.; McShane, L.M.; Sauerbrei, W.; Taube, S.E. Reporting recommendations for tumor marker prognostic studies (REMARK): Explanation and elaboration. BMC Med. 2012, 10, 1–39. [Google Scholar] [CrossRef]
- Huang, X.-J.; Choi, Y.-K.; Im, H.-S.; Yarimaga, O.; Yoon, E.; Kim, H.-S. Aspartate aminotransferase (AST/GOT) and alanine aminotransferase (ALT/GPT) detection techniques. Sensors 2006, 6, 756–782. [Google Scholar] [CrossRef]
- Pagana, K.D.; Pagana, T.J.; Pagana, T.N. Mosby’s® Diagnostic and Laboratory Test Reference-E-Book; Elsevier Health Sciences: Amsterdam, The Netherlands, 2022. [Google Scholar]
- Verrijken, A.; Francque, S.; Mertens, I.; Prawitt, J.; Caron, S.; Hubens, G.; Van Marck, E.; Staels, B.; Michielsen, P.; Van Gaal, L. Prothrombotic factors in histologically proven nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology 2014, 59, 121–129. [Google Scholar] [CrossRef]
- Matthews, D.R.; Hosker, J.P.; Rudenski, A.S.; Naylor, B.A.; Treacher, D.F.; Turner, R.C. Homeostasis model assessment: Insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985, 28, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Cajka, T.; Fiehn, O. Toward merging untargeted and targeted methods in mass spectrometry-based metabolomics and lipidomics. Anal. Chem. 2016, 88, 524–545. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Wu, H.; Tjeerdema, R.S.; Viant, M.R. Evaluation of metabolite extraction strategies from tissue samples using NMR metabolomics. Metabolomics 2007, 3, 55–67. [Google Scholar] [CrossRef]
- Aldana, J.; Romero-Otero, A.; Cala, M.P. Exploring the lipidome: Current lipid extraction techniques for mass spectrometry analysis. Metabolites 2020, 10, 231. [Google Scholar] [CrossRef] [PubMed]
- Menegollo, M.; Tessari, I.; Bubacco, L.; Szabadkai, G. Determination of ATP, ADP, and AMP levels by reversed-phase high-performance liquid chromatography in cultured cells. In Calcium Signalling; Springer: Berlin/Heidelberg, Germany, 2019; pp. 223–232. [Google Scholar]
- Bell, E.C.; John, M.; Hughes, R.J.; Pham, T. Ultra-performance liquid chromatographic determination of tocopherols and retinol in human plasma. J. Chromatogr. Sci. 2014, 52, 1065–1070. [Google Scholar] [CrossRef][Green Version]
- Zhang, Y.; Bilbao, A.; Bruderer, T.; Luban, J.; Strambio-De-Castillia, C.; Lisacek, F.d.r.; Hopfgartner, G.r.; Varesio, E. The use of variable Q1 isolation windows improves selectivity in LC–SWATH–MS acquisition. J. Proteome Res. 2015, 14, 4359–4371. [Google Scholar] [CrossRef]
- Bonner, R.; Hopfgartner, G. SWATH acquisition mode for drug metabolism and metabolomics investigations. Bioanalysis 2016, 8, 1735–1750. [Google Scholar] [CrossRef]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 2015, 12, 523–526. [Google Scholar] [CrossRef]
- Overmyer, K.A.; Shishkova, E.; Miller, I.J.; Balnis, J.; Bernstein, M.N.; Peters-Clarke, T.M.; Meyer, J.G.; Quan, Q.; Muehlbauer, L.K.; Trujillo, E.A. Large-scale multi-omic analysis of COVID-19 severity. Cell Syst. 2021, 12, 23–40.e27. [Google Scholar] [CrossRef]
