
Pharmacologic Tumor PDL1 Depletion with Cefepime or Ceftazidime Promotes DNA Damage and Sensitivity to DNA-Damaging Agents


 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
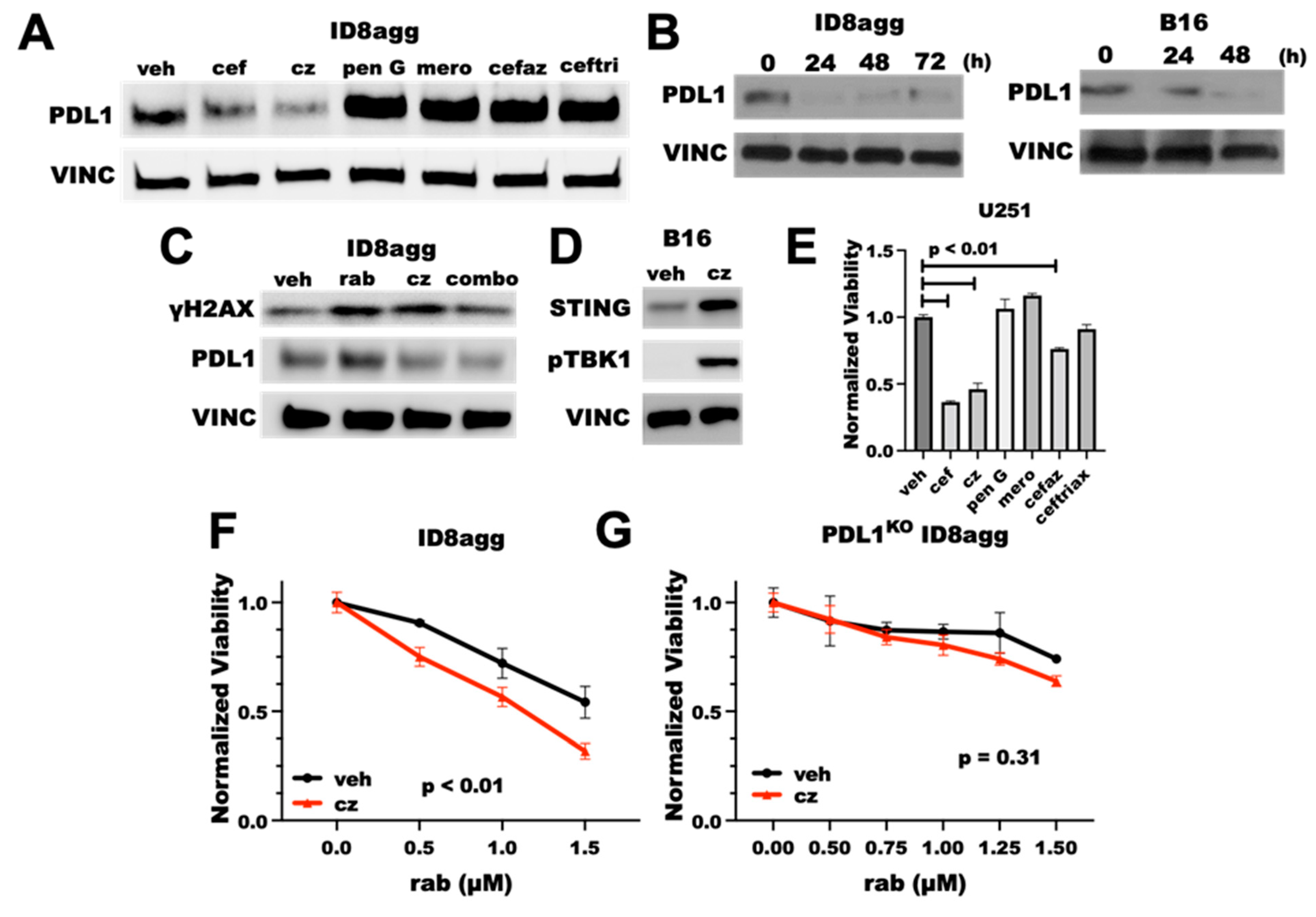
2.1. Cefepime Is a Pharmacologic Tumor PDL1-Depleting Drug
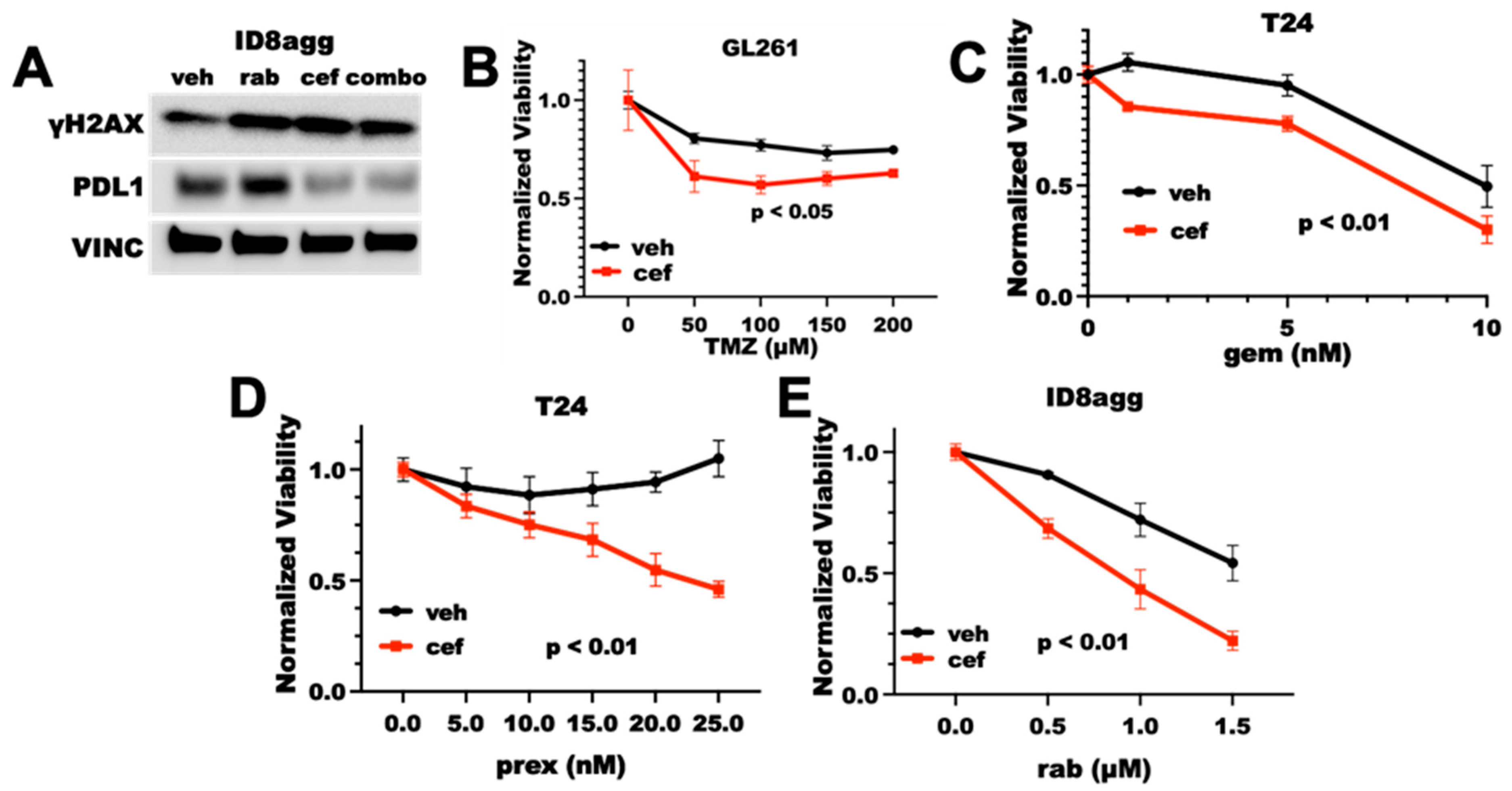
2.2. Cefepime Induces DNA Damage and Sensitizes to DNA-Damaging Agents In Vitro
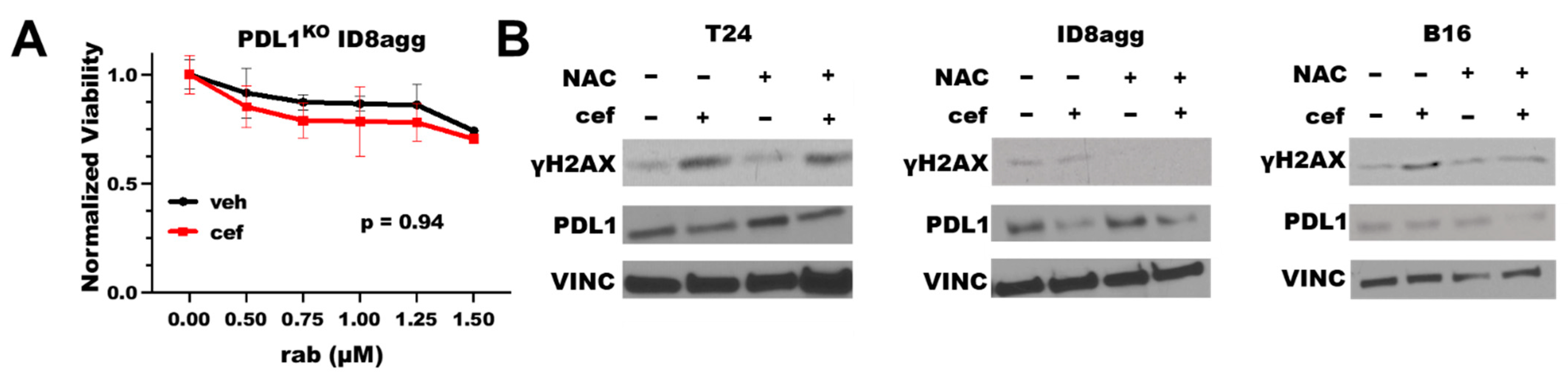
2.3. Cefepime-Induced DNA Damage and Synthetic Lethality Is Tumor-Cell-PDL1-Dependent and Can Include ROS Contributions
2.4. Cefepime Improves Rabusertib Sensitivity In Vivo and Skews towards TH1-Polarized Immunity
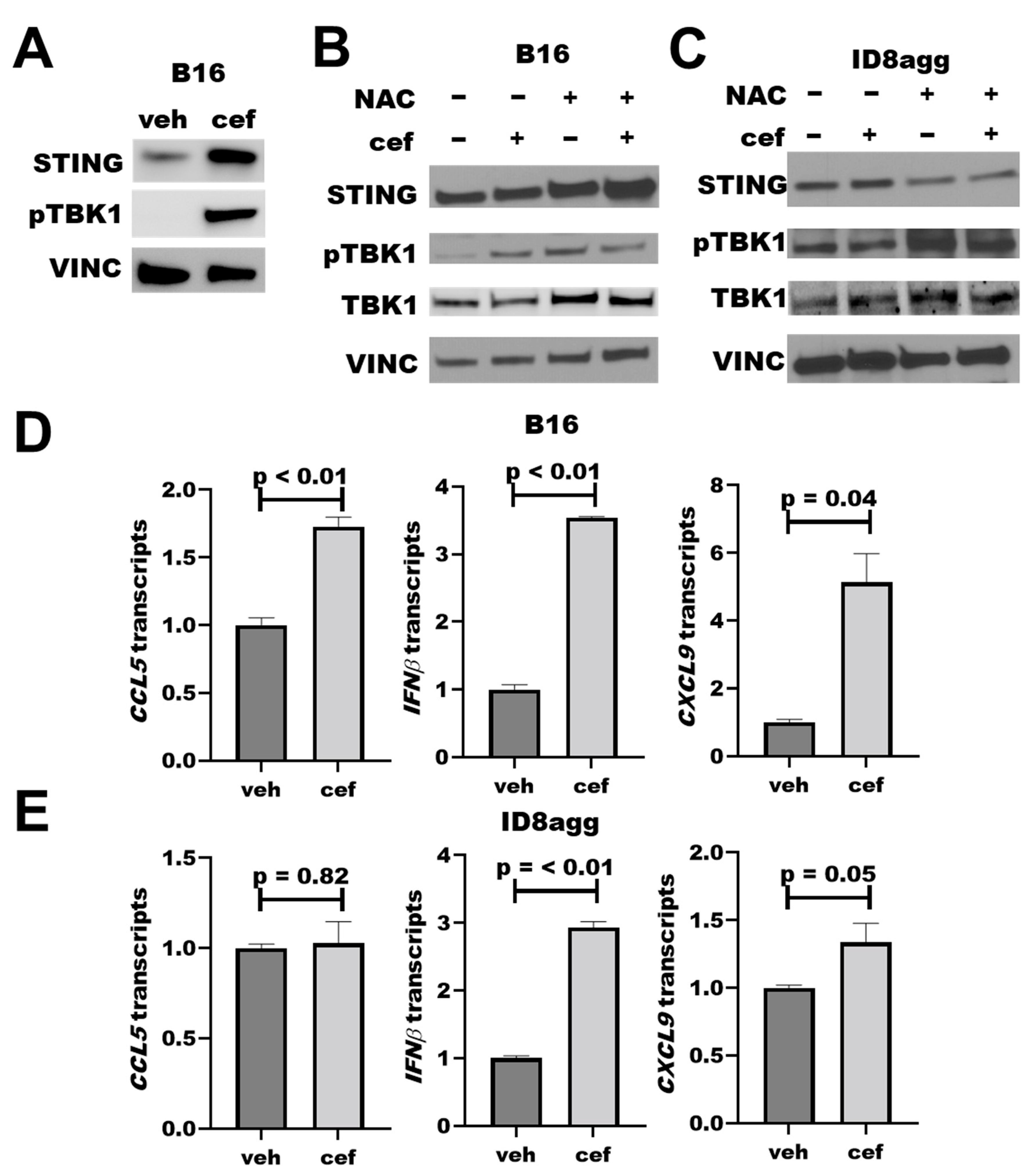
2.5. PDDs Promote Tumor STING Activation
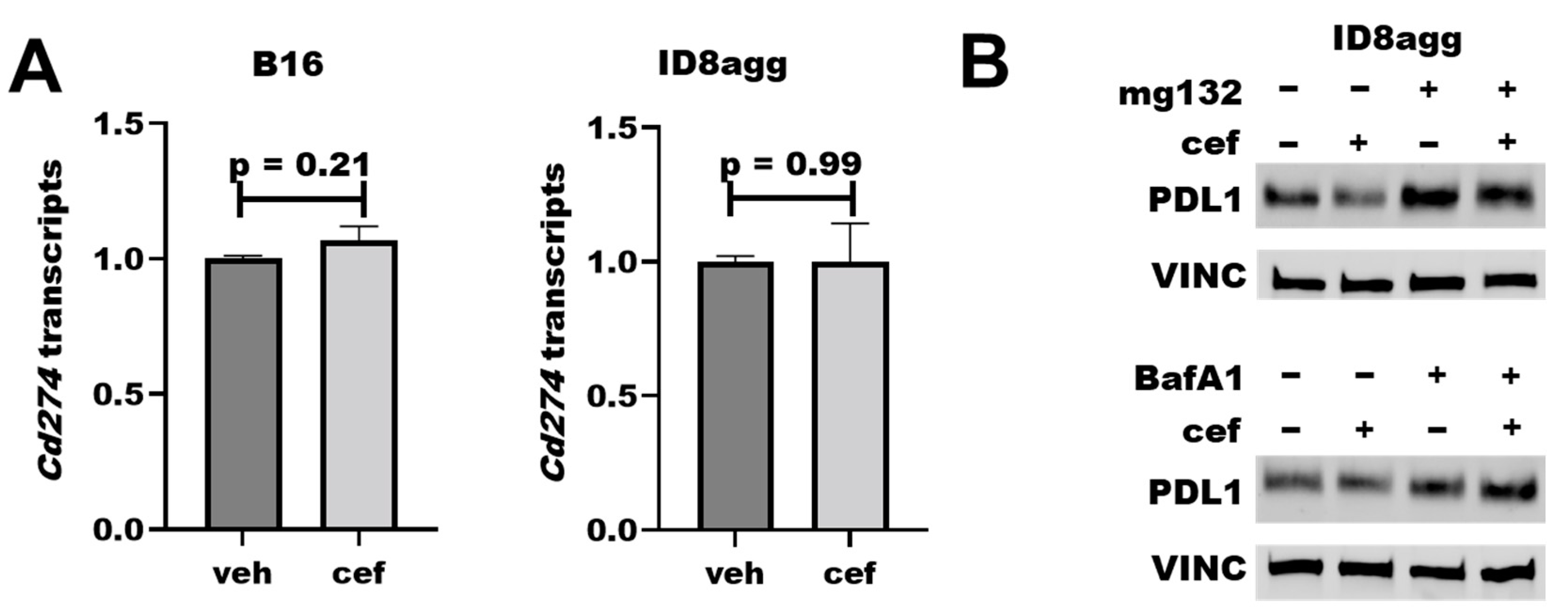
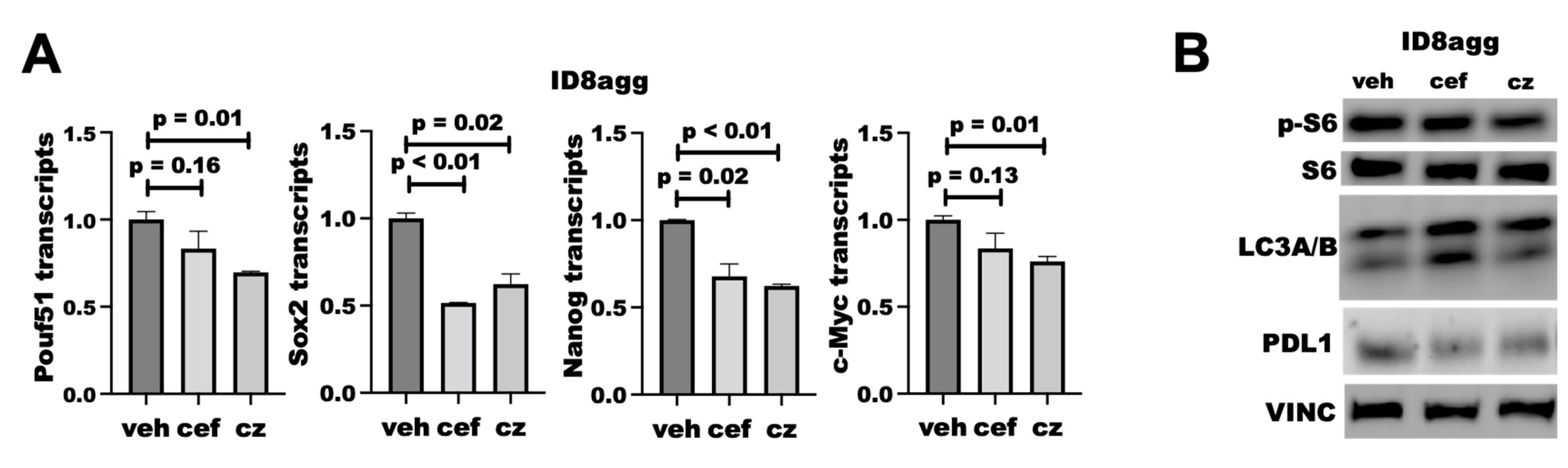
2.6. Cefepime Regulates Tumor PDL1 Post-Translationally
2.7. The Cefepime β-Lactam Ring Appears Dispensable for PDD and Cytotoxic Effects
2.8. PDDs Phenocopy Other Genetic Tumor-PDL1KO Outcomes
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hayashi, H.; Nakagawa, K. Combination therapy with PD-1 or PD-L1 inhibitors for cancer. Int. J. Clin. Oncol. 2020, 25, 818–830. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Taube, J.M.; Pardoll, D.M. Neoadjuvant checkpoint blockade for cancer immunotherapy. Science 2020, 367, 6477. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Liang, Y.; Anders, R.A.; Taube, J.M.; Qiu, X.; Mulgaonkar, A.; Liu, X.; Harrington, S.M.; Guo, J.; Xin, Y.; et al. PD-L1 on host cells is essential for PD-L1 blockade-mediated tumor regression. J. Clin. Investig. 