
NMR Relaxometry Accessing the Relaxation Spectrum in Molecular Glass Formers


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Spin-Lattice Relaxation and Dynamic Susceptibility
3. Experimental Results
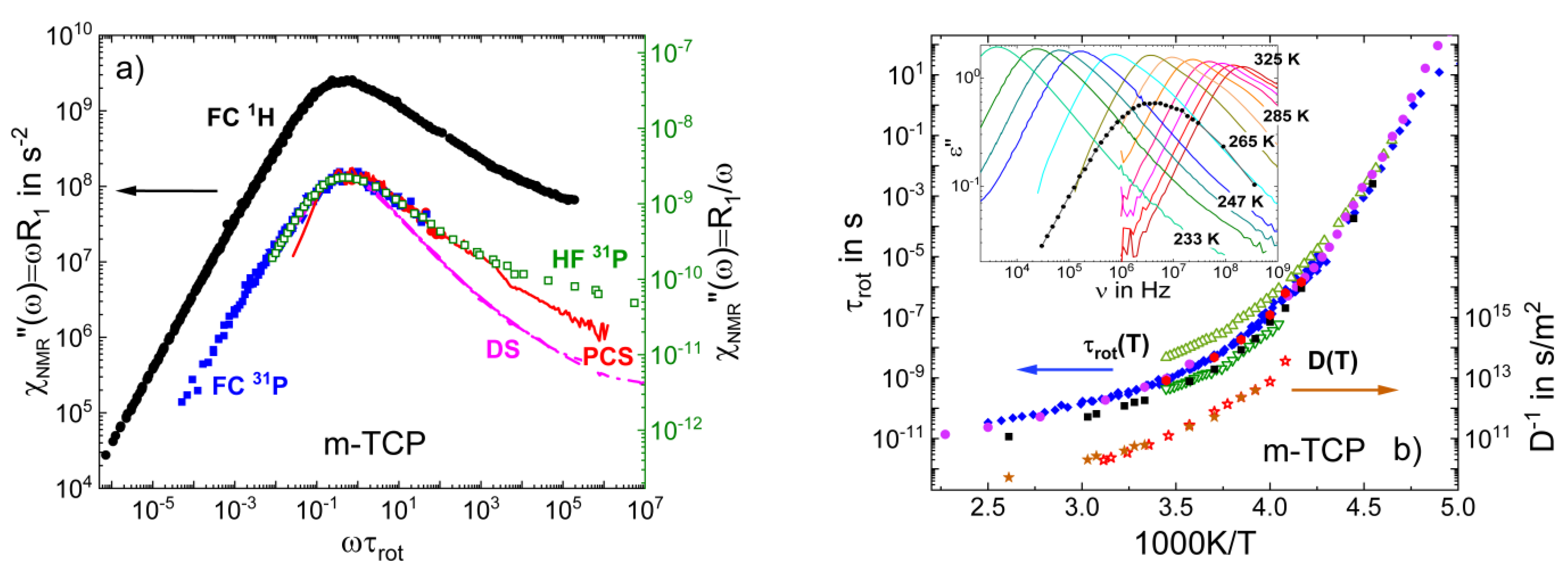
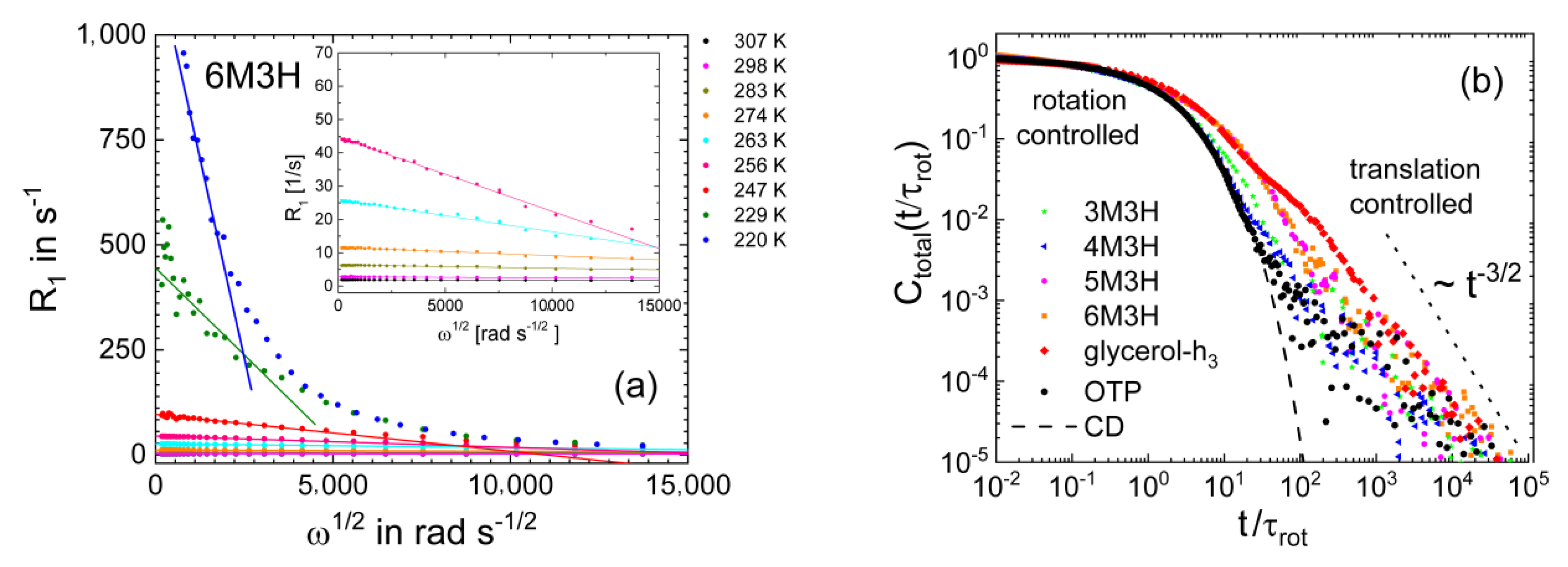
3.1. Simple Liquids
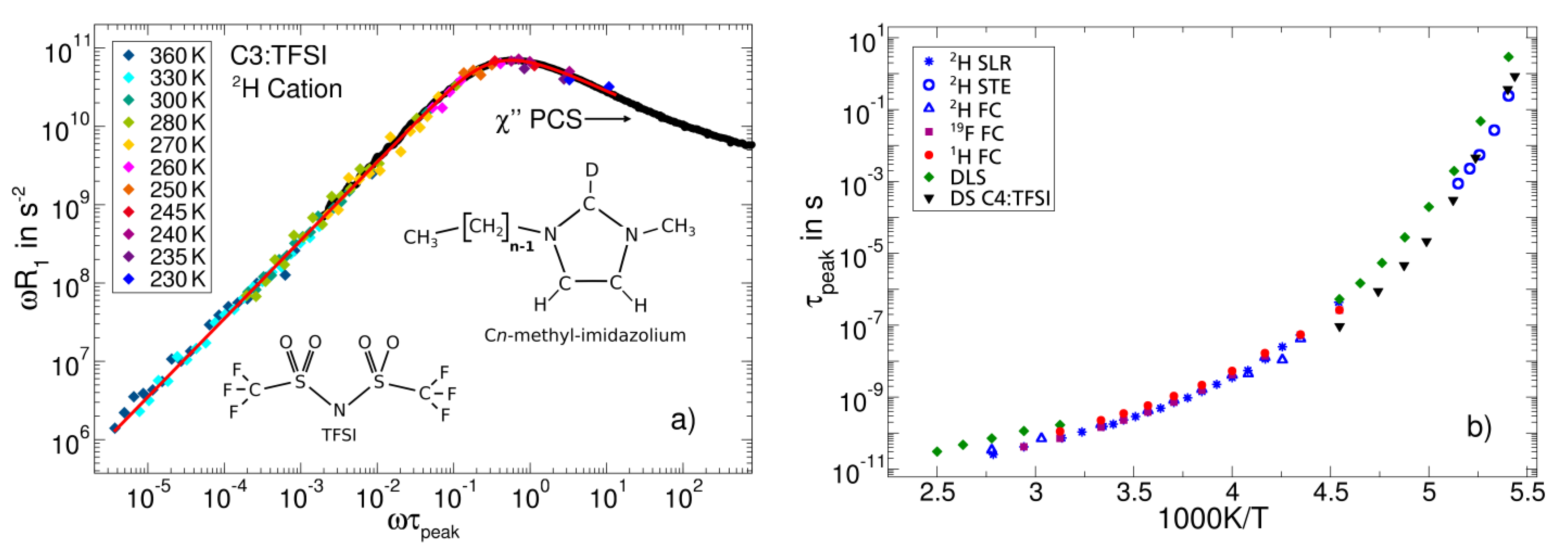
3.2. Ionic Liquids
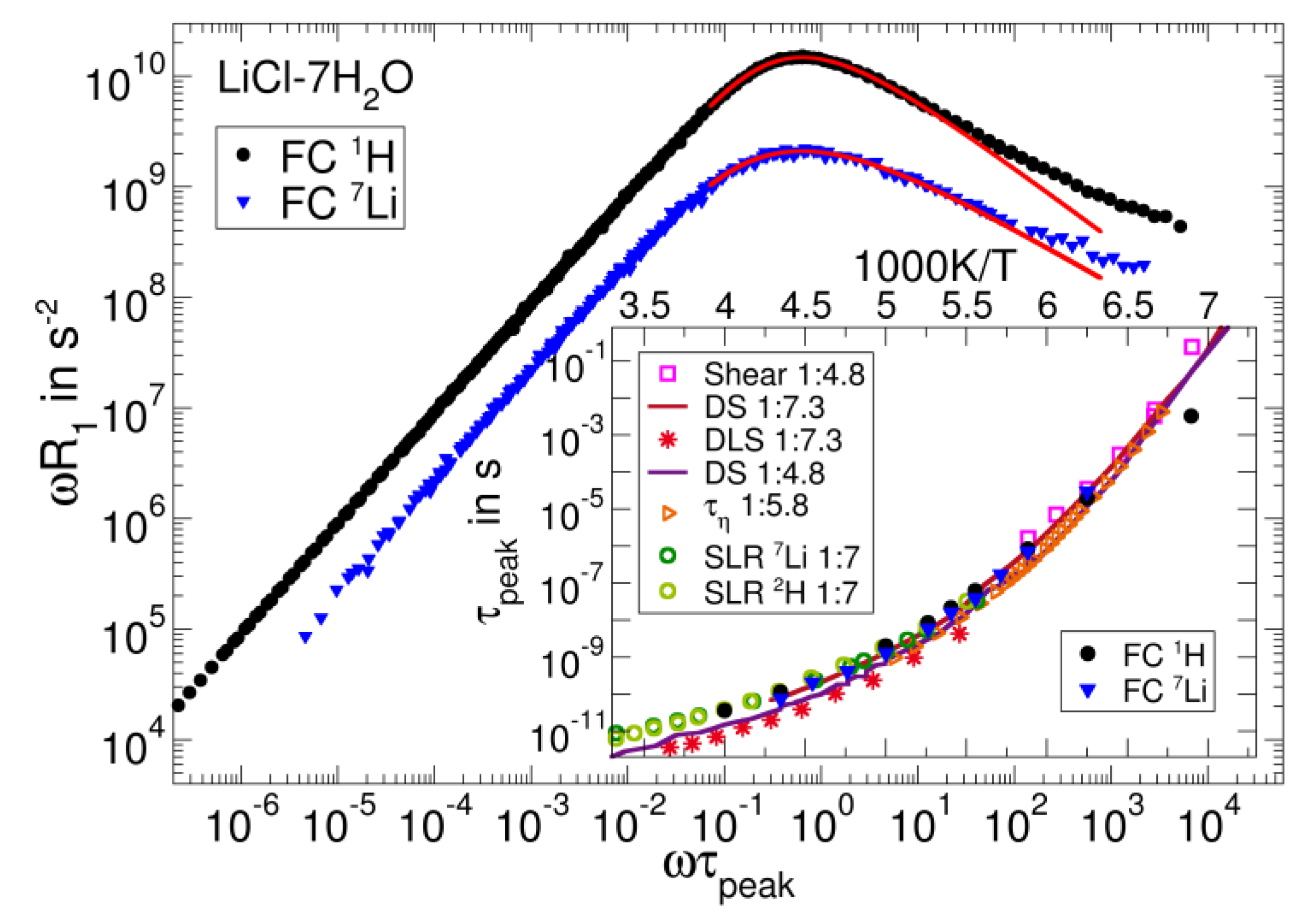
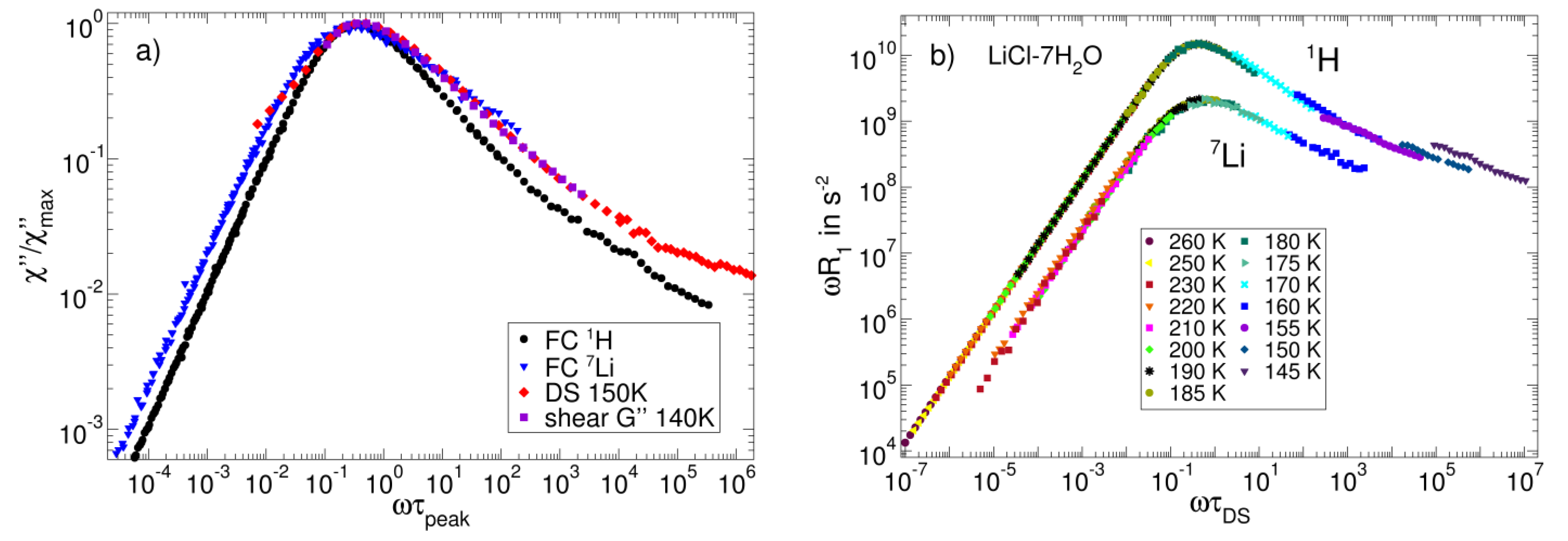
3.3. Salt Solutions
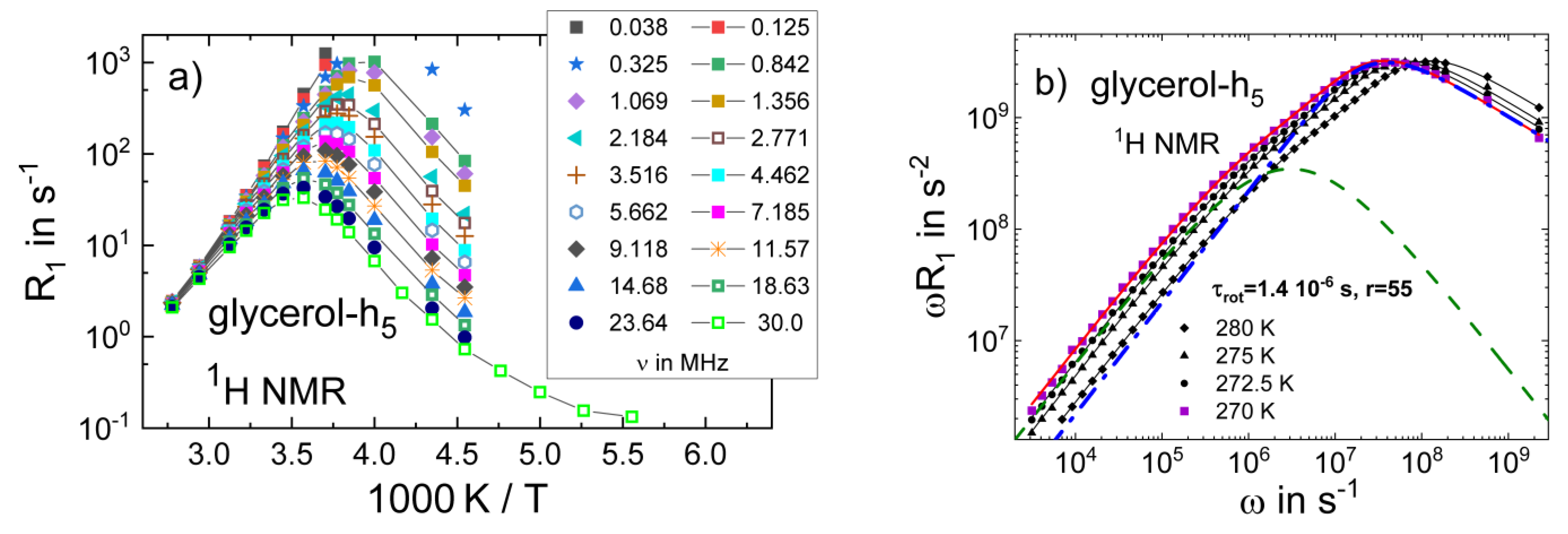
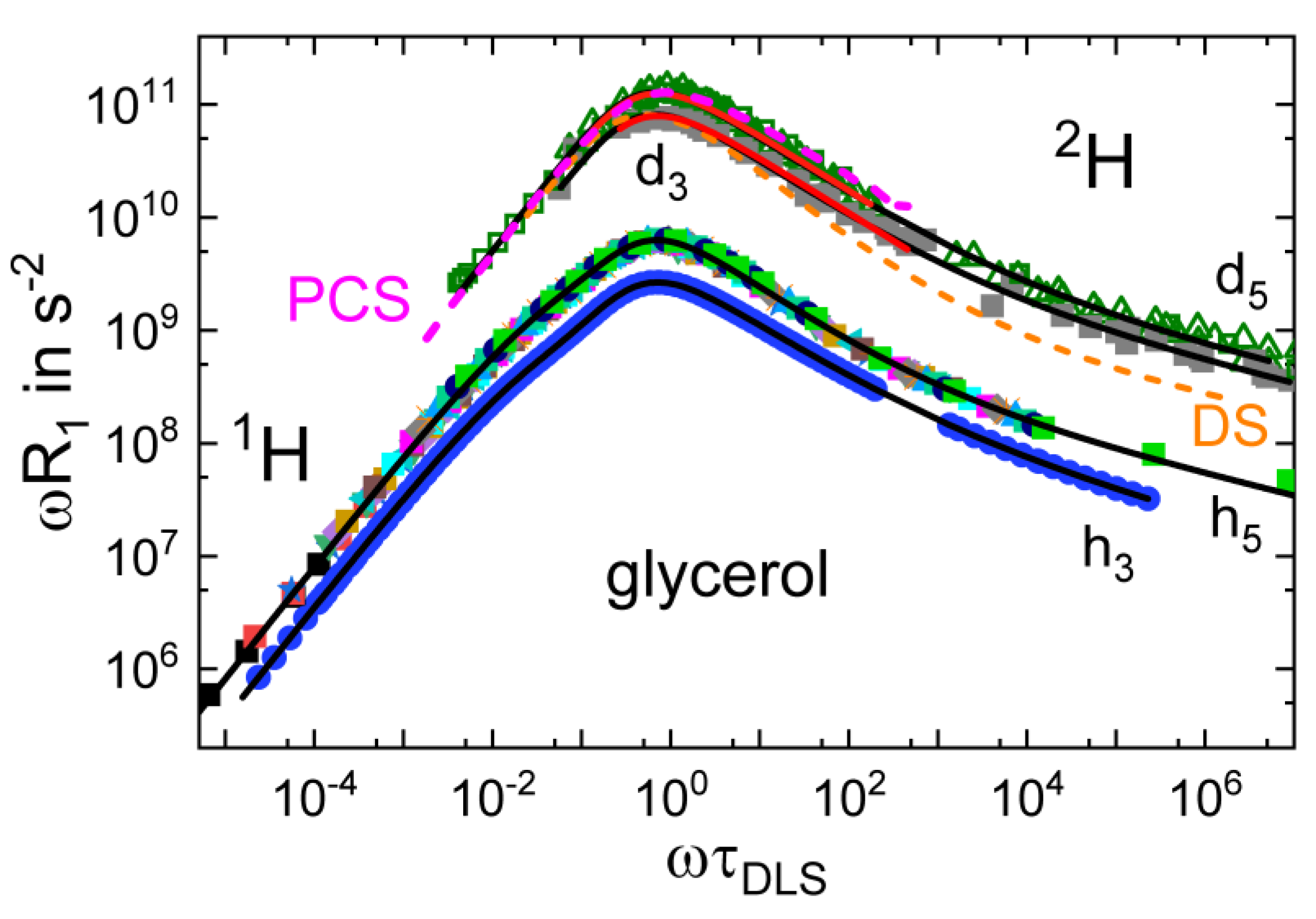
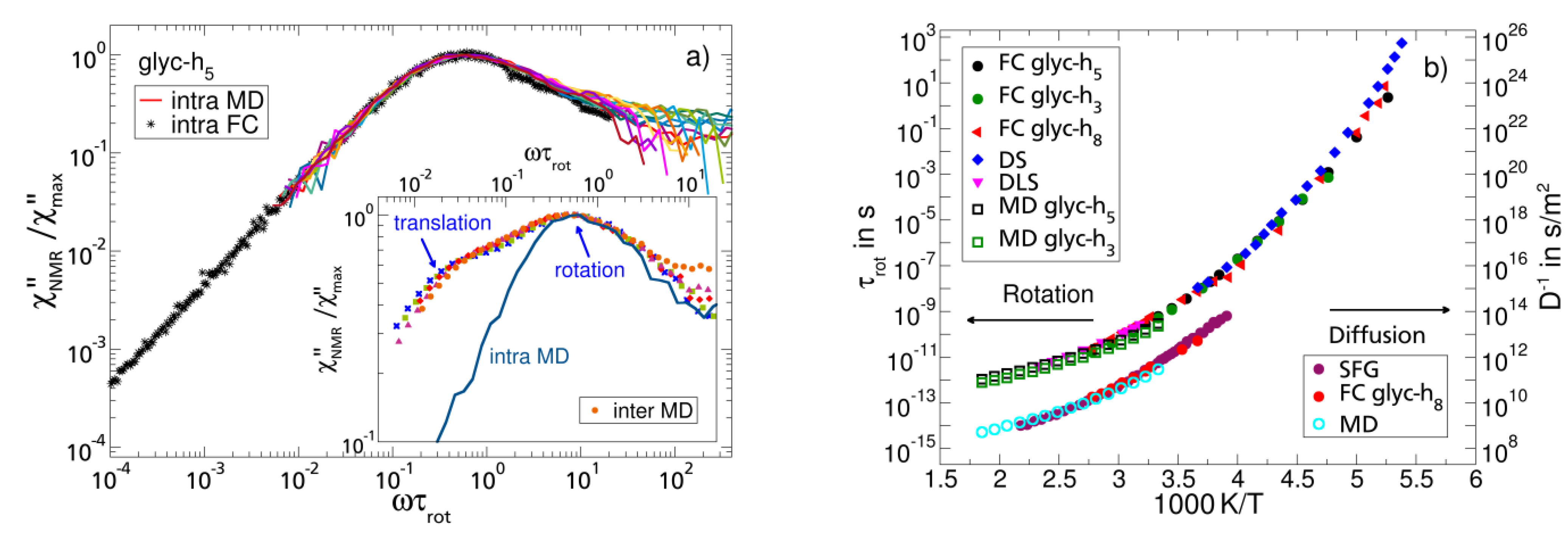
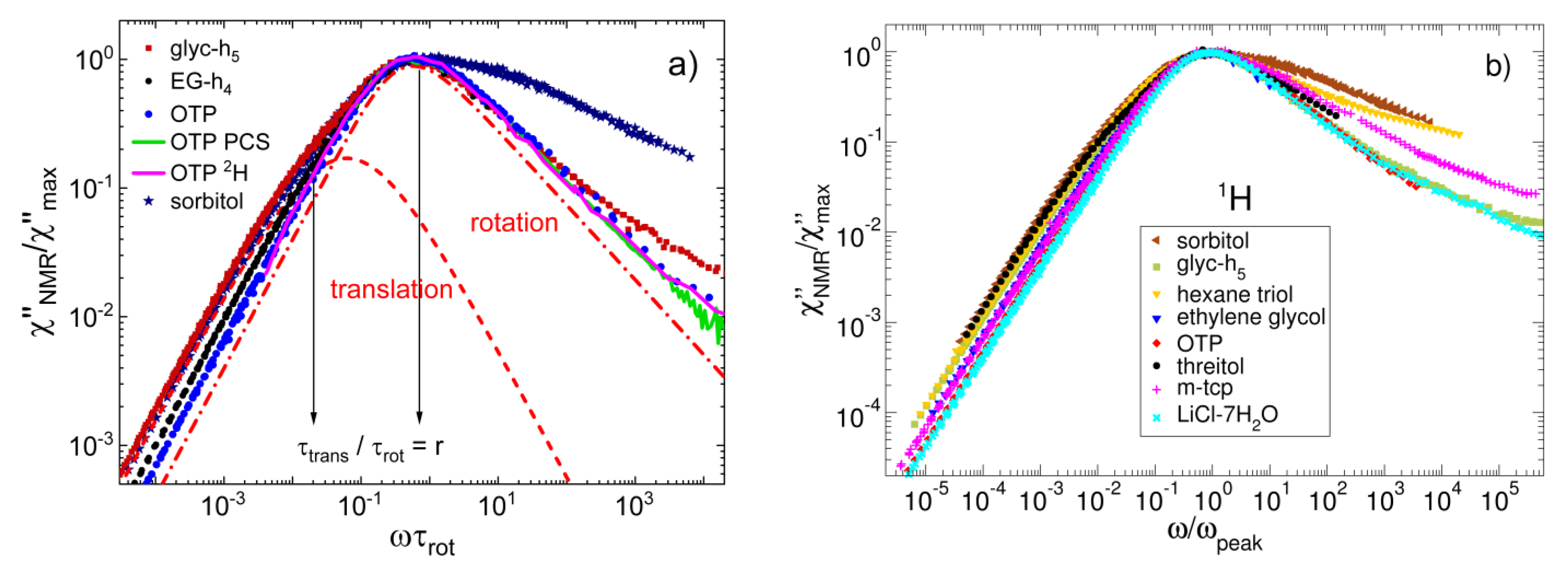
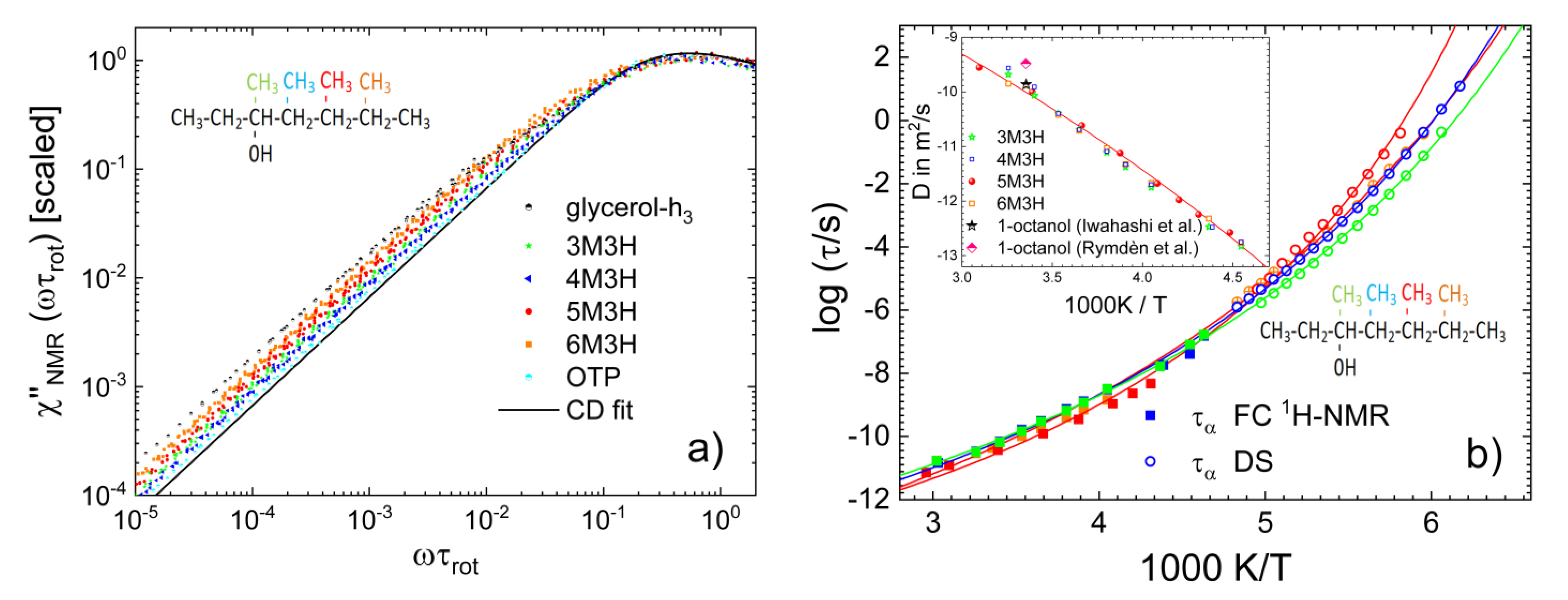
3.4. Monohydroxy Alcohols
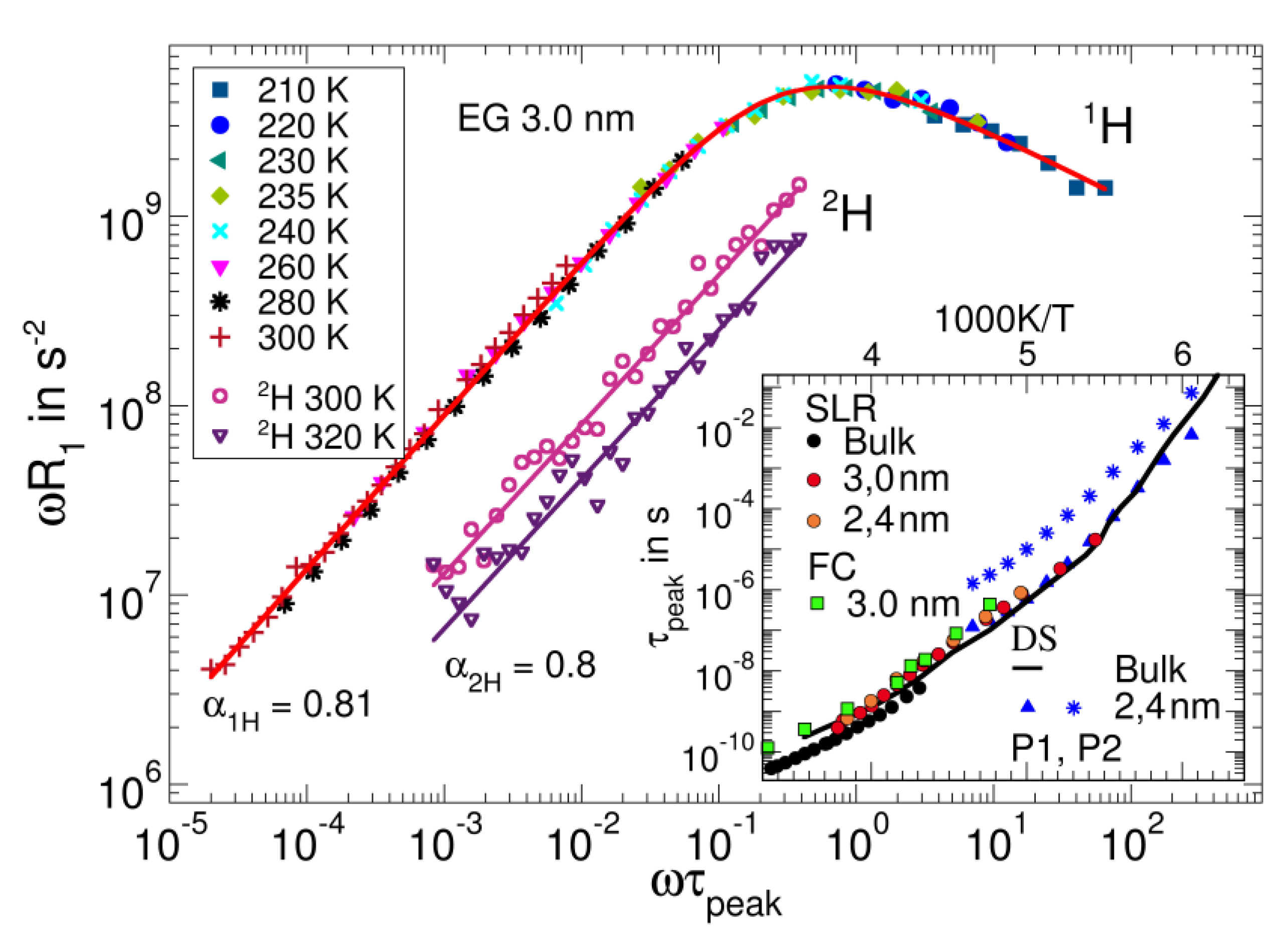
3.5. Confined Liquids
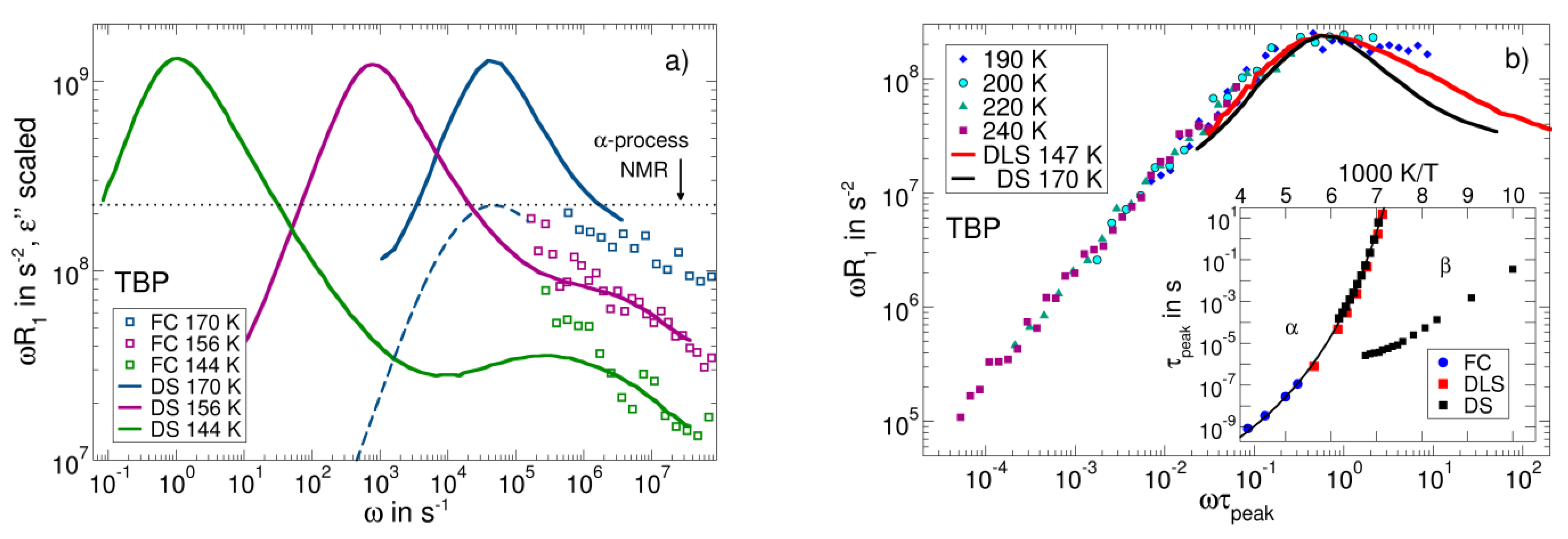
3.6. Field-Cycling NMR Experiments Addressing the β-Relaxation of Pure Molecular Liquids
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kudlik, A.; Benkhof, S.; Blochowicz, T.; Tschirwitz, C.; Rössler, E. The dielectric response of simple organic glass formers. J. Mol. Struct. 1999, 479, 201–218. [Google Scholar] [CrossRef]
- Lunkenheimer, P.; Schneider, U.; Brand, R.; Loidl, A. Glassy Dynamics. Contemp. Phys. 2000, 41, 15–36. [Google Scholar] [CrossRef]
- Blochowicz, T.; Brodin, A.; Rössler, E.A. Advances in Chemical Physics. Fractals, Diffusion, and Relaxation in Disordered Complex Systems; Coffey, W.T., Kalmykov, Y.P., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2006; Volume 133, pp. 127–256. [Google Scholar]
- Petzold, N.; Schmidtke, B.; Kahlau, R.; Bock, D.; Meier, R.; Micko, B.; Kruk, D.; Rössler, E.A. Evolution of the dynamic susceptibility in molecular glass formers: Results from light scattering, dielectric spectroscopy, and NMR. J. Chem. Phys. 2013, 138, 12A510. [Google Scholar] [CrossRef] [PubMed]
- Cummins, H.Z.; Li, G.; Hwang, Y.H.; Shen, G.Q.; Du, W.M.; Hernandez, J.; Tao, N.J. Dynamics of supercooled liquids and glasses: Comparison of experiments with theoretical predictions. Z. Phys. B 1997, 103, 501–519. [Google Scholar] [CrossRef]
- Patkowski, A.; Paluch, M.; Kriegs, H. Dynamic light scattering studies of supercooled phenylphthalein–dimethylether dynamics under high pressure. J. Chem. Phys. 2002, 117, 2192–2198. [Google Scholar] [CrossRef]
- Sidebottom, D.L.; Rodenburg, B.V. Changstrom, J.R. Connecting structure and dynamics in glass forming materials by photon correlation spectroscopy. Phys. Rev. B 2007, 75, 132201. [Google Scholar] [CrossRef]
- Torre, R.; Bartolini, P.; Pick, R.M. Time-resolved optical Kerr effect in a fragile glass-forming liquid, salol. Phys. Rev. E 1998, 57, 1912. [Google Scholar] [CrossRef]
- Hinze, G.; Brace, D.D.; Gottke, S.D.; Fayer, M.D. A detailed test of mode-coupling theory on all time scales: Time domain studies of structural relaxation in a supercooled liquid. J. Chem. Phys. 2000, 113, 3723–3733. [Google Scholar] [CrossRef][Green Version]
- Tölle, A. Neutron scattering studies of the model glass former ortho-terphenyl. Rep. Prog. Phys. 2001, 64, 1473. [Google Scholar] [CrossRef]
- Götze, W. Recent tests of the mode-coupling theory for glassy dynamics. J. Phys. Condens. Matter 1999, 11, A1–A45. [Google Scholar] [CrossRef]
- Binder, K.; Kob, W. Glassy Materials and Disordered Solids. An Introduction to Their Statistical Mechanics; World Scientific: Hackensack, NJ, USA, 2005. [Google Scholar]
