
Statins Inhibit the Gliosis of MIO-M1, a Müller Glial Cell Line Induced by TRPV4 Activation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
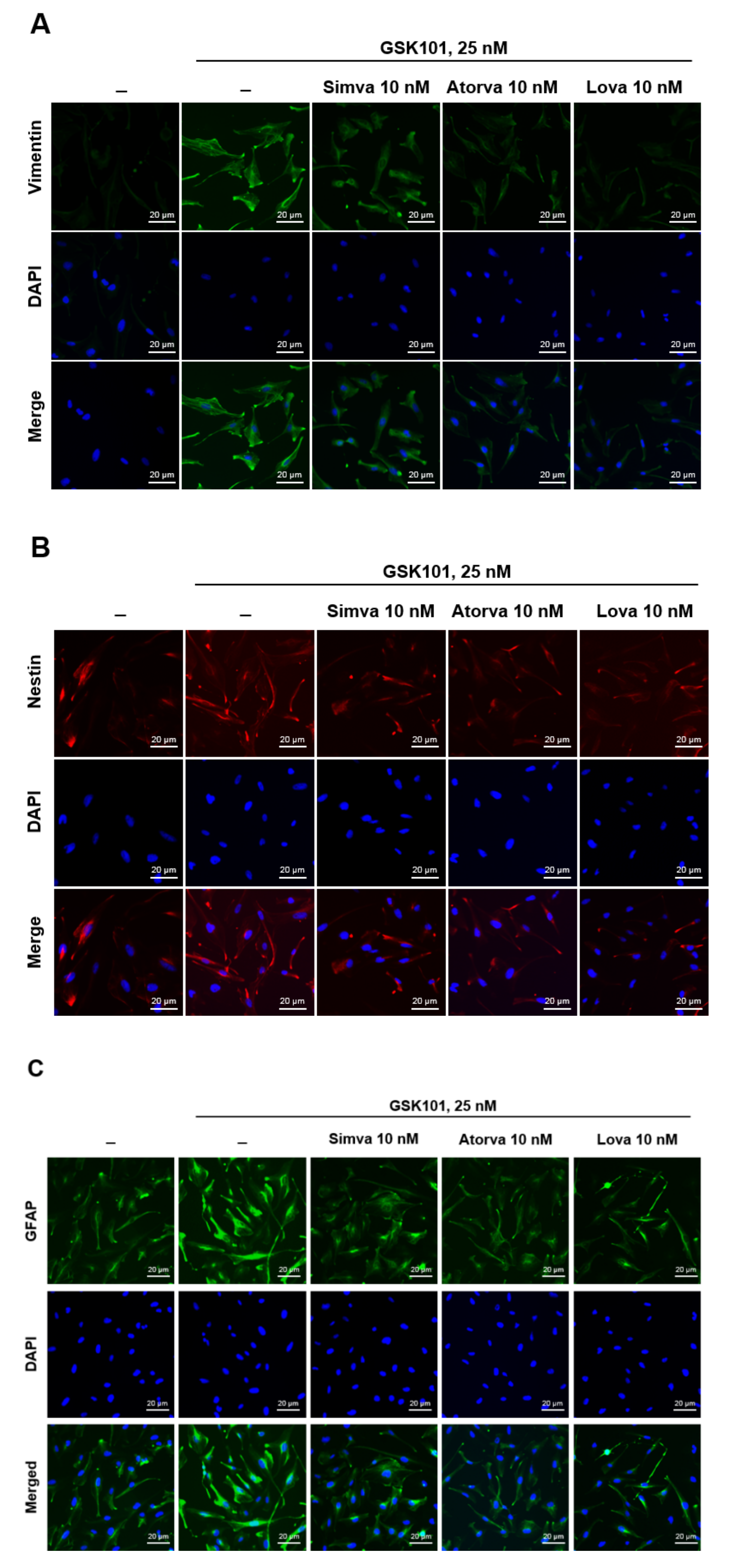
2.1. Evaluation of Müller Gliosis in MIO-M1 Cells and the Effect of Statins
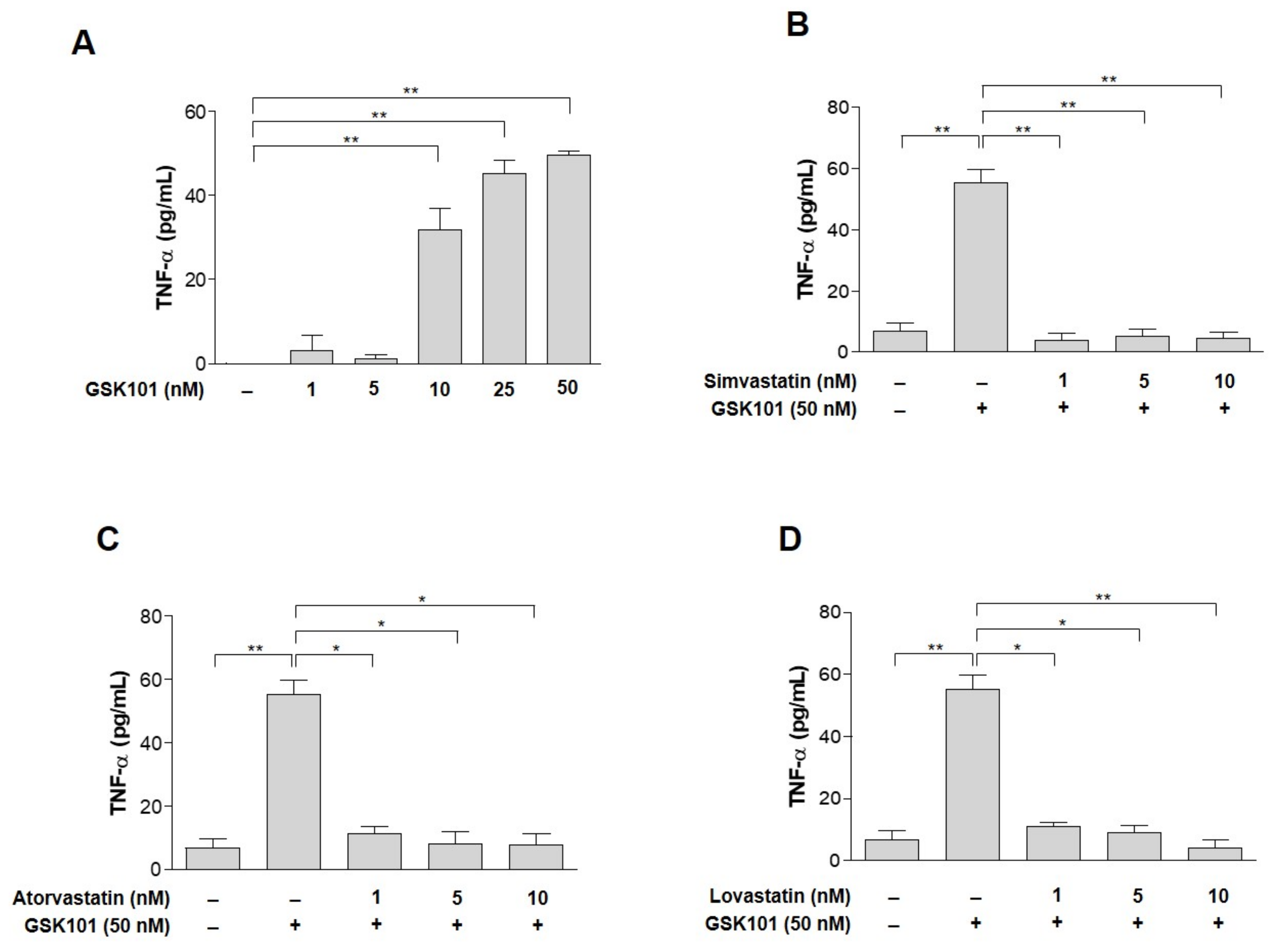
2.2. TNF-α as a Candidate to Induce Gliosis and Its Reduction by Statins
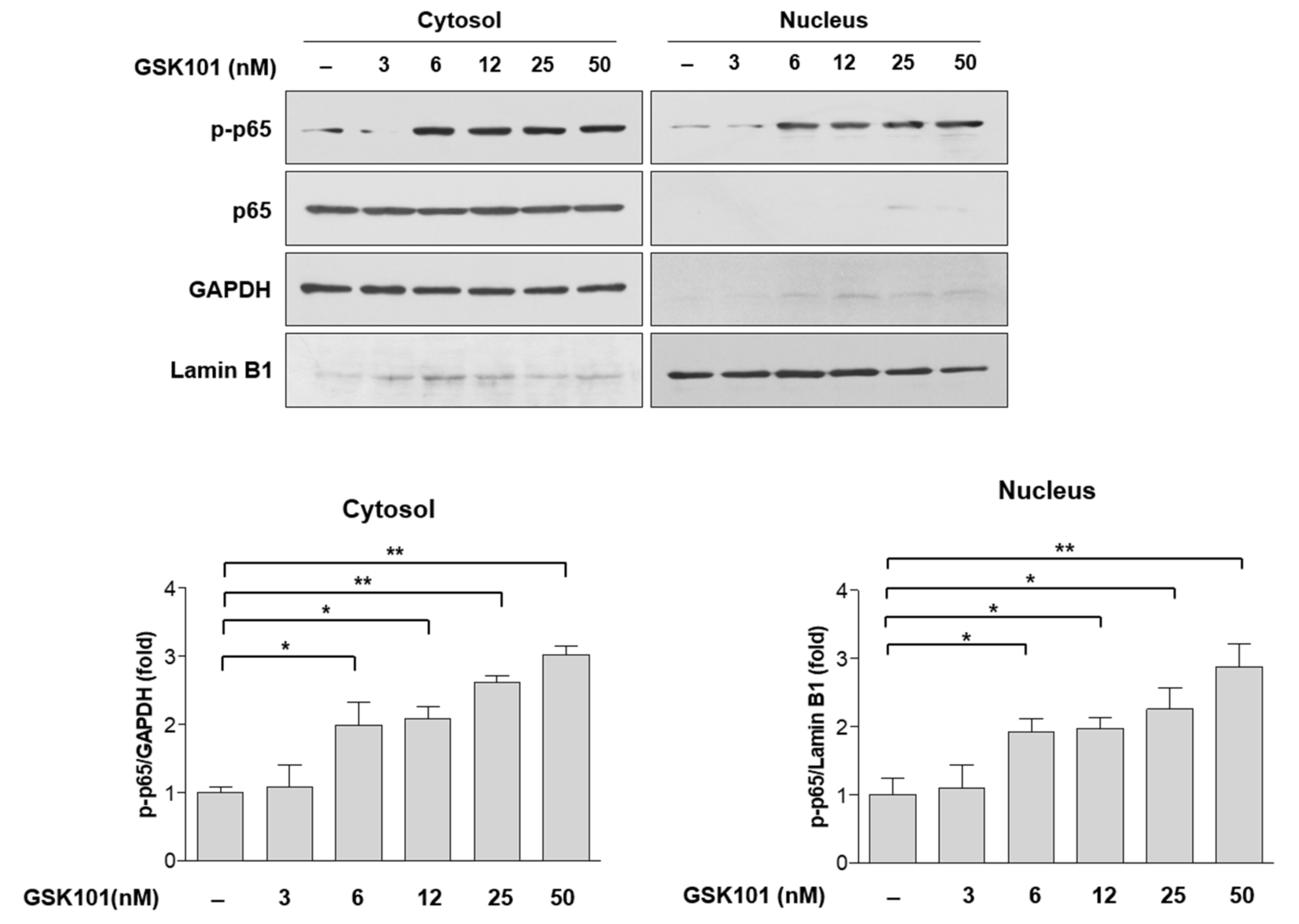
2.3. NF-κB Pathway in MIO-M1 Cells
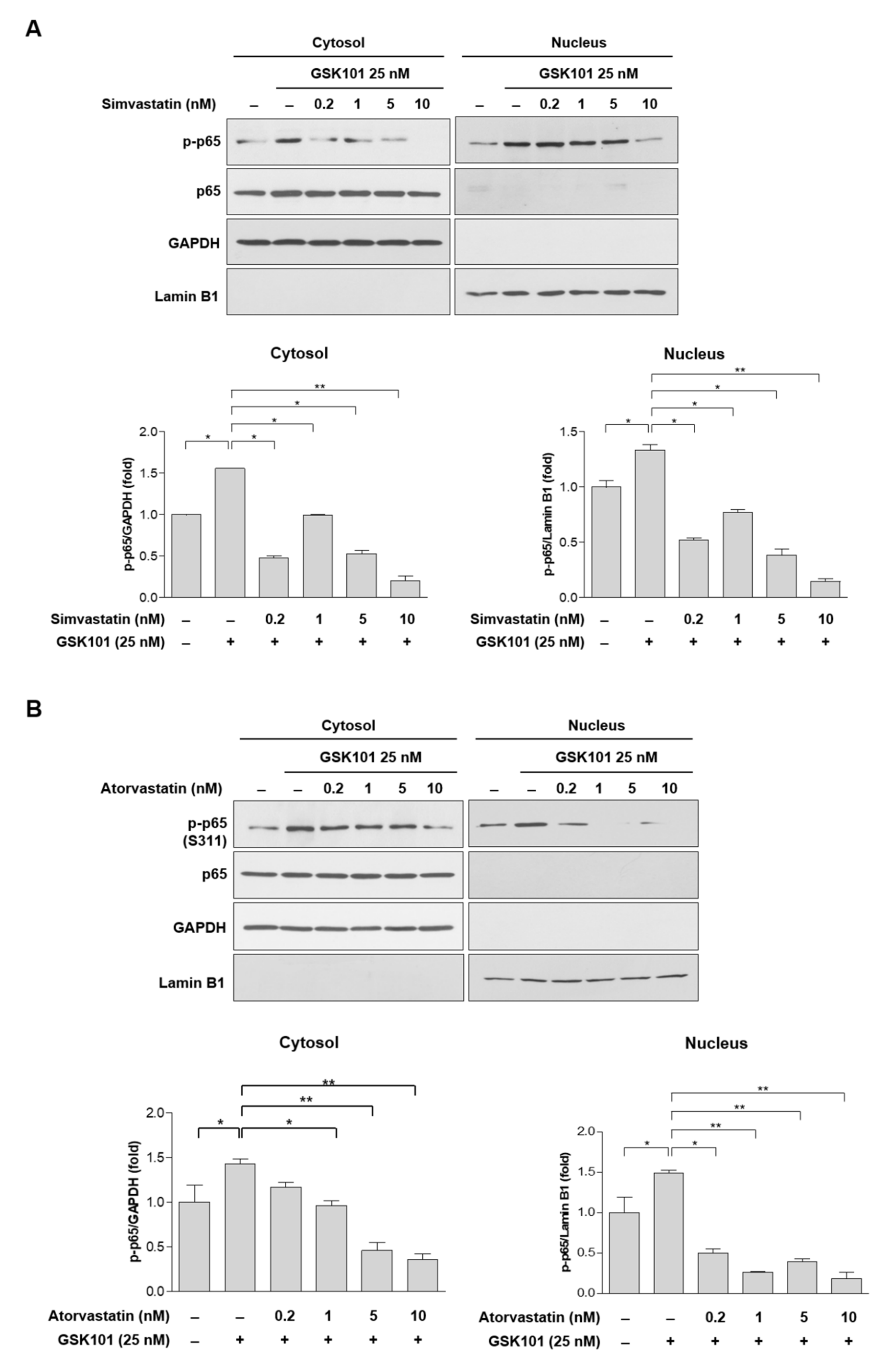
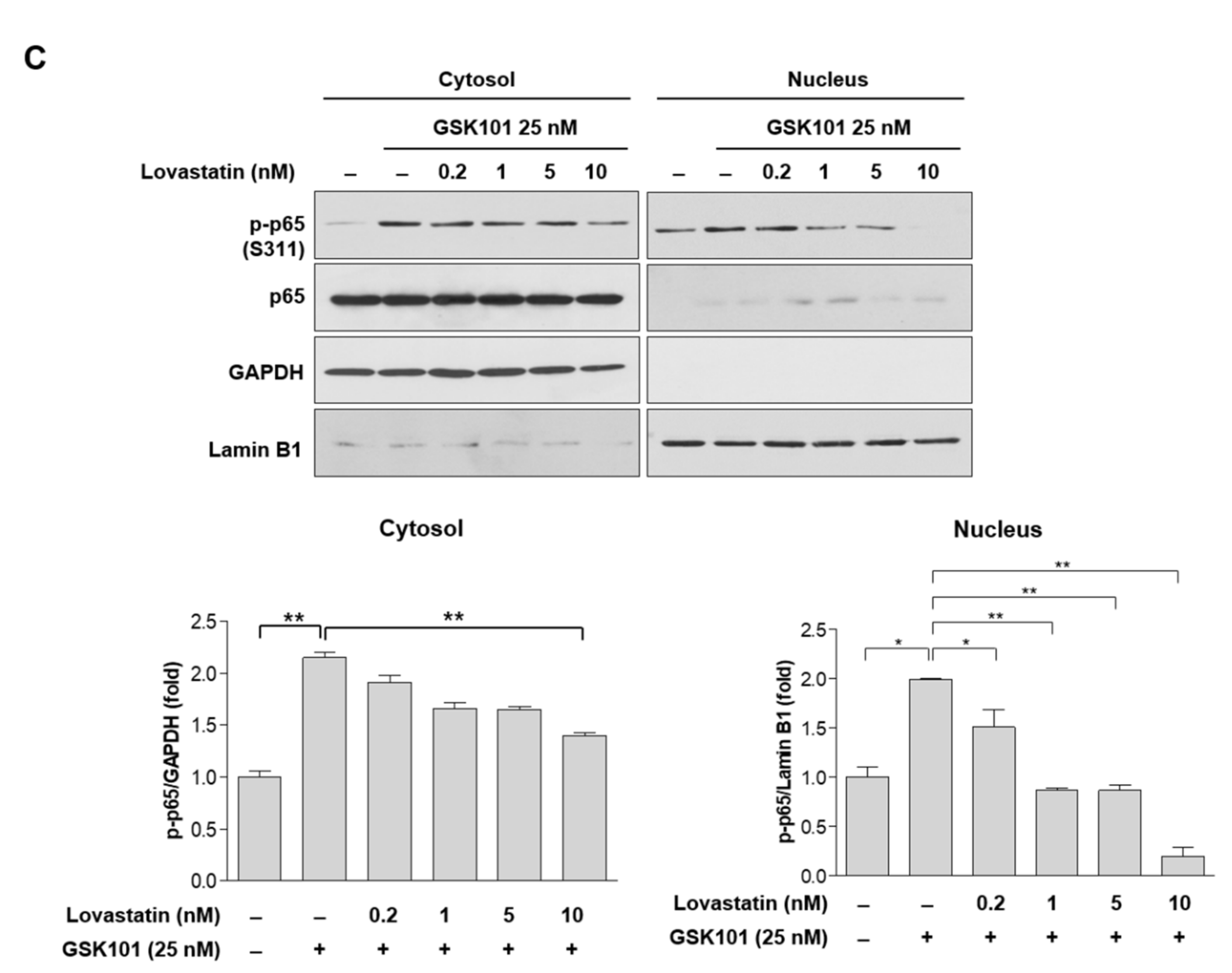
2.4. Statins Inhibit the NF-κB Pathway
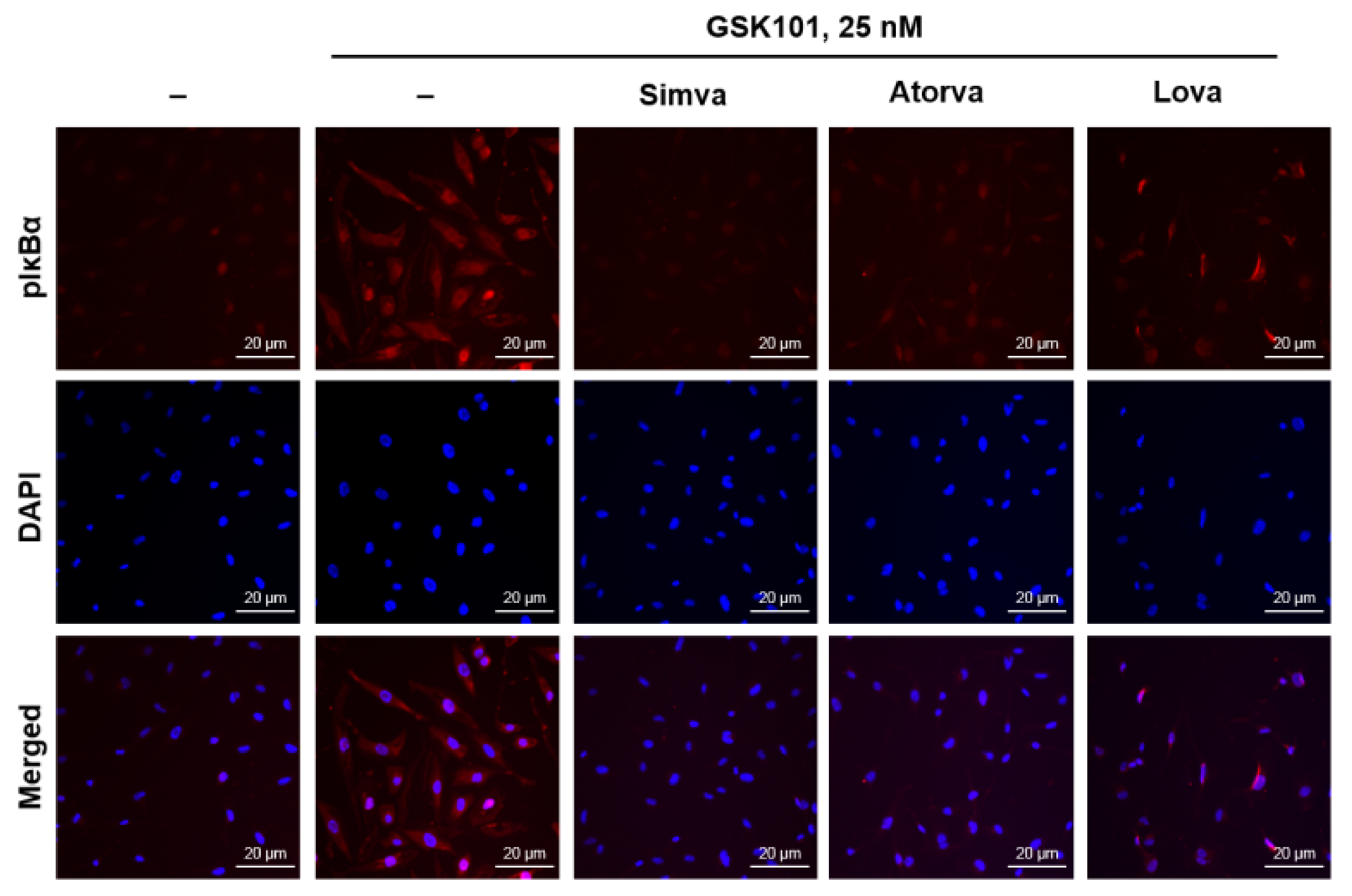
2.5. Statins Reduce IκBα Phosphorylation
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Reagents
4.3. Immunofluorescent Staining of Human Müller Glial (MIO-M1) Cells
4.4. Separation of Nuclei and Cytosol
4.5. Western Blot Analysis
4.6. Quantification of TNF-α
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wilson, A.; Di Polo, A. Gene therapy for retinal ganglion cell neuroprotection in glaucoma. Gene Ther. 2012, 19, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Almasieh, M.; Wilson, A.M.; Morquette, B.; Vargas, J.L.C.; Di Polo, A. The molecular basis of retinal ganglion cell death in glaucoma. Prog. Retin. Eye Res. 2012, 31, 152–181. [Google Scholar] [CrossRef] [PubMed]
- Bringmann, A.; Pannicke, T.; Grosche, J.; Francke, M.; Wiedemann, P.; Skatchkov, S.N.; Osborne, N.N.; Reichenbach, A. Müller cells in the healthy and diseased retina. Prog. Retin. Eye Res. 2006, 25, 397–424. [Google Scholar] [CrossRef] [PubMed]
- Bringmann, A.; Reichenbach, A.J.F.B. Role of Muller cells in retinal degenerations. Front. Biosci. 2001, 6, E72–E92. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.J.; Reh, T.A. Potential of Müller glia to become neurogenic retinal progenitor cells. Glia 2003, 43, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Cho, K.S.; Thee, E.F.; Jager, M.J.; Chen, D.F. Neuroinflammation and microglia in glaucoma: Time for a paradigm shift. J. Neurosci. Res. 2019, 97, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.-L.; Shi, J.-M. The role of microglia in the progression of glaucomatous neurodegeneration—A review. Int. J. Ophthalmol. 2018, 11, 143. [Google Scholar]
- Brian, C.T.; Dvoriantchikova, G.; Tao, W.; Gallo, R.A.; Lee, J.Y.; Pappas, S.; Brambilla, R.; Ivanov, D.; David, T.T.; Pelaez, D. Tumor necrosis factor inhibition in the acute management of traumatic optic neuropathy. Investig. Ophthalmol. Vis. Sci. 2018, 59, 2905–2912. [Google Scholar]
- Fuchs, C.; Forster, V.; Balse, E.; Sahel, J.-A.; Picaud, S.; Tessier, L.-H. Retinal-Cell–Conditioned Medium Prevents TNF-α-Induced Apoptosis of Purified Ganglion Cells. Investig. Ophthalmol. Vis. Sci. 2005, 46, 2983–2991. [Google Scholar] [CrossRef][Green Version]
- Kondkar, A.A.; Sultan, T.; Almobarak, F.A.; Kalantan, H.; Al-Obeidan, S.A.; Abu-Amero, K.K. Association of increased levels of plasma tumor necrosis factor alpha with primary open-angle glaucoma. Clin. Ophthalmol. 2018, 12, 701. [Google Scholar] [CrossRef]