2018, 128, 580–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizvi, N.A.; Cho, B.C.; Reinmuth, N.; Lee, K.H.; Luft, A.; Ahn, M.J.; van den Heuvel, M.M.; Cobo, M.; Vicente, D.; Smolin, A.; et al. Durvalumab with or without Tremelimumab vs Standard Chemotherapy in First-line Treatment of Metastatic Non-Small Cell Lung Cancer: The MYSTIC Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 661–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajan, A.; Heery, C.R.; Thomas, A.; Mammen, A.L.; Perry, S.; O’Sullivan Coyne, G.; Guha, U.; Berman, A.; Szabo, E.; Madan, R.A.; et al. Efficacy and tolerability of anti-programmed death-ligand 1 (PD-L1) antibody (Avelumab) treatment in advanced thymoma. J. Immunother. Cancer 2019, 7, 269. [Google Scholar] [CrossRef] [PubMed]
- Kornepati, A.V.R.; Vadlamudi, R.K.; Curiel, T.J. Programmed death ligand 1 signals in cancer cells. Nat. Rev. Cancer 2022, 22, 174–189. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.A.; Gupta, H.B.; Sareddy, G.; Pandeswara, S.; Lao, S.; Yuan, B.; Drerup, J.M.; Padron, A.; Conejo-Garcia, J.; Murthy, K.; et al. Tumor-Intrinsic PD-L1 Signals Regulate Cell Growth, Pathogenesis, and Autophagy in Ovarian Cancer and Melanoma. Cancer Res. 2016, 76, 6964–6974. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Reyes, R.M.; Osta, E.; Kari, S.; Gupta, H.B.; Padron, A.S.; Kornepati, A.V.R.; Kancharla, A.; Sun, X.; Deng, Y.; et al. Bladder cancer cell-intrinsic PD-L1 signals promote mTOR and autophagy activation that can be inhibited to improve cytotoxic chemotherapy. Cancer Med. 2021, 10, 2137–2152. [Google Scholar] [CrossRef]