- Wolynes, P.G.; Lubchenko, V. Structural Glasses and Supercooled Liquids. Theory, Experiment, and Applications; Wiley: Hoboken, NJ, USA, 2012. [Google Scholar]
- Johari, G.P.; Goldstein, M. Viscous liquids and the glass transition. II. Secondary relaxations in glasses of rigid molecules. J. Chem. Phys. 1970, 53, 2372–2388. [Google Scholar] [CrossRef]
- Ngai, K.L.; Paluch, M. Classification of secondary relaxation in glass-formers based on dynamic properties. J. Chem. Phys. 2004, 120, 857–873. [Google Scholar] [CrossRef] [PubMed]
- Gainaru, C.; Kahlau, R.; Rössler, E.A.; Böhmer, R. Evolution of excess wing and β-process in simple glass formers. J. Chem. Phys. 2009, 131, 184510. [Google Scholar] [CrossRef] [PubMed]
- Floudas, G.; Paluch, M.; Grzybowski, A.; Ngai, K. Molecular Dynamics of Glass-Forming Systems. Effects of Pressure; Springer: Berlin/Heidelberg, Germany, 2011. [Google Scholar]
- Schneider, U.; Brand, R.; Lunkenheimer, P.; Loidl, A. Excess wing in the dielectric loss of glass formers: A Johari-Goldstein β relaxation? Phys. Rev. Lett. 2000, 84, 5560–5563. [Google Scholar] [CrossRef] [PubMed]
- Dixon, P.K.; Wu, L.; Nagel, S.R.; Williams, B.D.; Carini, J.P. Scaling in the relaxation of supercooled liquids. Phys. Rev. Lett. 1990, 65, 1108–1111. [Google Scholar] [CrossRef]
- Kudlik, A.; Benkhof, S.; Lenk, S.; Rössler, E. Spectral Shape of the α-Process in Supercooled Liquids Revisited. Europhys. Lett. 1995, 32, 511–516. [Google Scholar] [CrossRef]
- Kudlik, A.; Blochowicz, T.; Benkhof, S.; Rössler, E. Reply to Comment on “Spectral shape of the α-process in supercooled liquids revisited”. Europhys. Lett. 1996, 36, 475–476. [Google Scholar]
- Leheny, R.L.; Menon, N.; Nagel, S.R. Comment on “Spectral shape of the α-process in supercooled liquids revisited”. Europhys. Lett. 1996, 36, 473–474. [Google Scholar] [CrossRef]
- Körber, T.; Stäglich, R.; Gainaru, C.; Böhmer, R.; Rössler, E.A. Systematic differences in the relaxation stretching of polar molecular liquids probed by dielectric vs magnetic resonance and photon correlation spectroscopy. J. Chem. Phys. 2020, 153, 124510. [Google Scholar] [CrossRef]
- Nielsen, A.I.; Christensen, T.; Jakobsen, B.; Niss, K.; Olsen, N.B.; Richert, R.; Dyre, J.C. Prevalence of approximate √t relaxation for the dielectric α process in viscous organic liquids. J. Chem. Phys. 2009, 130, 154508. [Google Scholar] [CrossRef]
- Paluch, M.; Knapik, J.; Wojnarowska, Z.; Grzybowski, A.; Ngai, K.L. Universal behavior of dielectric responses of glass formers: Role of dipole-dipole interactions. Phys. Rev. Lett. 2016, 116, 25702. [Google Scholar] [CrossRef] [PubMed]
- Pabst, F.; Gabriel, J.P.; Böhmer, T.; Weigl, P.; Helbling, A.; Richter, T.; Zourchang, P.; Walther, T.; Blochowicz, T. Generic Structural Relaxation in Supercooled Liquids. J. Phys. Chem. Lett. 2021, 12, 3685–3690. [Google Scholar] [CrossRef] [PubMed]
- Pabst, F.; Helbling, A.; Gabriel, J.; Weigl, P.; Blochowicz, T. Dipole-dipole correlations and the Debye process in the dielectric response of nonassociating glass forming liquids. Phys. Rev. E 2020, 102, 10606. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, J.P.; Zourchang, P.; Pabst, F.; Helbling, A.; Weigl, P.; Böhmer, T.; Blochowicz, T. Intermolecular cross-correlations in the dielectric response of glycerol. Phys. Chem. Chem. Phys. 2020, 22, 11644–11651. [Google Scholar] [CrossRef]
