
Dilated-Left Ventricular Non-Compaction Cardiomyopathy in a Pediatric Case with SPEG Compound Heterozygous Variants

 , ,
, , 
Abstract
:1. Introduction
2. Results
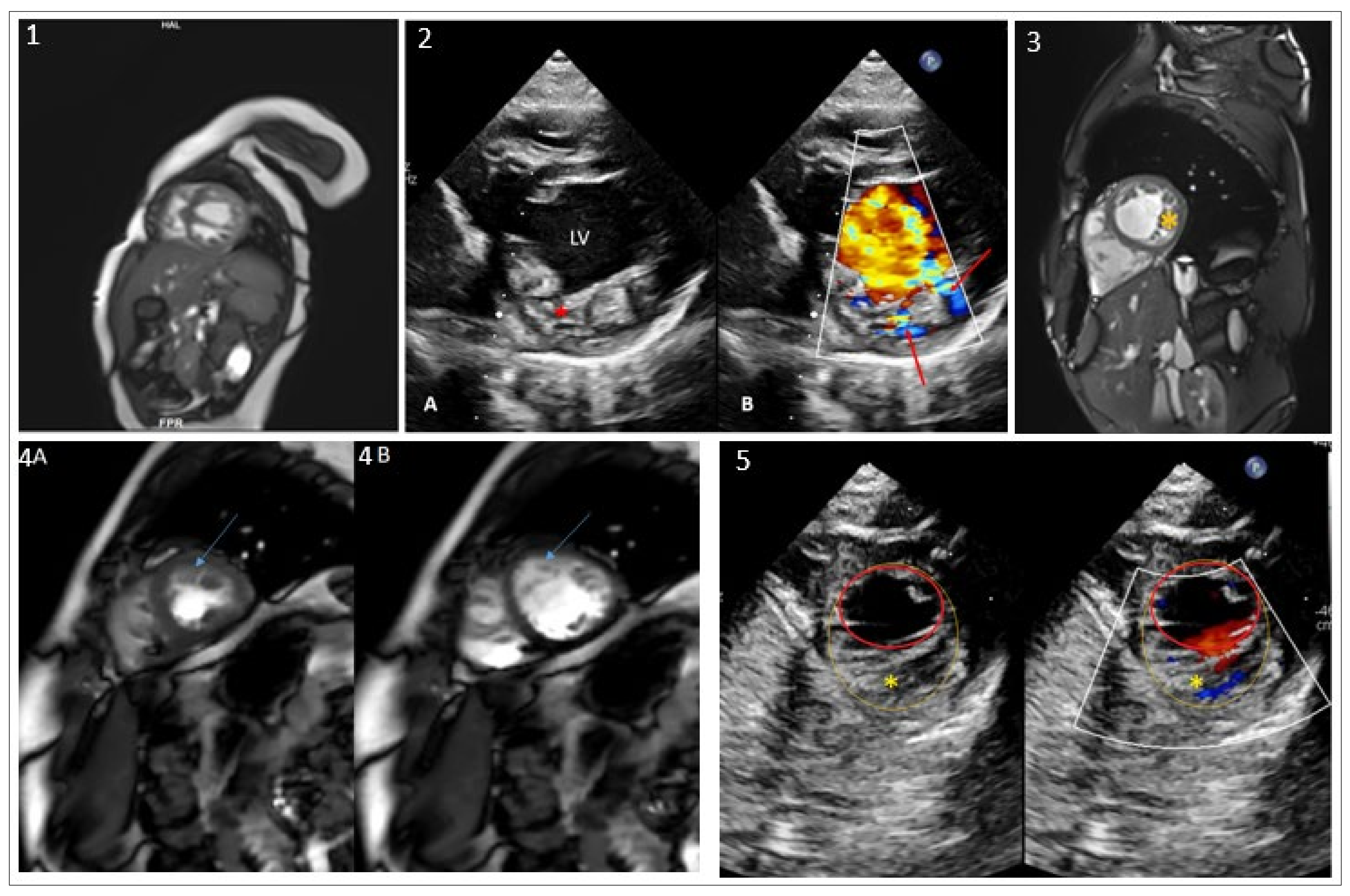
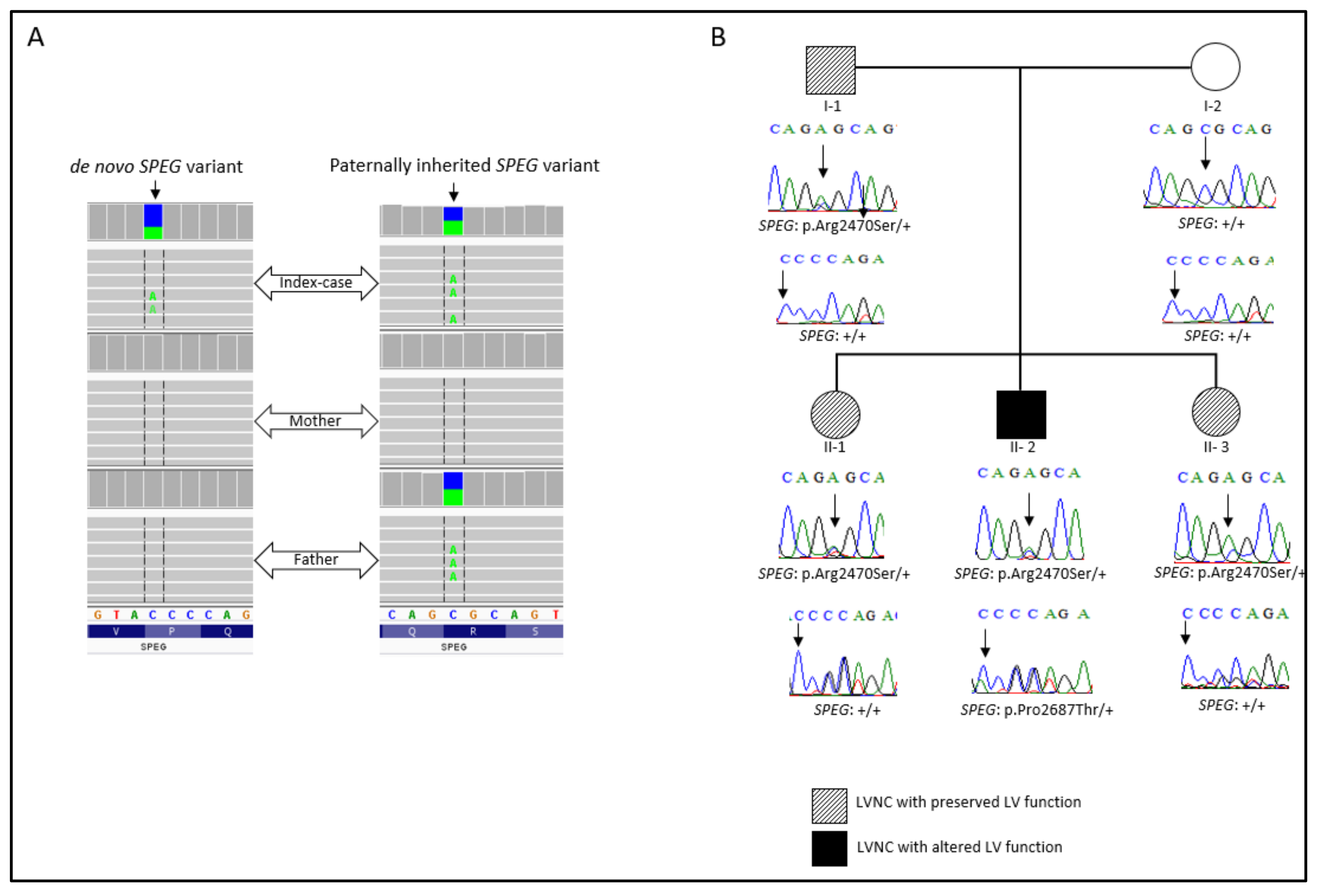
2.1. Clinical Presentation and WES Findings
2.2. In Silico Analysis
3. Discussion
4. Materials and Methods
4.1. Whole-Exome Sequencing (WES)
4.2. In Silico Analysis Tools
4.2.1. Variant Prioritization
4.2.2. Residue Conservation Analysis
4.2.3. Protein Physical Properties, Stability, and Interaction Assessment
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lorca, R.; Martín, M.; Pascual, I.; Astudillo, A.; Molina, B.D.; Cigarrán, H.; Cuesta-Llavona, E.; Avanzas, P.; Reguero, J.R.; Coto, E.; et al. Characterization of Left Ventricular Non-Compaction Cardiomyopathy. J. Clin. Med. 2020, 9, 2524. [Google Scholar] [CrossRef] [PubMed]
- Towbin, J.A.; Lorts, A.; Jefferies, J.L. Left ventricular non-compaction cardiomyopathy. Lancet 2015, 386, 813–825. [Google Scholar] [CrossRef]
- Di Toro, A.; Giuliani, L.; Smirnova, A.; Favalli, V.; Serio, A.; Urtis, M.; Grasso, M.; Arbustini, E. Myths to debunk: The non-compacted myocardium. Eur. Heart J. Suppl. 2020, 22 (Suppl. L), L6–L10. [Google Scholar] [CrossRef] [PubMed]
- Petersen, S.E.; Selvanayagam, J.B.; Wiesmann, F.; Robson, M.D.; Francis, J.M.; Anderson, R.H.; Watkins, H.; Neubauer, S. Left ven-tricular non-compaction: Insights from cardiovascular magnetic resonance imaging. J. Am. Coll. Cardiol. 2005, 46, 101–105. [Google Scholar]
- Nugent, A.W.; Daubeney, P.E.; Chondros, P.; Carlin, J.; Cheung, M.; Wilkinson, L.C.; Davis, A.; Kahler, S.G.; Chow, C.; Wilkinson, J.L.; et al. The Epidemiology of Childhood Cardiomyopathy in Australia. N. Engl. J. Med. 2003, 348, 1639–1646. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.Y.; Moreno-Betancur, M.; Nugent, A.W.; Cheung, M.; Colan, S.; Turner, C.; Sholler, G.F.; Robertson, T.; Justo, R.; Bullock, A.; et al. Long-Term Outcomes of Childhood Left Ventricular Noncompaction Cardiomyopathy: Results from a National Population-Based Study. Circulation 2018, 138, 367–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jefferies, J.L.; Wilkinson, J.D.; Sleeper, L.A.; Colan, S.D.; Lu, M.; Pahl, E.; Kantor, P.F.; Everitt, M.D.; Webber, S.A.; Kaufman, B.D.; et al. Cardiomyopathy phenotypes and outcomes for children with left ventricular myocardial noncompaction: Results from the pediatric cardiomyopathy registry. J. Card. Fail. 2015, 21, 877–884. [Google Scholar] [CrossRef] [Green Version]
- Ichida, F. Left ventricular noncompaction—Risk stratification and genetic consideration. J. Cardiol. 2020, 75, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Takasaki, A.; Ozawa, S.W.; Nakaoka, H.; Okabe, M.; Miyao, N.; Saito, K.; Ibuki, K.; Hirono, K.; Yoshimura, N.; et al. Long-Term Prognosis of Patients with Left Ventricular Noncompaction—Comparison Between Infantile and Juvenile Types. Circ. J. 2017, 81, 694–700. [Google Scholar] [CrossRef] [Green Version]
- Ross, S.B.; Singer, E.S.; Driscoll, E.; Nowak, N.; Yeates, L.; Puranik, R.; Sy, R.W.; Rajagopalan, S.; Barratt, A.; Ingles, J.; et al. Genetic architecture of left ventricular noncompaction in adults. Hum. Genome Var. 2020, 7, 33. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Lindenfeld, J.; Mestroni, L.; Seidman, C.E.; Taylor, M.R.; Towbin, J.A. Genetic Evaluation of Cardiomyopathy—A Heart Failure Society of America Practice Guideline. J. Card. Fail. 2009, 15, 83–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005, 33, W306–W310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, S.; Rosen, S.; Li, Q.; Agrawal, P. Striated Preferentially Expressed Protein Kinase (SPEG) in Muscle Development, Function, and Disease. Int. J. Mol. Sci. 2021, 22, 5732. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ramjiganesh, T.; Chen, Y.