- Liu, S.-T.; Zhong, S.-M.; Li, X.-Y.; Gao, F.; Li, F.; Zhang, M.-L.; Zhu, K.; Sun, X.-H.; Wang, X.; Miao, Y.; et al. EphrinB/EphB forward signaling in Müller cells causes apoptosis of retinal ganglion cells by increasing tumor necrosis factor alpha production in rat experimental glaucomatous model. Acta Neuropathol. Commun. 2018, 6, 111. [Google Scholar] [CrossRef]
- Limb, G.A.; Salt, T.E.; Munro, P.M.; Moss, S.E.; Khaw, P.T. In vitro characterization of a spontaneously immortalized human Müller cell line (MIO-M1). Investig. Ophthalmol. Vis. Sci. 2002, 43, 864–869. [Google Scholar]
- Hollborn, M.; Jahn, K.; Limb, G.A.; Kohen, L.; Wiedemann, P.; Bringmann, A. Characterization of the basic fibroblast growth factor-evoked proliferation of the human Müller cell line, MIO-M1. Graefe’s Arch. Clin. Exp. Ophthalmol. 2004, 242, 414–422. [Google Scholar] [CrossRef]
- Ramírez, C.; Pham, K.; Franco, M.F.E.; Chwa, M.; Limb, A.; Kuppermann, B.D.; Kenney, M.C. Hydroquinone induces oxidative and mitochondrial damage to human retinal Müller cells (MIO-M1). NeuroToxicology 2013, 39, 102–108. [Google Scholar] [CrossRef]
- Baratchi, S.; Keov, P.; Darby, W.G.; Lai, A.; Khoshmanesh, K.; Thurgood, P.; Vahidi, P.; Ejendal, K.; McIntyre, P. The TRPV4 agonist GSK1016790A regulates the membrane expression of TRPV4 channels. Front. Pharmacol. 2019, 10, 6. [Google Scholar] [CrossRef]
- Li, Q.; Cheng, Y.; Zhang, S.; Sun, X.; Wu, J. TRPV4-induced Müller cell gliosis and TNF-α elevation-mediated retinal ganglion cell apoptosis in glaucomatous rats via JAK2/STAT3/NF-κB pathway. J. Neuroinflamm. 2021, 18, 271. [Google Scholar] [CrossRef]
- Okada, M.; Matsumura, M.; Ogino, N.; Honda, Y. Müller cells in detached human retina express glial fibrillary acidic protein and vimentin. Graefe’s Arch. Clin. Exp. Ophthalmol. 1990, 228, 467–474. [Google Scholar] [CrossRef]
- Walcott, J.C.; Provis, J.M. Müller cells express the neuronal progenitor cell marker nestin in both differentiated and undifferentiated human foetal retina. Clin. Exp. Ophthalmol. 2003, 31, 246–249. [Google Scholar] [CrossRef]
- Nork, T.M.; Ghobrial, M.W.; Peyman, G.A.; Tso, M.O. Massive retinal gliosis: A reactive proliferation of Müller cells. Arch. Ophthalmol. 1986, 104, 1383–1389. [Google Scholar] [CrossRef]
- Mizutani, M.; Gerhardinger, C.; Lorenzi, M. Müller cell changes in human diabetic retinopathy. Diabetes 1998, 47, 445–449. [Google Scholar] [CrossRef]
- Hotchkiss, R.S.; Karl, I.E. The pathophysiology and treatment of sepsis. N. Engl. J. Med. 2003, 348, 138–150. [Google Scholar] [CrossRef]
- Arnalich, F.; Garcia-Palomero, E.; López, J.; Jiménez, M.; Madero, R.; Renart, J.; Vázquez, J.J.; Montiel, C. Predictive value of nuclear factor κB activity and plasma cytokine levels in patients with sepsis. Infect. Immun. 2000, 68, 1942–1945. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.K.; Laufs, U. Pleiotropic effects of statins. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 89–118. [Google Scholar] [CrossRef] [PubMed]