- Böhmer, T.; Horstmann, R.; Gabriel, J.P.; Pabst, F.; Vogel, M.; Blochowicz, T. Origin of Apparent Slow Solvent Dynamics in Concentrated Polymer Solutions. Macromolecules 2021, 54, 10340–10349. [Google Scholar] [CrossRef]
- Körber, T.; Pötzschner, B.; Krohn, F.; Rössler, E.A. Reorientational dynamics in highly asymmetric binary low-molecular mixtures-A quantitative comparison of dielectric and NMR spectroscopy results. J. Chem. Phys. 2021, 155, 024504. [Google Scholar] [CrossRef]
- Kivelson, D.; Madden, P. Theory of dielectric relaxation. Mol. Phys. 1975, 30, 1749–1780. [Google Scholar] [CrossRef]
- Bauer, D.R.; Alms, G.R.; Brauman, J.I.; Pecora, R. Depolarized Rayleigh scattering and 13C NMR studies of anisotropic molecular reorientation of aromatic compounds in solution. J. Chem. Phys. 1974, 61, 2255–2261. [Google Scholar] [CrossRef]
- Spiess, H.W. Rotation of Molecules and Nuclear Spin Relaxation. In Dynamic NMR Spectroscopy; Diehl, P., Fluck, E., Kosfeld, R., Eds.; NMR Basic Principles and Progress; Springer: Berlin/Heidelberg, Germany, 1978; Volume 15, pp. 55–214. [Google Scholar]
- Dorfmüller, T.; Pecora, R. Rotational Dynamics of Small and Macromolecules; Springer-Verlag: Berlin, Germany, 1987; Volume 293. [Google Scholar]
- Becher, M.; Körber, T.; Döß, A.; Hinze, G.; Gainaru, C.; Böhmer, R.; Vogel, M.; Rössler, E.A. Nuclear spin relaxation in viscous liquids: Relaxation stretching of single-particle probes. J. Phys. Chem. B 2021, 125, 13519–13532. [Google Scholar] [CrossRef]
- Becher, M.; Wohlfromm, T.; Rössler, E.A.; Vogel, M. Molecular dynamics simulations vs field-cycling NMR relaxometry: Structural relaxation mechanisms in the glass-former glycerol revisited. J. Chem. Phys. 2021, 154, 124503. [Google Scholar] [CrossRef]
- Rössler, E. Corresponding States Concept for Simple Supercooled Liquids Identifying a Change of Diffusion Mechanism Above the Glass Transition Temperature. Ber. Bunsenges. Phys. Chem. 1990, 94, 392–399. [Google Scholar] [CrossRef]
- Hinze, G.; Sillescu, H.; Fujara, F. Anisotropic motion of toluene above and below the glass transition studied by 2H NMR. Chem. Phys. Lett. 1995, 232, 154–158. [Google Scholar] [CrossRef]
- Cang, H.; Novikov, V.N.; Fayer, M.D. Logarithmic decay of the orientational correlation function in supercooled liquids on the Ps to Ns time scale. J. Chem. Phys. 2003, 118, 2800–2807. [Google Scholar] [CrossRef]
- Brodin, A.; Bergman, R.; Mattsson, J.; Rössler, E.A. Light scattering and dielectric manifestations of secondary relaxations in molecular glassformers. Eur. Phys. J. B 2003, 36, 349–357. [Google Scholar] [CrossRef]
- Noack, F. Nuclear Magnetic Relaxation Spectroscopy. In NMR: Basic Principles and Progress Grundlagen und Fortschritte; Diehl, P., Fluck, E., Kosfeld, R., Eds.; Springer Verlag: Berlin, Germany, 1971; Volume 3, pp. 83–144. [Google Scholar]
- Anoardo, E.; Galli, G.; Ferrante, G. Fast-field-cycling NMR: Applications and instrumentation. Appl. Magn. Reson. 2001, 20, 365–404. [Google Scholar] [CrossRef]
- Kimmich, R.; Anoardo, E. Field-cycling NMR relaxometry. Prog. Nucl. Magn. Reson. Spectrosc. 2004, 44, 257–320. [Google Scholar] [CrossRef]
- Meier, R.; Kruk, D.; Rössler, E.A. Intermolecular spin relaxation and translation diffusion in liquids and polymer melts: Insight from field-cycling 1H NMR relaxometry. ChemPhysChem 2013, 14, 3071–3081. [Google Scholar] [CrossRef]
- Flämig, M.; Hofmann, M.; Lichtinger, A.; Rössler, E.A. Application of proton field-cycling NMR relaxometry for studying translational diffusion in simple liquids and polymer melts. Magn. Reson. Chem. 2019, 57, 805–817. [Google Scholar] [CrossRef]