- Wu, X.; Li, Y.; Liu, X.; Chen, C.; Harrington, S.M.; Cao, S.; Xie, T.; Orzechowski, A.; Pham, T.; Mansfield, A.S.; et al. Targeting B7-H1 (PD-L1) sensitizes cancer cells to chemotherapy. Heliyon 2018, 4, e01039. [Google Scholar] [CrossRef] [Green Version]
- Peng, S.; Wang, R.; Zhang, X.; Ma, Y.; Zhong, L.; Li, K.; Nishiyama, A.; Arai, S.; Yano, S.; Wang, W. EGFR-TKI resistance promotes immune escape in lung cancer via increased PD-L1 expression. Mol. Cancer 2019, 18, 165. [Google Scholar] [CrossRef]
- Gao, Y.; Nihira, N.T.; Bu, X.; Chu, C.; Zhang, J.; Kolodziejczyk, A.; Fan, Y.; Chan, N.T.; Ma, L.; Liu, J.; et al. Acetylation-dependent regulation of PD-L1 nuclear translocation dictates the efficacy of anti-PD-1 immunotherapy. Nat. Cell. Biol. 2020, 22, 1064–1075. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Zhao, R.; Xia, W.; Chang, C.W.; You, Y.; Hsu, J.M.; Nie, L.; Chen, Y.; Wang, Y.C.; Liu, C.; et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat. Cell Biol. 2020, 22, 1264–1275. [Google Scholar] [CrossRef] [PubMed]
- Gupta, H.B.; Clark, C.A.; Yuan, B.; Sareddy, G.; Pandeswara, S.; Padron, A.S.; Hurez, V.; Conejo-Garcia, J.; Vadlamudi, R.; Li, R.; et al. Tumor cell-intrinsic PD-L1 promotes tumor-initiating cell generation and functions in melanoma and ovarian cancer. Signal Transduct. Target. Ther. 