-H.; Chung, S.W.; Hall, S.R.; Schissel, S.L.; Padera, R.F., Jr.; Liao, R.; Ackerman, K.G.; Kajstura, J.; et al. Disruption of Striated Preferentially Expressed Gene Locus Leads to Dilated Cardiomyopathy in Mice. Circulation 2009, 119, 261–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, J.L.; Triantaphyllopoulos, K.; Todd, H.; Raguz, S.; de Wit, T.; Morgan, J.E.; Partridge, T.A.; Makrinou, E.; Grosveld, F.; Antoniou, M. The human desmin locus: Gene organization and LCR-mediated transcriptional control. Genomics 2006, 87, 733–746. [Google Scholar] [CrossRef] [Green Version]
- Goebel, H.H. Desmin-related myopathies. Curr. Opin. Neurol. 1997, 10, 426–429. [Google Scholar]
- Huang, Y.-S.; Xing, Y.-L.; Li, H.-W. Heterozygous desmin gene (DES) mutation contributes to familial dilated cardiomyopathy. J. Int. Med. Res. 2021, 49, 03000605211006598. [Google Scholar] [CrossRef]
- Klauke, B.; Kossmann, S.; Gaertner, A.; Brand, K.; Stork, I.; Brodehl, A.; Dieding, M.; Walhorn, V.; Anselmetti, D.; Gerdes, D.; et al. De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Hum. Mol. Genet. 2010, 19, 4595–4607. [Google Scholar] [CrossRef] [Green Version]
- Lorenzon, A.; Beffagna, G.; Bauce, B.; De Bortoli, M.; Mura, I.E.L.; Calore, M.; Dazzo, E.; Basso, C.; Nava, A.; Thiene, G.; et al. Desmin Mutations and Arrhythmogenic Right Ventricular Cardiomyopathy. Am. J. Cardiol. 2013, 111, 400–405. [Google Scholar] [CrossRef]
- Tamiya, R.; Saito, Y.; Fukamachi, D.; Nagashima, K.; Aizawa, Y.; Ohkubo, K.; Hatta, T.; Sezai, A.; Tanaka, M.; Ishikawa, T.; et al. Desmin-related myopathy characterized by non-compaction cardiomyopathy, cardiac conduction defect, and coronary artery dissection. ESC Heart Fail. 2020, 7, 1338–1343. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, P.B.; Pierson, C.R.; Joshi, M.; Liu, X.; Ravenscroft, G.; Moghadaszadeh, B.; Talabere, T.; Viola, M.; Swanson, L.C.; Haliloğlu, G.; et al. SPEG Interacts with Myotubularin, and Its Deficiency Causes Centronuclear Myopathy with Dilated Cardiomyopathy. Am. J. Hum. Genet. 2014, 95, 218–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.; Ma, W.; Chen, Y.; Jiang, R.; Zeng, Q.; Tan, J.; Jiang, H.; Li, Q.; Zhang, V.W.; Wang, J.; et al. Novel SPEG variant cause centronuclear myopathy in China. J. Clin. Lab. Anal. 2020, 34, e23054. [Google Scholar] [CrossRef] [PubMed]