- Fujara, F.; Kruk, D.; Privalov, A.F. Solid state field-cycling NMR relaxometry: Instrumental improvements and new applications. Prog. Nucl. Magn. Reson. Spectrosc. 2014, 82, 39–69. [Google Scholar] [CrossRef]
- Kresse, B.; Becher, M.; Privalov, A.F.; Hofmann, M.; Rössler, E.A.; Vogel, M.; Fujara, F. 1H NMR at Larmor frequencies down to 3 Hz by means of Field-Cycling techniques. J. Magn. Reson. 2017, 277, 79–85. [Google Scholar] [CrossRef]
- Kimmich, R.; Fatkullin, N. Self-diffusion studies by intra- and inter-molecular spin-lattice relaxometry using field-cycling: Liquids, plastic crystals, porous media, and polymer segments. Prog. Nucl. Magn. Reson. Spectrosc. 2017, 101, 18–50. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, M.; Kresse, B.; Heymann, L.; Privalov, A.F.; Willner, L.; Fatkullin, N.; Aksel, N.; Fujara, F.; Rössler, E.A. Dynamics of a Paradigmatic Linear Polymer: A Proton Field-Cycling NMR Relaxometry Study on Poly(ethylene−propylene). Macromolecules 2016, 49, 8622–8632. [Google Scholar] [CrossRef]
- Flämig, M.; Hofmann, M.; Fatkullin, N.; Rössler, E.A. NMR relaxometry: The canonical case glycerol. J. Phys. Chem. B 2020, 124, 1557–1570. [Google Scholar] [CrossRef]
- Kruk, D.; Meier, R.; Rachocki, A.; Korpała, A.; Singh, R.K.; Rössler, E.A. Determining diffusion coefficients of ionic liquids by means of field cycling nuclear magnetic resonance relaxometry. J. Chem. Phys. 2014, 140, 244509. [Google Scholar] [CrossRef] [PubMed]
- Flämig, M.; Becher, M.; Hofmann, M.; Körber, T.; Kresse, B.; Privalov, A.F.; Willner, L.; Kruk, D.; Fujara, F.; Rössler, E.A. Perspectives of deuteron field-cycling NMR relaxometry for probing molecular dynamics in soft matter. J. Phys. Chem. B 2016, 120, 7754–7766. [Google Scholar] [CrossRef] [PubMed]
- Becher, M.; Flämig, M.; Rössler, E.A. Field-cycling 31P and 1H NMR relaxometry studying the reorientational dynamics of glass forming organophosphates. J. Chem. Phys. 2022, 156, 074502. [Google Scholar] [CrossRef] [PubMed]
- Böhmer, R.; Diezemann, G.; Hinze, G.; Rössler, E. Dynamics of supercooled liquids and glassy solids. Prog. Nucl. Magn. Reson. Spectrosc. 2001, 39, 191–267. [Google Scholar] [CrossRef]
- Vogel, M.; Medick, P.; Rössler, E.A. Secondary relaxation processes in molecular glasses studied by nuclear magnetic resonance spectroscopy. In Annual Reports on NMR Spectroscopy; Webb, G.A., Ed.; Academic Press: Cambridge, MA, USA, 2005; pp. 231–299. [Google Scholar]
- Bock, D.; Kahlau, R.; Micko, B.; Pötzschner, B.; Schneider, G.J.; Rössler, E.A. On the cooperative nature of the β-process in neat and binary glasses: A dielectric and nuclear magnetic resonance spectroscopy study. J. Chem. Phys. 2013, 139, 64508. [Google Scholar] [CrossRef]
- Micko, B.; Kruk, D.; Rössler, E.A. Primary and secondary relaxation process in plastically crystalline cyanocyclohexane studied by 2H nuclear magnetic resonance. II. Quantitative analysis. J. Chem. Phys. 2013, 138, 074504. [Google Scholar] [CrossRef]
- Körber, T.; Mohamed, F.; Hofmann, M.; Lichtinger, A.; Willner, L.; Rössler, E.A. The Nature of Secondary Relaxations: The Case of Poly(ethylene-alt-propylene) Studied by Dielectric and Deuteron NMR Spectroscopy. Macromolecules 2017, 50, 1554–1568. [Google Scholar] [CrossRef]