- Davignon, J. Beneficial cardiovascular pleiotropic effects of statins. Circulation 2004, 109 (Suppl. 1), III-39–III-43. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Liao, J.K. Pleiotropic effects of statins. Circ. J. 2010, 74, 818–826. [Google Scholar] [CrossRef] [PubMed]
- Schmeer, C.; Kretz, A.; Isenmann, S. Statin-mediated protective effects in the central nervous system: General mechanisms and putative role of stress proteins. Restor. Neurol. Neurosci. 2006, 24, 79–95. [Google Scholar] [PubMed]
- Stein, J.D.; Newman-Casey, P.A.; Talwar, N.; Nan, B.; Richards, J.E.; Musch, D.C. The relationship between statin use and open-angle glaucoma. Ophthalmology 2012, 119, 2074–2081. [Google Scholar] [CrossRef] [PubMed]
- McGwin, G.; McNeal, S.; Owsley, C.; Girkin, C.; Epstein, D.; Lee, P.P. Statins and Other Cholesterol-Lowering Medications and the Presenceof Glaucoma. Arch. Ophthalmol. 2004, 122, 822–826. [Google Scholar] [CrossRef]
- De Castro, D.K.; Punjabi, O.S.; Bostrom, A.G.; Stamper, R.L.; Lietman, T.M.; Ray, K.; Lin, S.C. Effect of statin drugs and aspirin on progression in open-angle glaucoma suspects using confocal scanning laser ophthalmoscopy. Clin. Exp. Ophthalmol. 2007, 35, 506–513. [Google Scholar] [CrossRef]
- Shon, K.; Sung, K.R. Dyslipidemia, Dyslipidemia Treatment, and Open-angle Glaucoma in the Korean National Health and Nutrition Examination Survey. J. Glaucoma 2019, 28, 550–556. [Google Scholar] [CrossRef]
- Marcus, M.W.; Müskens, R.P.; Ramdas, W.D.; Wolfs, R.C.; De Jong, P.T.; Vingerling, J.R.; Hofman, A.; Stricker, B.H.; Jansonius, N.M. Cholesterol-lowering drugs and incident open-angle glaucoma: A population-based cohort study. PLoS ONE 2012, 7, e29724. [Google Scholar] [CrossRef]
- Kang, J.H.; Boumenna, T.; Stein, J.D.; Khawaja, A.; Rosner, B.A.; Wiggs, J.L.; Pasquale, L.R. Association of statin use and high serum cholesterol levels with risk of primary open-angle glaucoma. JAMA Ophthalmol. 2019, 137, 756–765. [Google Scholar] [CrossRef]
- Ooba, N.; Iwahashi, R.; Nogami, A.; Nakayama, T.; Kanno, A.; Tochikura, N.; Ootsuka, S.; Fukuoka, N. Comparison between high and low potency statins in the incidence of open-angle glaucoma: A retrospective cohort study in Japanese working-age population. PLoS ONE 2020, 15, e0237617. [Google Scholar] [CrossRef]
- Chen, H.-Y.; Hsu, S.-Y.; Chang, Y.-C.; Lin, C.-C.; Sung, F.-C.; Chen, W.-C.; Kao, C.-H. Association between statin use and open-angle glaucoma in hyperlipidemia patients: A Taiwanese population-based case-control study. Medicine 2015, 94, e2018. [Google Scholar] [CrossRef]
- Villarreal, G.; Chatterjee, A.; Oh, S.S.; Oh, D.-J.; Rhee, D.J. Pharmacological regulation of SPARC by lovastatin in human trabecular meshwork cells. Investig. Ophthalmol. Vis. Sci. 2014, 55, 1657–1665. [Google Scholar] [CrossRef]
- Kim, M.-L.; Sung, K.R.; Shin, J.A.; Yoon, J.Y.; Jang, J. Statins reduce TGF-beta2-modulation of the extracellular matrix in cultured astrocytes of the human optic nerve head. Exp. Eye Res. 2017, 164, 55–63. [Google Scholar] [CrossRef]