- Lornage, X.; Sabouraud, P.; Lannes, B.; Gaillard, D.; Schneider, R.; Deleuze, J.-F.; Boland, A.; Thompson, J.; Böhm, J.; Biancalana, V.; et al. Novel SPEG Mutations in Congenital Myopathy without Centralized Nuclei. J. Neuromuscul. Dis. 2018, 5, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Almannai, M.; Luo, S.; Faqeih, E.; Al Mutairi, F.; Li, Q.; Agrawal, P.B. Homozygous SPEG Mutation Is Associated with Isolated Dilated Cardiomyopathy. Circ. Genom. Precis. Med. 2021, 14, e003310. [Google Scholar] [CrossRef]
- Levitas, A.; Muhammad, E.; Zhang, Y.; Perea Gil, I.; Serrano, R.; Diaz, N.; Arafat, M.; Gavidia, A.A.; Kapiloff, M.S.; Mercola, M.; et al. A novel recessive mutation in SPEG causes early onset dilated cardiomyopathy. PLoS Genet. 2020, 16, e1009000. [Google Scholar] [CrossRef]
- Wang, H.; Schänzer, A.; Kampschulte, B.; Daimagüler, H.-S.; Logeswaran, T.; Schlierbach, H.; Petzinger, J.; Ehrhardt, H.; Hahn, A.; Cirak, S. A novel SPEG mutation causes non-compaction cardiomyopathy and neuropathy in a floppy infant with centronuclear myopathy. Acta Neuropathol. Commun. 2018, 6, 83. [Google Scholar] [CrossRef]
- Quan, C.; Li, M.; Du, Q.; Chen, Q.; Wang, H.; Campbell, D.; Fang, L.; Xue, B.; MacKintosh, C.; Gao, X.; et al. SPEG Controls Calcium Reuptake into the Sarcoplasmic Reticulum Through Regulating SERCA2a by Its Second Kinase-Domain. Circ. Res. 2019, 124, 712–726. [Google Scholar] [CrossRef] [Green Version]
- Campbell, H.; Aguilar-Sanchez, Y.; Quick, A.P.; Dobrev, D.; Wehrens, X.H. SPEG: A key regulator of cardiac calcium homeostasis. Cardiovasc. Res. 2020, 117, 2175–2185. [Google Scholar] [CrossRef] [PubMed]
- Sedaghat-Hamedani, F.; Haas, J.; Zhu, F.; Geier, C.; Kayvanpour, E.; Liss, M.; Lai, A.; Frese, K.; Pribe-Wolferts, R.; Amr, A.; et al. Clinical genetics and outcome of left ventricular non-compaction cardiomyopathy. Eur. Heart J. 2017, 38, 3449–3460. [Google Scholar]