- Winter, E.; Seipel, P.; Miß, V.; Spannenberger, S.; Roling, B.; Vogel, M. 7Li NMR Studies of Short-Range and Long-Range Lithium Ion Dynamics in a Heat-Treated Lithium Iodide-Doped Lithium Thiophosphate Glass Featuring High Ion Conductivity. Phys. Chem. C 2020, 124, 28614–28622. [Google Scholar] [CrossRef]
- Haaks, M.; Martin, S.W.; Vogel, M. Relation of short-range and long-range lithium ion dynamics in glass-ceramics: Insights from 7Li NMR field-cycling and field-gradient studies. Phys. Rev. B 2017, 96, 104301. [Google Scholar] [CrossRef]
- Abragam, A. The Principles of Nuclear Magnetism; Oxford University Press: Oxford, UK, 1961; Volume 32. [Google Scholar]
- Kariyo, S.; Herrmann, A.; Gainaru, C.; Schick, H.; Brodin, A.; Novikov, V.N.; Rössler, E.A. From a simple liquid to a polymer melt: NMR relaxometry study of polybutadiene. Phys. Rev. Lett. 2006, 97, 207803, Erratum in Phys. Rev. Lett. 2008, 100, 109901. [Google Scholar] [CrossRef] [PubMed]
- Böhmer, R.; Jeffrey K., R.; Vogel, M. Solid-state Li NMR with applications to the translational dynamics in ion conductors. Progr. Magn. Reson. Spectrosc. 2007, 50, 87–174. [Google Scholar] [CrossRef]
- Böhmer, R.; Storek, M.; Vogel, M. NMR studies of ionic dynamics in solids. In Modern Methods in Solid-State NMR: A Practitioner’s Guide; The Royal Society of Chemistry: London, UK, 2018; pp. 193–230. [Google Scholar]
- Böttcher, C.J.F.; Bordewijk, P. Dielectrics in Time-Dependent Fields, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 1978. [Google Scholar]
- Sholl, C.A. Nuclear Spin Relaxation by Translational Diffusion in Liquids and Solids: High-and Low-Frequency Limits. J. Phys. C Solid State Phys. 1981, 14, 447–464. [Google Scholar] [CrossRef]
- Kruk, D.; Meier, R.; Roessler, E.A. Nuclear Magnetic Resonance Relaxometry as a Method of Measuring Translational Diffusion Coefficients in Liquids. Phys. Rev. E 2012, 85, 020201. [Google Scholar] [CrossRef] [PubMed]
- Rössler, E.A.; Hofmann, M.; Fatkullin, N. Application of Field-cycling 1H NMR Relaxometry to the Study of Translational and Rotational Dynamics in Liquids and Polymers. In Field-Cycling NMR Relaxometry: Instrumentation, Model Theories and Applications; Kimmich, R., Ed.; RSC: Cambridge, UK, 2019. [Google Scholar]
- Hwang, L.-P.; Freed, J.H. Dynamic effects of pair correlation functions on spin relaxation by translational diffusion in liquids. J. Chem. Phys. 1975, 63, 4017–4025. [Google Scholar] [CrossRef]
- Ayant, Y.; Belorizky, E.; Aluzon, J.; Gallice, J. Calcul des densités spectrales résultant d’un mouvement aléatoire de translation en relaxation par interaction dipolaire magnétique dans les liquides. J. Phys. France 1975, 36, 991–1004. [Google Scholar] [CrossRef]
- Henritzi, P.; Bormuth, A.; Vogel, M. Interpretation of H-1 and H-2 spin-lattice relaxation dispersions: Insights from molecular dynamics simulations of polymer melts. Solid State NMR 2013, 54, 32–40. [Google Scholar] [CrossRef]
- Meier, R.; Kruk, D.; Gmeiner, J.; Rössler, E.A. Intermolecular relaxation in glycerol as revealed by field cycling 1H NMR relaxometry dilution experiments. J. Chem. Phys. 2012, 136, 34508. [Google Scholar] [CrossRef]
- Chang, I.; Sillescu, H. Heterogeneity at the Glass Transition: Translational and Rotational Self-Diffusion. J. Phys. Chem. B 1997, 101, 8794–8801. [Google Scholar] [CrossRef]
- Meier, R.; Schneider, E.; Rössler, E.A. Change of translational-rotational coupling in liquids revealed by field-cycling 1H NMR. J. Chem. Phys. 2015, 142, 34503. [Google Scholar] [CrossRef] [PubMed]