2016, 1, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornepati, A.V.R.; Boyd, J.T.; Murray, C.E.; Saifetiarova, J.; Avalos, B.d.l.P.; Rogers, C.M.; Bai, H.; Padron, A.S.; Liao, Y.; Ontiveros, C.; et al. Tumor-intrinsic programmed death-ligand 1 promotes DNA repair in distinct cancers and can suppress PARP inhibitor synthetic lethality. Cancer Res. 2022. online ahead of print. [Google Scholar]
- Chapman, T.M.; Perry, C.M. Cefepime: A review of its use in the management of hospitalized patients with pneumonia. Am. J. Respir. Med. 2003, 2, 75–107. [Google Scholar] [CrossRef]
- von der Maase, H.; Sengelov, L.; Roberts, J.T.; Ricci, S.; Dogliotti, L.; Oliver, T.; Moore, M.J.; Zimmermann, A.; Arning, M. Long-term survival results of a randomized trial comparing gemcitabine plus cisplatin, with methotrexate, vinblastine, doxorubicin, plus cisplatin in patients with bladder cancer. J. Clin. Oncol. 2005, 23, 4602–4608. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Qiu, Z.; Oleinick, N.L.; Zhang, J. ATR/CHK1 inhibitors and cancer therapy. Radiother. Oncol. 2018, 126, 450–464. [Google Scholar] [CrossRef]
- Sato, H.; Jeggo, P.A.; Shibata, A. Regulation of programmed death-ligand 1 expression in response to DNA damage in cancer cells: Implications for precision medicine. Cancer Sci. 2019, 110, 3415–3423. [Google Scholar] [CrossRef] [Green Version]
- Sen, T.; Rodriguez, B.L.; Chen, L.; Corte, C.M.D.; Morikawa, N.; Fujimoto, J.; Cristea, S.; Nguyen, T.; Diao, L.; Li, L.; et al. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discov. 2019, 9, 646–661. [Google Scholar] [CrossRef] [Green Version]