- Gromski, P.S.; Xu, Y.; Kotze, H.L.; Correa, E.; Ellis, D.I.; Armitage, E.G.; Turner, M.L.; Goodacre, R. Influence of missing values substitutes on multivariate analysis of metabolomics data. Metabolites 2014, 4, 433–452. [Google Scholar] [CrossRef]
- Li, B.; Tang, J.; Yang, Q.; Cui, X.; Li, S.; Chen, S.; Cao, Q.; Xue, W.; Chen, N.; Zhu, F. Performance evaluation and online realization of data-driven normalization methods used in LC/MS based untargeted metabolomics analysis. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Mondul, A.M.; Weinstein, S.J.; Koutros, S.; Derkach, A.; Karoly, E.; Sampson, J.N.; Moore, S.C.; Berndt, S.I.; Albanes, D. Serum metabolomic profiling of prostate cancer risk in the prostate, lung, colorectal, and ovarian cancer screening trial. Br. J. Cancer 2016, 115, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Aredo, J.V.; Purington, N.; Su, L.; Luo, S.J.; Diao, N.; Christiani, D.C.; Wakelee, H.A.; Han, S.S. Metabolomic profiling for second primary lung cancer: A pilot case-control study. Lung Cancer 2021, 155, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Henglin, M.; Claggett, B.L.; Antonelli, J.; Alotaibi, M.; Magalang, G.A.; Watrous, J.D.; Lagerborg, K.A.; Ovsak, G.; Musso, G.; Demler, O.V. Quantitative Comparison of Statistical Methods for Analyzing Human Metabolomics Data. Metabolites 2022, 12, 519. [Google Scholar] [CrossRef]
- Chong, J.; Xia, J. MetaboAnalystR: An R package for flexible and reproducible analysis of metabolomics data. Bioinformatics 2018, 34, 4313–4314. [Google Scholar] [CrossRef]
- Cavill, R.; Kamburov, A.; Ellis, J.K.; Athersuch, T.J.; Blagrove, M.S.; Herwig, R.; Ebbels, T.M.; Keun, H.C. Consensus-phenotype integration of transcriptomic and metabolomic data implies a role for metabolism in the chemosensitivity of tumour cells. PLoS Comput. Biol. 2011, 7, e1001113. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | NASH | NASH-HCC | p Value |
|---|---|---|---|
| Age, median (25th–75th), (years) | 62 (56.5–67.5) | 63 (58–66) | |
| Gender distribution [n (%)] | |||
| Male | 24 (~77) | 25 (~83) | |
| Female | 7 (~23) | 5 (~16) | |
| BMI, Median (25th–75th), kg/m2 | 30 (28–33.4) | 28.5 (25.7–32) | |
| a TAG, Median (25th–75th), mg/dL | 185 (143–231) | 169 (155–189) | |
| b FBG, Median (25th–75th), mg/dL | 98 (88–104) | 106 (93–140) | |
| Insulin, Median (25th–75th), µU/mL | 12 (10.2–13.6) | 10.9 (7.8–13.1) | |
| c HOMA-IR, median (25th–75th) | 3 (2.4–3.5) | 2.9 (1.9–3.5) | |
| d AST, median (25th–75th), U/L | 55 (44.5–65.5) | 67 (59.7–67.7) | 0.034 |
| e ALT, median (25th–75th), U/L | 37 (26–54.5) | 45 (35.5–55.5) | |
| Albumin, median (25th–75th), g/dL | 3.7 (3.4–3.9) | 3.78 (2.9–3.8) | |
| Bilirubin, Median (25th–75th), mg/dL | 1 (0.9–1.7) | 1.1 (0.9–1.5) | |
| Hemoglobin, median (25th–75th), g/dL | 11.3 (10.5–13.2) | 12 (11.3–13.3) | |