- Ferry, J.D. Viscoelastic Properties of Polymers, 3rd ed.; Wiley: New York, NY, USA, 1980. [Google Scholar]
- Brodin, A.; Gainaru, C.; Porokhonskyy, V.; Rössler, E.A. Evolution of dynamic susceptibility in molecular glass formers—a critical assessment. J. Phys. Condens. Matter 2007, 19, 205104. [Google Scholar] [CrossRef]
- Herrmann, A.; Kresse, B.; Gmeiner, J.; Privalov, A.F.; Kruk, D.; Fujara, F.; Rössler, E.A. Protracted Crossover to Reptation Dynamics: A Field Cycling 1H NMR Study Including Extremely Low Frequencies. Macromolecules 2012, 45, 1408–1416. [Google Scholar] [CrossRef]
- Schnauss, W.; Fujara, F.; Sillescu, H. The molecular dynamics around the glass transition and in the glassy state of molecular organic systems: A 2H−nuclear magnetic resonance (NMR) study. J. Chem. Phys. 1992, 97, 1378–1389. [Google Scholar] [CrossRef]
- Blochowicz, T.; Tschirwitz, C.; Benkhof, S.; Rössler, E.A. Susceptibility functions for slow relaxation processes in supercooled liquids and the search for universal relaxation patterns. J. Chem. Phys. 2003, 118, 7544–7555. [Google Scholar] [CrossRef]
- Gainaru, C.; Lips, O.A.; Troshagina, A.; Kahlau, R.; Brodin, A.; Fujara, F.; Rössler, E.A. On the nature of the high-frequency relaxation in a molecular glass former: A joint study of glycerol by field cycling NMR, dielectric spectroscopy, and light scattering. J. Chem. Phys. 2008, 128, 174505. [Google Scholar] [CrossRef]
- Brodin, A.; Rössler, E.A. Depolarized light scattering study of glycerol. Eur. Phys. J. B 2005, 44, 3–14. [Google Scholar] [CrossRef]
- Chang, I.; Fujara, F.; Geil, B.; Hinze, G.; Sillescu, H.; Tölle, A. New Perspectives of NMR in Ultrahigh Static Magnetic-Field Gradients. J. Non-Cryst. Solids 1994, 172, 674–681. [Google Scholar] [CrossRef]
- Edward, J.T. Molecular Volumes and the Stokes-Einstein-Equation. J. Chem. Educ. 1970, 47, 261–270. [Google Scholar] [CrossRef]
- Becher, M.; Horstmann, R.; Kloth, S.; Rössler, E.A.; Vogel, M. A relation between the formation of a hydrogen-bond network and a time-scale separation of translation and rotation in molecular liquids. J. Phys. Chem. Lett. 2022. submitted. [Google Scholar]
- Meier, R.; Kahlau, R.; Kruk, D.; Rössler, E.A. Comparative Studies of the Dynamics in Viscous Liquids by Means of Dielectric Spectroscopy and Field Cycling NMR. J. Phys. Chem. A 2010, 114, 7847–7855. [Google Scholar] [CrossRef] [PubMed]
- Dries, T.; Fujara, F.; Kiebel, M.; Rössler, E.; Sillescu, H. 2H-NMR study of the glass transition in supercooled ortho-terphenyl. J. Chem. Phys. 1988, 88, 2139–2147. [Google Scholar] [CrossRef]
- Petzold, N.; Rössler, E.A. Light scattering study on the glass former o-terphenyl. J. Chem. Phys. 2010, 133, 124512. [Google Scholar] [CrossRef]
- Roed, A.L.; Dyre, J.C.; Niss, C.; Hecksher, T.; Riechers, B. Time-scale ordering in hydrogen- and van der Waals-bonded liquids. J. Chem. Phys. 2021, 154, 184508. [Google Scholar] [CrossRef]
- Döß, A.; Paluch, M.; Sillescu, H.; Hinze, G. Dynamics in supercooled polyalcohols: Primary and secondary relaxation. J. Chem. Phys. 2002, 117, 6582–6589. [Google Scholar] [CrossRef]
- Rössler, E.A.; Eiermann, P. Reorientational dynamics in supercooled m-tricresyl phosphate: Its relation to main and secondary relaxation—31P nuclear magnetic resonance study of relaxation, line shape, and stimulated echo. J. Chem. Phys. 1990, 100, 5237–5248. [Google Scholar] [CrossRef]