- Kalghatgi, S.; Spina, C.S.; Costello, J.C.; Liesa, M.; Morones-Ramirez, J.R.; Slomovic, S.; Molina, A.; Shirihai, O.S.; Collins, J.J. Bactericidal antibiotics induce mitochondrial dysfunction and oxidative damage in Mammalian cells. Sci. Transl. Med. 2013, 5, 192ra185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helmink, B.A.; Khan, M.A.W.; Hermann, A.; Gopalakrishnan, V.; Wargo, J.A. The microbiome, cancer, and cancer therapy. Nat. Med. 2019, 25, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Elkrief, A.; Derosa, L.; Kroemer, G.; Zitvogel, L.; Routy, B. The negative impact of antibiotics on outcomes in cancer patients treated with immunotherapy: A new independent prognostic factor? Ann. Oncol. 2019, 30, 1572–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sallusto, F.; Lenig, D.; Mackay, C.R.; Lanzavecchia, A. Flexible programs of chemokine receptor expression on human polarized T helper 1 and 2 lymphocytes. J. Exp. Med. 1998, 187, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 2021, 21, 548–569. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Wang, L.; Wang, C.; Shen, J.; Su, B.; Marisetty, A.L.; Fang, D.; Kassab, C.; Jeong, K.J.; Zhao, W.; et al. Verteporfin Inhibits PD-L1 through Autophagy and the STAT1-IRF1-TRIM28 Signaling Axis, Exerting Antitumor Efficacy. Cancer Immunol. Res. 2020, 8, 952–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.O.; Li, C.W.; Xia, W.; Cha, J.H.; Chan, L.C.; Wu, Y.; Chang, S.S.; Lin, W.C.; Hsu, J.M.; Hsu, Y.H.; et al. Deubiquitination and Stabilization of PD-L1 by CSN5. Cancer Cell 2016, 30, 925–939. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Jiang, L.; Li, S.C.; He, Q.J.; Yang, B.; Cao, J. Small molecule inhibitors targeting the PD-1/PD-L1 signaling pathway. Acta Pharmacol. Sin. 2021, 42, 1–9. [Google Scholar] [CrossRef]
- Liu, C.; Seeram, N.P.; Ma, H. Small molecule inhibitors against PD-1/PD-L1 immune checkpoints and current methodologies for their development: A review. Cancer Cell Int. 2021, 21, 239. [Google Scholar] [CrossRef]
- Tu, X.; Qin, B.; Zhang, Y.; Zhang, C.; Kahila, M.; Nowsheen, S.; Yin, P.; Yuan, J.; Pei, H.; Li, H.; et al. PD-L1 (B7-H1) Competes with the RNA Exosome to Regulate the DNA Damage Response and Can Be Targeted to Sensitize to Radiation or Chemotherapy. Mol. Cell 2019, 74, 1215–1226.e4. [Google Scholar] [CrossRef]
- Ciccolini, J.; Serdjebi, C.; Peters, G.J.; Giovannetti, E. Pharmacokinetics and pharmacogenetics of Gemcitabine as a mainstay in adult and pediatric oncology: An EORTC-PAMM perspective. Cancer Chemother. Pharmacol. 2016, 78, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwala, S.S.; Kirkwood, J.M. Temozolomide, a Novel Alkylating Agent with Activity in the Central Nervous System, May Improve the Treatment of Advanced Metastatic Melanoma. Oncologist 2000, 5, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Kornepati, A.; Murray, C.; Avalos, B.; Rogers, C.; Ramkumar, K.; Gupta, H.; Deng, Y.; Liu, Z.; Padron, A.; Vadlamudi, R.; et