- Nozaki, Y.; Kato, Y.; Uike, K.; Yamamura, K.; Kikuchi, M.; Yasuda, M.; Ohno, S.; Horie, M.; Murayama, T.; Kurebayashi, N.; et al. Co-Phenotype of Left Ventricular Non-Compaction Cardiomyopathy and Atypical Catecholaminergic Polymorphic Ventricular Tachycardia in Association with R169Q, a Ryanodine Receptor Type 2 Missense Mutation. Circ. J. 2020, 84, 226–234. [Google Scholar] [CrossRef] [Green Version]
- Quick, A.P.; Wang, Q.; Philippen, L.E.; Barreto-Torres, G.; Chiang, D.; Beavers, D.; Wang, G.; Khalid, M.; Reynolds, J.O.; Campbell, H.M.; et al. SPEG (Striated Muscle Preferentially Expressed Protein Kinase) Is Essential for Cardiac Function by Regulating Junctional Membrane Complex Activity. Circ. Res. 2017, 120, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Qualls, A.; Donkervoort, S.; Herkert, J.C.; D’Gama, A.M.; Bharucha-Goebel, D.; Collins, J.; Bs, K.R.C.; Foley, A.R.; Schoots, M.H.; Jongbloed, J.D.; et al. Novel SPEG mutations in congenital myopathies: Genotype–phenotype correlations. Muscle Nerve 2019, 59, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Castiglioni, C.; Bayram, A.K.; Fattori, F.; Pekuz, S.; Araneda, D.; Per, H.; Erazo, R.; Gümüş, H.; Zorludemir, S.; et al. Insights from genotype–phenotype correlations by novel SPEG mutations causing centronuclear myopathy. Neuromuscul. Disord. 2017, 27, 836–842. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazy, H.; Erez, E.; Martz, E.; Pupko, T.; Ben-Tal, N. ConSurf 2010: Calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010, 38, W529–W533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venselaar, H.; Te Beek, T.A.; Kuipers, R.K.; Hekkelman, M.L.; Vriend, G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinform. 2010, 11, 1–10. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Gene | cDNA_Change | Protein_Change | Cardiac Phenotype | Myopathy | Reference |
|---|---|---|---|---|---|
| SPEG | c.1071_1074dup; c.4399C>T | Lys359Valfs*35; Arg1467* | Reduced myocardial contraction | Present | [23] |
| SPEG | c.2183delT; c.8962_8963insCGGG GCGAACGTTCGTG GCCAAGAT | Leu728Argfs*82; Val2997Glyfs*52 | Sinus tachycardia | Present | [32] |