- Adishchev, S.; Bock, D.; Gainaru, C.; Kahlau, R.; Micko, B.; Petzold, N.; Pötzschner, B.; Rössler, E.A. Reorientational dynamics of organophosphate glass formers–a joint study by 31P NMR, dielectric spectroscopy and light scattering. Z. Phys. Chem. 2012, 226, 1149–1168. [Google Scholar] [CrossRef]
- Schmidtke, B.; Petzold, N.; Kahlau, R.; Rössler, E.A. Reorientational dynamics in molecular liquids as revealed by dynamic light scattering: From boiling point to glass transition temperature. J. Chem. Phys. 2013, 139, 84504. [Google Scholar] [CrossRef]
- Kudlik, A. Dielektrische Spektroskopie an Organischen Glasbildnern: Ein Beitrag zur Linienform der Dynamischen Suszeptibilität. Ph.D. Thesis, Universität Bayreuth, Bayreuth, Germany, 1997. [Google Scholar]
- Blochowicz, T.; Gainaru, C.; Medick, P.; Tschirwitz, C.; Rössler, E.A. The dynamic susceptibility in glass forming molecular liquids: The search for universal relaxation patterns II. J. Chem. Phys. 2006, 124, 134503. [Google Scholar] [CrossRef]
- Rams-Baron, M.; Yao, B.; Cheng, S.; Dulski, M.; Paluch, M. Complex Reorientation Dynamics of Sizable Glass-Formers with Polar Rotors Revealed by Dielectric Spectroscopy. J. Phys. Chem. Lett. 2021, 12, 11303. [Google Scholar] [CrossRef] [PubMed]
- Weingärtner, H. Zum Verständnis ionischer Flüssigkeiten auf molekularer Ebene: Fakten, Probleme und Kontroversen. Angew. Chem. Int. Ed. 2008, 120, 664–682. [Google Scholar] [CrossRef]
- Rivera, A.; Brodin, A.; Pugachev, A.; Rössler, E.A. Orientational and translational dynamics in room temperature ionic liquids. J. Chem. Phys. 2007, 126, 114503. [Google Scholar] [CrossRef] [PubMed]
- Griffin, P.J.; Holt, A.P.; Tsunashima, K.; Sangoro, J.R.; Friedrich Kremer, F.; Sokolov, A.P. Ion transport and structural dynamics in homologous ammonium and phosphonium-based room temperature ionic liquids. J. Chem. Phys. 2015, 142, 084501. [Google Scholar] [CrossRef]
- Becher, M.; Steinrücken, E.; Vogel, M. On the relation between reorientation and diffusion in glass-forming ionic liquids with micro-heterogeneous structures. J. Chem. Phys. 2019, 151, 194503. [Google Scholar] [CrossRef]
- Steinrücken, E.; Becher, M.; Vogel, M. On the molecular mechanisms of α and β relaxations in ionic liquids. J. Chem. Phys. 2020, 153, 104507. [Google Scholar] [CrossRef]
- Pabst, F.; Gabriel, J.; Blochowicz, T. Mesoscale Aggregates and Dynamic Asymmetry in Ionic Liquids: Evidence from Depolarized Dynamic Light Scattering. J. Phys. Chem. Lett. 2019, 10, 2130–2134. [Google Scholar] [CrossRef]
- Sangoro, J.R.; Kremer, F. Charge Transport and Glassy Dynamics in Ionic Liquids. Acc. Chem. Res. 2012, 45, 525–532. [Google Scholar] [CrossRef]
- Sangoro, J.R.; Serghei, A.; Naumov, S.; Galvosas, P.; Kärger, J.; Wespe, C.; Bordusa, F.; Kremer, F. Charge transport and mass transport in imidazolium-based ionic liquids. Phys. Rev. E 2008, 77, 051202. [Google Scholar] [CrossRef]
- Schmidtke, B.; Petzold, N.; Pötzschner, B.; Weingärtner, W.; Rössler, E.A. Relaxation Stretching, Fast Dynamics, and Activation Energy: A Comparison of Molecular and Ionic Liquids as Revealed by Depolarized Light Scattering. J. Phys. Chem. 2014, 118, 7108–7118. [Google Scholar] [CrossRef]
- Russina, O.; Beiner, M.; Pappas, C.; Russina, M.; Arrighi, V.; Unruh, T.; Mullan, C.L.; Hardacre, C.; Triolo, A. Temperature Dependence of the Primary Relaxation in 1-Hexyl-3-methylimidazolium bis{(trifluoromethyl)sulfonyl}imide. J. Phys. Chem. B 2009, 113, 8469–8474. [Google Scholar] [CrossRef] [PubMed]