- Kim, M.-L.; Sung, K.R.; Kwon, J.; Shin, J.A. Statins Suppress TGF-β2-Mediated MMP-2 and MMP-9 Expression and Activation through RhoA/ROCK Inhibition in Astrocytes of the Human Optic Nerve Head. Investig. Ophthalmol. Vis. Sci. 2020, 61, 29. [Google Scholar] [CrossRef]
- Taylor, S.; Calder, C.J.; Albon, J.; Erichsen, J.T.; Boulton, M.E.; Morgan, J.E. Involvement of the CD200 receptor complex in microglia activation in experimental glaucoma. Exp. Eye Res. 2011, 92, 338–343. [Google Scholar] [CrossRef]
- Howell, G.R.; Libby, R.T.; Jakobs, T.C.; Smith, R.S.; Phalan, F.C.; Barter, J.W.; Barbay, J.M.; Marchant, J.K.; Mahesh, N.; Porciatti, V.; et al. Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2J glaucoma. J. Cell Biol. 2007, 179, 1523–1537. [Google Scholar] [CrossRef]
- Kaur, C.; Foulds, W.S.; Ling, E.-A. Hypoxia-ischemia and retinal ganglion cell damage. Clin. Ophthalmol. 2008, 2, 879. [Google Scholar] [CrossRef]
- Tezel, G.; Yang, X.; Luo, C.; Cai, J.; Kain, A.D.; Powell, D.W.; Kuehn, M.H.; Pierce, W.M. Hemoglobin expression and regulation in glaucoma: Insights into retinal ganglion cell oxygenation. Investig. Ophthalmol. Vis. Sci. 2010, 51, 907–919. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G.; Yang, X.; Luo, C.; Peng, Y.; Sun, S.L.; Sun, D. Mechanisms of immune system activation in glaucoma: Oxidative stress-stimulated antigen presentation by the retina and optic nerve head glia. Investig. Ophthalmol. Vis. Sci. 2007, 48, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Gendaszewska-Darmach, E.; Garstka, M.A.; Błażewska, K.M. Targeting Small GTPases and Their Prenylation in Diabetes Mellitus. J. Med. Chem. 2021, 64, 9677–9710. [Google Scholar] [CrossRef]
- Wang, C.Y.; Liu, P.Y.; Liao, J.K. Pleiotropic effects of statin therapy: Molecular mechanisms and clinical results. Trends Mol. Med. 2008, 14, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Laufs, U.; Custodis, F.; Böhm, M. HMG-CoA reductase inhibitors in chronic heart failure: Potential mechanisms of benefit and risk. Drugs 2006, 66, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, J.; Steinman, L.; Zamvil, S.S. Statin therapy and autoimmune disease: From protein prenylation to immunomodulation. Nat. Rev. Immunol. 2006, 6, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Alt, A.; Hilgers, R.D.; Tura, A.; Nassar, K.; Schneider, T.; Hueber, A.; Januschowski, K.; Grisanti, S.; Lüke, J.; Lüke, M. The Neuroprotective Potential of Rho-Kinase Inhibition in Promoting Cell Survival and Reducing Reactive Gliosis in Response to Hypoxia in Isolated Bovine Retina. Cell. Physiol. Biochem. 2013, 32, 218–234. [Google Scholar] [CrossRef]
- Leung, D.Y.; Li, F.C.; Kwong, Y.Y.; Tham, C.C.; Chi, S.C.; Lam, D.S. Simvastatin and disease stabilization in normal tension glaucoma: A cohort study. Ophthalmology 2010, 117, 471–476. [Google Scholar] [CrossRef]