al. 900 Depleting non-canonical, cell-intrinsic PD-L1 signals induces synthetic lethality to small molecule DNA damage response inhibitors in an immune independent and dependent manner. J. Immunother. Cancer 2021, 9, A944. [Google Scholar] [CrossRef]
- Yu, J.; Qin, B.; Moyer, A.M.; Nowsheen, S.; Tu, X.; Dong, H.; Boughey, J.C.; Goetz, M.P.; Weinshilboum, R.; Lou, Z.; et al. Regulation of sister chromatid cohesion by nuclear PD-L1. Cell Res. 2020, 30, 590–601. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Jin, J.; Wang, Y.; Fang, L.; Min, L.; Wang, X.; Ding, L.; Weng, L.; Xiao, T.; Zhou, T.; et al. PD-L1 regulates genomic stability via interaction with cohesin-SA1 in the nucleus. Signal Transduct. Target. Ther. 2021, 6, 81. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Padrón, Á.; Deng, Y.; Polusan, S.R.; Kornepati, A.; Kari, S.; Kancharla, A.; Garcia, M.; Reyes, R.M.; Ji, N.; et al. Pharmacologic tumor PD-L1 depletion with chlorambucil treats ovarian cancer and melanomas in a tumor PD-L1-dependent manner and renders α PD-L1-resistant tumors into α PD-L1-sensitive. Soc. Immunother. Cancer 2021, 9, A261. [Google Scholar] [CrossRef]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef]
- Parsa, A.T.; Waldron, J.S.; Panner, A.; Crane, C.A.; Parney, I.F.; Barry, J.J.; Cachola, K.E.; Murray, J.C.; Tihan, T.; Jensen, M.C.; et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 2007, 13, 84–88. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murray, C.; Galvan, E.; Ontiveros, C.; Deng, Y.; Bai, H.; Padron, A.S.; Hinchee-Rodriguez, K.; Garcia, M.G.; Kornepati, A.; Conejo-Garcia, J.; et al. Pharmacologic Tumor PDL1 Depletion with Cefepime or Ceftazidime Promotes DNA Damage and Sensitivity to DNA-Damaging Agents. Int. J. Mol. Sci. 2022, 23, 5129. https://doi.org/10.3390/ijms23095129
Murray C, Galvan E, Ontiveros C, Deng Y, Bai H, Padron AS, Hinchee-Rodriguez K, Garcia MG, Kornepati A, Conejo-Garcia J, et al. Pharmacologic Tumor PDL1 Depletion with Cefepime or Ceftazidime Promotes DNA Damage and Sensitivity to DNA-Damaging Agents. International Journal of Molecular Sciences. 2022; 23(9):5129. https://doi.org/10.3390/ijms23095129
Chicago/Turabian StyleMurray, Clare, Eva Galvan, Carlos Ontiveros, Yilun Deng, Haiyan Bai, Alvaro Souto Padron, Kathryn Hinchee-Rodriguez, Myrna G. Garcia, Anand Kornepati, Jose Conejo-Garcia, and et al. 2022. "Pharmacologic Tumor PDL1 Depletion with Cefepime or Ceftazidime Promotes DNA Damage and Sensitivity to DNA-Damaging Agents" International Journal of Molecular Sciences 23, no. 9: 5129. https://doi.org/10.3390/ijms23095129
APA StyleMurray, C., Galvan, E., Ontiveros, C., Deng, Y., Bai, H., Padron, A. S., Hinchee-Rodriguez, K., Garcia, M. G., Kornepati, A., Conejo-Garcia, J., & Curiel, T. J. (2022). Pharmacologic Tumor PDL1 Depletion with Cefepime or Ceftazidime Promotes DNA Damage and Sensitivity to DNA-Damaging Agents. International Journal of Molecular Sciences, 23(9), 5129. https://doi.org/10.3390/ijms23095129