| Platelets, median (25th–75th), 109/L | 178 (115–212) | 107 (62–194) | 0.025 |
| f TLC, median (25th–75th), 109/µL | 6.5 (5.2–8) | 6 (4.4–6) | 0.041 |
| Prothrombin, Median (25th–75th), % | 88 (85–95) | 70.9 (66.4–84) | 0.001 |
| g AFP, Median (25th–75th), ng/mL | 9 (5.7–12) | 312 (5.1–882) | 0.033 |
| h CEA, Mean ± SD, ng/mL | 2.93 ± 0.1 | 2.94 ± 0.98 | |
| i CA 19-9, Mean ± SD, U/mL | 31.1 ± 3.6 | 43.2 ± 4.3 |
| Metabolite | p Ajusted | Log 2 (Fold Change) | Regulation |
|---|---|---|---|
| Linoleic acid | 0.096819 | −2.06 | Down |
| Indole-3-propionic acid | 0.008281 | −1.59 | Down |
| (D/L)-leucine | 0.001103 | −1.46 | Down |
| 8(R)-Hydroperoxylinoleic acid | 0.096819 | 0.61 | Up |
| LysoPC 22:6/0:0 | 0.099088 | 0.91 | Up |
| N-ethyl arachidonoyl amine | 0.081234 | 1.38 | Up |
| Cortisol | 0.096819 | 1.38 | Up |
| LysoPC O-15:1 | 0.026366 | 1.41 | Up |
| 13-HODE | 0.052795 | 1.48 | Up |
| 9,10-DiHOME | 0.004044 | 1.52 | Up |
| Metabolite | p Value | Log 2 (Fold Change) | Regulation | |
|---|---|---|---|---|
| Targeted Non-quantified | 17:0 Lyso PE-d5 | 0.016402 | −0.7244 | Down |
| 19:0 Lyso PE-d5 | 0.014073 | −0.67317 | Down | |
| 18:1 SM (d18:1/18:1)-d9 | 0.037147 | −1.0716 | Down | |
| 20:1 SM (d18:1/20:1)-d9 | 1.92 × 10−6 | −2.3629 | Down | |
| 22:1 SM (d18:1/22:1)-d9 | 1.41 × 10−10 | −2.5234 | Down | |
| Targeted quantified | Ergocalciferol | 0.018409 | −1.1810 | Down |
| alpha-Tocopherol | 0.015821 | −0.9796 | Down | |
| Folic acid | 0.040606 | −1.0113 | Down | |
| Pantothenic acid | 0.043206 | −0.7311 | Down | |
| Phylloquinone | 0.006148 | 1.0917 | Up |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, E.A.; El-Derany, M.O.; Anwar, A.M.; Saied, E.M.; Magdeldin, S. Metabolomics and Lipidomics Screening Reveal Reprogrammed Signaling Pathways toward Cancer Development in Non-Alcoholic Steatohepatitis. Int. J. Mol. Sci. 2023, 24, 210. https://doi.org/10.3390/ijms24010210
Ahmed EA, El-Derany MO, Anwar AM, Saied EM, Magdeldin S. Metabolomics and Lipidomics Screening Reveal Reprogrammed Signaling Pathways toward Cancer Development in Non-Alcoholic Steatohepatitis. International Journal of Molecular Sciences. 2023; 24(1):210. https://doi.org/10.3390/ijms24010210
Chicago/Turabian StyleAhmed, Eman A., Marwa O. El-Derany, Ali Mostafa Anwar, Essa M. Saied, and Sameh Magdeldin. 2023. "Metabolomics and Lipidomics Screening Reveal Reprogrammed Signaling Pathways toward Cancer Development in Non-Alcoholic Steatohepatitis" International Journal of Molecular Sciences 24, no. 1: 210. https://doi.org/10.3390/ijms24010210
APA StyleAhmed, E. A., El-Derany, M. O., Anwar, A. M., Saied, E. M., & Magdeldin, S. (2023). Metabolomics and Lipidomics Screening Reveal Reprogrammed Signaling Pathways toward Cancer Development in Non-Alcoholic Steatohepatitis. International Journal of Molecular Sciences, 24(1), 210. https://doi.org/10.3390/ijms24010210