- McCann, P.; Hogg, R.E.; Fallis, R.; Azuara-Blanco, A. The effect of statins on intraocular pressure and on the incidence and progression of glaucoma: A systematic review and meta-analysis. Investig. Ophthalmol. Vis. Sci. 2016, 57, 2729–2748. [Google Scholar] [CrossRef]
- Lee, E.J.; Kim, T.-W.; Kim, J.-A.; Kim, J.-A. Parapapillary Deep-Layer microvasculature dropout in primary open-angle glaucoma eyes with a parapapillary γ-Zone. Investig. Ophthalmol. Vis. Sci. 2017, 58, 5673–5680. [Google Scholar] [CrossRef]
- Toft-Kehler, A.; Skytt, D.; Kolko, M. A perspective on the Müller cell-neuron metabolic partnership in the inner retina. Mol. Neurobiol. 2018, 55, 5353–5361. [Google Scholar] [CrossRef]
- Shinozaki, Y.; Koizumi, S. Pathogenic roles of retinal glia in glaucoma. Folia Pharmacol. Jpn. 2020, 155, 87–92. [Google Scholar] [CrossRef]
- Seitz, R.; Ohlmann, A.; Tamm, E.R. The role of Müller glia and microglia in glaucoma. Cell Tissue Res. 2013, 353, 339–345. [Google Scholar] [CrossRef]
- De Hoz, R.; Rojas, B.; Ramírez, A.I.; Salazar, J.J.; Gallego, B.I.; Triviño, A.; Ramírez, J.M. Retinal macroglial responses in health and disease. BioMed Res. Int. 2016, 2016, 2954721. [Google Scholar] [CrossRef]
- Baumann, B.; Sterling, J.; Song, Y.; Song, D.; Fruttiger, M.; Gillies, M.; Shen, W.; Dunaief, J.L. Conditional Müller cell ablation leads to retinal iron accumulation. Investig. Ophthalmol. Vis. Sci. 2017, 58, 4223–4234. [Google Scholar] [CrossRef]
- Johnson, E.C.; Morrison, J.C. Friend or foe? Resolving the impact of glial responses in glaucoma. J. Glaucoma 2009, 18, 341. [Google Scholar] [CrossRef]
- Gallego, B.I.; Salazar, J.J.; de Hoz, R.; Rojas, B.; Ramírez, A.I.; Salinas-Navarro, M.; Ortín-Martínez, A.; Valiente-Soriano, F.J.; Avilés-Trigueros, M.; Villegas-Perez, M.P.; et al. IOP induces upregulation of GFAP and MHC-II and microglia reactivity in mice retina contralateral to experimental glaucoma. J. Neuroinflamm. 2012, 9, 92. [Google Scholar] [CrossRef]
- Gao, F.; Yang, Z.; Jacoby, R.A.; Wu, S.M.; Pang, J.-J. The expression and function of TRPV4 channels in primate retinal ganglion cells and bipolar cells. Cell Death Dis. 2019, 10, 364. [Google Scholar] [CrossRef]
- Sappington, R.M.; Sidorova, T.; Ward, N.J.; Chakravarthy, R.; Ho, K.W.; Calkins, D.J. Activation of transient receptor potential vanilloid-1 (TRPV1) influences how retinal ganglion cell neurons respond to pressure-related stress. Channels 2015, 9, 102–113. [Google Scholar] [CrossRef]
- Lee, J.C.; Choe, S.Y. Age-related changes in the distribution of transient receptor potential vanilloid 4 channel (TRPV4) in the central nervous system of rats. J. Mol. Histol. 2014, 45, 497–505. [Google Scholar] [CrossRef]
- Butenko, O.; Dzamba, D.; Benesova, J.; Honsa, P.; Benfenati, V.; Rusnakova, V.; Ferroni, S.; Anderova, M. The increased activity of TRPV4 channel in the astrocytes of the adult rat hippocampus after cerebral hypoxia/ischemia. PLoS ONE 2012, 7, e39959. [Google Scholar]