| SPEG | c.2915_2916delCCinsA; c.8270G>T | Ala972Aspfs*79; Gly2757Val | DCM; Mitral insufficiency | Present | [21] |
| SPEG | c.3709_3715 + 29del36; c.4276C>T | Thr1237Serfs*46; Arg1426* | DCM | Present | [21] |
| SPEG | c. 5038G>A | Glu1680Lys | DCM | Absent | [25] |
| SPEG | c.6697C>T | Gln2233* | NA | Present | [21] |
| SPEG | c.7119C>A | Tyr2373* | LVNC/enlarged atria | Present | [26] |
| SPEG | c.7408C>A; c.8059C>A | Arg2470Ser; Pro2687Thr | DCM-LVNC | Absent | Present study |
| SPEG | c.8710A>G | Thr2904Ala | DCM | Present | [22] |
| SPEG | c.9028_9030delGAG | Glu3010del | DCM | Absent | [24] |
| SPEG | c.9185_9187delTGG | Val3062del | DCM/Severe mitral valve insufficiency | Present | [32] |
| SPEG | c.9586C>T | Arg3196* | Fetal bradycardia, DCM, Mild mitral insufficiency | Mild | [33] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaouadi, H.; El Louali, F.; Wanert, C.; Cano, A.; Ovaert, C.; Zaffran, S. Dilated-Left Ventricular Non-Compaction Cardiomyopathy in a Pediatric Case with SPEG Compound Heterozygous Variants. Int. J. Mol. Sci. 2022, 23, 5205. https://doi.org/10.3390/ijms23095205
Jaouadi H, El Louali F, Wanert C, Cano A, Ovaert C, Zaffran S. Dilated-Left Ventricular Non-Compaction Cardiomyopathy in a Pediatric Case with SPEG Compound Heterozygous Variants. International Journal of Molecular Sciences. 2022; 23(9):5205. https://doi.org/10.3390/ijms23095205
Chicago/Turabian StyleJaouadi, Hager, Fedoua El Louali, Chloé Wanert, Aline Cano, Caroline Ovaert, and Stéphane Zaffran. 2022. "Dilated-Left Ventricular Non-Compaction Cardiomyopathy in a Pediatric Case with SPEG Compound Heterozygous Variants" International Journal of Molecular Sciences 23, no. 9: 5205. https://doi.org/10.3390/ijms23095205
APA StyleJaouadi, H., El Louali, F., Wanert, C., Cano, A., Ovaert, C., & Zaffran, S. (2022). Dilated-Left Ventricular Non-Compaction Cardiomyopathy in a Pediatric Case with SPEG Compound Heterozygous Variants. International Journal of Molecular Sciences, 23(9), 5205. https://doi.org/10.3390/ijms23095205