- Ryskamp, D.A.; Witkovsky, P.; Barabas, P.; Huang, W.; Koehler, C.; Akimov, N.P.; Lee, S.H.; Chauhan, S.; Xing, W.; Rentería, R.C.; et al. The polymodal ion channel transient receptor potential vanilloid 4 modulates calcium flux, spiking rate, and apoptosis of mouse retinal ganglion cells. J. Neurosci. 2011, 31, 7089–7101. [Google Scholar] [CrossRef] [PubMed]
- Ryskamp, D.A.; Jo, A.O.; Frye, A.M.; Vazquez-Chona, F.; MacAulay, N.; Thoreson, W.B.; Križaj, D. Swelling and eicosanoid metabolites differentially gate TRPV4 channels in retinal neurons and glia. J. Neurosci. 2014, 34, 15689–15700. [Google Scholar] [CrossRef]
- Taylor, L.; Arnér, K.; Ghosh, F. Specific inhibition of TRPV4 enhances retinal ganglion cell survival in adult porcine retinal explants. Exp. Eye Res. 2017, 154, 10–21. [Google Scholar] [CrossRef]
- Liu, M.; Liu, X.; Wang, L.; Wang, Y.; Dong, F.; Wu, J.; Qu, X.; Liu, Y.; Liu, Z.; Fan, H.; et al. TRPV4 Inhibition Improved Myelination and Reduced Glia Reactivity and Inflammation in a Cuprizone-Induced Mouse Model of Demyelination. Front. Cell. Neurosci. 2018, 12, 392. [Google Scholar] [CrossRef] [PubMed]
- Ryskamp, D.A.; Frye, A.M.; Phuong, T.T.; Yarishkin, O.; Jo, A.O.; Xu, Y.; Lakk, M.; Iuso, A.; Redmon, S.N.; Ambati, B.; et al. TRPV4 regulates calcium homeostasis, cytoskeletal remodeling, conventional outflow and intraocular pressure in the mammalian eye. Sci. Rep. 2016, 6, 30583. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.D.; Chen, Y.-L.; Kasetti, R.B.; Maddineni, P.; Mayhew, W.; Millar, J.C.; Ellis, D.Z.; Sonkusare, S.K.; Zode, G.S. Impaired TRPV4-eNOS signaling in trabecular meshwork elevates intraocular pressure in glaucoma. Proc. Natl. Acad. Sci. USA 2021, 118, e2022461118. [Google Scholar] [CrossRef] [PubMed]
- Hiscott, J.; Marois, J.; Garoufalis, J.; D’addario, M.; Roulston, A.; Kwan, I.; Pepin, N.; Lacoste, J.; Nguyen, H.; Bensi, G. Characterization of a functional NF-kappa B site in the human interleukin 1 beta promoter: Evidence for a positive autoregulatory loop. Mol. Cell. Biol. 1993, 13, 6231–6240. [Google Scholar]
- Mori, N.; Prager, D. Transactivation of the interleukin-1alpha promoter by human T-cell leukemia virus type I and type II Tax proteins. Blood 1996, 87, 3410–3417. [Google Scholar] [CrossRef]
- Shakhov, A.N.; Collart, M.; Vassalli, P.; Nedospasov, S.; Jongeneel, C.V. Kappa B-type enhancers are involved in lipopolysaccharide-mediated transcriptional activation of the tumor necrosis factor alpha gene in primary macrophages. J. Exp. Med. 1990, 171, 35–47. [Google Scholar] [CrossRef]
- Tezel, G. TNF-α signaling in glaucomatous neurodegeneration. Prog. Brain Res. 2008, 173, 409–421. [Google Scholar]
- Kitaoka, Y.; Kitaoka, Y.; Kwong, J.M.; Ross-Cisneros, F.N.; Wang, J.; Tsai, R.K.; Sadun, A.A.; Lam, T.T. TNF-α-induced optic nerve degeneration and nuclear factor-κB p65. Investig. Ophthalmol. Vis. Sci. 2006, 47, 1448–1457. [Google Scholar] [CrossRef]
- Shi, Z.; Rudzinski, M.; Meerovitch, K.; Lebrun-Julien, F.; Birman, E.; Di Polo, A.; Saragovi, H.U. α2-Macroglobulin is a mediator of retinal ganglion cell death in glaucoma. J. Biol. Chem. 2008, 283, 29156–29165. [Google Scholar] [CrossRef]
- Vargas, J.L.C.; Osswald, I.K.; Unsain, N.; Aurousseau, M.R.; Barker, P.A.; Bowie, D.; Di Polo, A. Soluble tumor necrosis factor alpha promotes retinal ganglion cell death in glaucoma via calcium-permeable AMPA receptor activation. J. Neurosci. 2015, 35, 12088–12102. [Google Scholar] [CrossRef]
- Nakazawa, T.; Nakazawa, C.; Matsubara, A.; Noda, K.; Hisatomi, T.; She, H.; Michaud, N.; Hafezi-Moghadam, A.; Miller, J.W.; Benowitz, L.I. Tumor necrosis factor-α mediates oligodendrocyte death and delayed retinal ganglion cell loss in a mouse model of glaucoma. J. Neurosci. 2006, 26, 12633–12641. [Google Scholar] [CrossRef]
- Wang, X.; Tay, S.S.-W.; Ng, Y.-K. An immunohistochemical study of neuronal and glial cell reactions in retinae of rats with experimental glaucoma. Exp. Brain Res. 2000, 132, 476–484. [Google Scholar] [CrossRef]
- Tezel, G.; Li, L.Y.; Patil, R.V.; Wax, M.B. TNF-α and TNF-α receptor-1 in the retina of normal and glaucomatous eyes. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1787–1794. [Google Scholar]
- Yan, X.; Tezel, G.; Wax, M.B.; Edward, D.P. Matrix metalloproteinases and tumor necrosis factor α in glaucomatous optic nerve head. Arch. Ophthalmol. 2000, 118, 666–673. [Google Scholar] [CrossRef]
- Yuan, L.; Neufeld, A.H. Tumor necrosis factor-α: A potentially neurodestructive cytokine produced by glia in the human glaucomatous optic nerve head. Glia 2000, 32, 42–50. [Google Scholar] [CrossRef]
- Lebrun-Julien, F.; Bertrand, M.J.; De Backer, O.; Stellwagen, D.; Morales, C.R.; Di Polo, A.; Barker, P.A. ProNGF induces TNFα-dependent death of retinal ganglion cells through a p75NTR non-cell-autonomous signaling pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 3817–3822. [Google Scholar] [CrossRef]
- Roh, M.; Zhang, Y.; Murakami, Y.; Thanos, A.; Lee, S.C.; Vavvas, D.G.; Benowitz, L.I.; Miller, J.W. Etanercept, a widely used inhibitor of tumor necrosis factor-α (TNF-α), prevents retinal ganglion cell loss in a rat model of glaucoma. PLoS ONE 2012, 7, e40065. [Google Scholar] [CrossRef] [PubMed]
- Livne-Bar, I.; Lam, S.; Chan, D.; Guo, X.; Askar, I.; Nahirnyj, A.; Flanagan, J.G.; Sivak, J.M. Pharmacologic inhibition of reactive gliosis blocks TNF-α-mediated neuronal apoptosis. Cell Death Dis. 2016, 7, e2386. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.S.; Sethi, G.; Aggarwal, B.B. Reversal of chemoresistance and enhancement of apoptosis by statins through down-regulation of the NF-kappaB pathway. Biochem. Pharmacol. 2008, 75, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Tu, J.; Li, W.; Zhang, Y.; Wu, X.; Song, Y.; Kang, L.; Liu, W.; Wang, K.; Li, S.; Hua, W.; et al. Simvastatin Inhibits IL-1 [beta]-Induced Apoptosis and Extracellular Matrix Degradation by Suppressing the NF-kB and MAPK Pathways in Nucleus Pulposus Cells. Inflammation 2017, 40, 725. [Google Scholar] [CrossRef]
- Reiss, A.B.; Wirkowski, E. Statins in neurological disorders: Mechanisms and therapeutic value. Sci. World J. 2009, 9, 1242–1259. [Google Scholar] [CrossRef]
- Undela, K.; Gudala, K.; Malla, S.; Bansal, D. Statin use and risk of Parkinson’s disease: A meta-analysis of observational studies. J. Neurol. 2013, 260, 158–165. [Google Scholar] [CrossRef]
- Carroll, J.A.; Race, B.; Phillips, K.; Striebel, J.F.; Chesebro, B. Statins are ineffective at reducing neuroinflammation or prolonging survival in scrapie-infected mice. J. Gen. Virol. 2017, 98, 2190–2199. [Google Scholar] [CrossRef]
- Tison, F.; Nègre-Pagès, L.; Meissner, W.G.; Dupouy, S.; Li, Q.; Thiolat, M.-L.; Thiollier, T.; Galitzky, M.; Ory-Magne, F.; Milhet, A.; et al. Simvastatin decreases levodopa-induced dyskinesia in monkeys, but not in a randomized, placebo-controlled, multiple cross-over (“n-of-1”) exploratory trial of simvastatin against levodopa-induced dyskinesia in Parkinson’s disease patients. Parkinsonism Relat. Disord. 2013, 19, 416–421. [Google Scholar] [CrossRef]
- Ma, T.; Zhao, Y.; Kwak, Y.D.; Yang, Z.; Thompson, R.; Luo, Z.; Xu, H.; Liao, F.F. Statin’s excitoprotection is mediated by sAPP and the subsequent attenuation of calpain-induced truncation events, likely via rho-ROCK signaling. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 11226–11236. [Google Scholar] [CrossRef]
- Lawrence, J.; Singhal, S.; Bhatia, B.; Keegan, D.; Reh, T.; Luthert, P.; Khaw, P.; Limb, G.A. MIO-M1 cells and similar muller glial cell lines derived from adult human retina exhibit neural stem cell characteristics. Stem Cells 2007, 25, 2033–2043. [Google Scholar] [CrossRef]
- Vohra, R.; Gurubaran, I.S.; Henriksen, U.; Bergersen, L.H.; Rasmussen, L.J.; Desler, C.; Skytt, D.M.; Kolko, M. Disturbed mitochondrial function restricts glutamate uptake in the human Müller glia cell line, MIO-M1. Mitochondrion 2017, 36, 52–59. [Google Scholar] [CrossRef]
- Jin, M.; Wu, Z.; Chen, L.; Jaimes, J.; Collins, D.; Walters, E.T.; O’Neil, R.G. Determinants of TRPV4 activity following selective activation by small molecule agonist GSK1016790A. PLoS ONE 2011, 6, e16713. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jo, Y.H.; Choi, G.W.; Kim, M.-L.; Sung, K.R. Statins Inhibit the Gliosis of MIO-M1, a Müller Glial Cell Line Induced by TRPV4 Activation. Int. J. Mol. Sci. 2022, 23, 5190. https://doi.org/10.3390/ijms23095190
Jo YH, Choi GW, Kim M-L, Sung KR. Statins Inhibit the Gliosis of MIO-M1, a Müller Glial Cell Line Induced by TRPV4 Activation. International Journal of Molecular Sciences. 2022; 23(9):5190. https://doi.org/10.3390/ijms23095190
Chicago/Turabian StyleJo, Youn Hye, Go Woon Choi, Mi-Lyang Kim, and Kyung Rim Sung. 2022. "Statins Inhibit the Gliosis of MIO-M1, a Müller Glial Cell Line Induced by TRPV4 Activation" International Journal of Molecular Sciences 23, no. 9: 5190. https://doi.org/10.3390/ijms23095190
APA StyleJo, Y. H., Choi, G. W., Kim, M.-L., & Sung, K. R. (2022). Statins Inhibit the Gliosis of MIO-M1, a Müller Glial Cell Line Induced by TRPV4 Activation. International Journal of Molecular Sciences, 23(9), 5190. https://doi.org/10.3390/ijms23095190