- Annapureddy, H.V.R.; Kashyap, H.K.; De Biase, P.M.; Margulis, C.J. What is the Origin of the Prepeak in the X-ray Scattering of Imidazolium-Based Room-Temperature Ionic Liquids? J. Phys. Chem. B 2010, 114, 16838–16846. [Google Scholar] [CrossRef] [PubMed]
- Russina, O.; Triolo, A.; Gontrani, L. Caminiti, R. Mesoscopic structural heterogeneities in room-temperature ionic liquids. J. Phys. Chem. Lett. 2012, 3, 27–33. [Google Scholar] [CrossRef]
- Galitskaya, E.A.; Privalov, A.F.; Vogel, M.; Ryzhkin, I.A.; Sinitsyn, V.V. Self-diffusion micromechanism in Nafion studied by 2H NMR relaxation dispersion. J. Chem. Phys. 2021, 154, 034904. [Google Scholar] [CrossRef]
- Kehr, M.; Fatkullin, N.; Kimmich, R. Deuteron and proton spin-lattice relaxation dispersion of polymer melts: Intrasegment, intrachain, and interchain contributions. J. Chem. Phys. 2007, 127, 084911. [Google Scholar] [CrossRef]
- Qvist, J.; Persson, E.; Mattea, C.; Halle, B. Time scales of water dynamics at biological interfaces: Peptides, proteins and cells. Faraday Discuss. 2009, 141, 131–144. [Google Scholar] [CrossRef]
- Pabst, F. Understanding the Relaxation Spectra of Neat and Mixed Ionic Liquids. Ph.D. Thesis, TU Darmstadt, Darmstadt, Germany, 2022. [Google Scholar]
- Rivera, A.; Rössler, E.A. Evidence of secondary relaxations in the dielectric spectra of ionic liquids. Phys. Rev. B 2006, 73, 212201. [Google Scholar] [CrossRef]
- Angell, C.A. Liquid Fragility and the Glass Transition in Water and Aqueous Solutions. Chem. Rev. 2002, 102, 2627–2650. [Google Scholar] [CrossRef]
- Kobayashi, M.; Tanaka, H. Possible link of the V-shaped phase diagram to the glass-forming ability and fragility in a water-salt mixture. Phys. Rev. Lett. 2011, 106, 125703. [Google Scholar] [CrossRef]
- Turton, D.A.; Corsaro, C.; Martin, D.F.; Mallamace, F.; Wynne, K. The dynamic crossover in water does not require bulk water. Phys. Chem. Chem. Phys. 2012, 14, 8067–8073. [Google Scholar] [CrossRef][Green Version]
- Moynihan, C.T.; Balitactac, N.; Boone, L.; Litovitz, T.A. Comparison of shear and conductivity relaxation times for concentrated lithium chloride solutions. J. Chem. Phys. 1971, 55, 3013–3019. [Google Scholar] [CrossRef]
- Nakanishi, M.; Griffin, P.; Mamontov, E.; Sokolov, A.P. No fragile-to-strong crossover in LiCl-H2O solution. J. Chem. Phys. 2012, 136, 124512. [Google Scholar] [CrossRef]
- Schneider, S.; Vogel, M. NMR studies on the coupling of ion and water dynamics on various time and length scales in glass-forming LiCl aqueous solutions. J. Chem. Phys. 2018, 149, 104501. [Google Scholar] [CrossRef] [PubMed]
- Münzner, P.; Hoffmann, L.; Böhmer, R.; Gainaru, C. Deeply supercooled aqueous LiCl solution studied by frequency-resolved shear rheology. J. Chem. Phys. 2019, 150, 234505. [Google Scholar] [CrossRef]
- Capaccioli, S.; Ngai, K.L.; Shinyashiki, N. The Johari- Goldstein β-relaxation of water. J. Chem Phys. 2007, 111, 8197–8209. [Google Scholar] [CrossRef] [PubMed]
- Gainaru, C.; Meier, R.; Schildmann, S.; Lederle, C.; Hiller, W.; Rössler, E.A.; Böhmer, R. Nuclear-magnetic-resonance measurements reveal the origin of the Debye process in monohydroxy alcohols. Phys. Rev. Lett. 2010, 105, 258303. [Google Scholar] [CrossRef] [PubMed]
- Gainaru, C.; Figuli, R.; Hecksher, T.; Jakobsen, B.; Dyre, J.C.; Wilhelm, M.; Böhmer, R. Shear-Modulus Investigations of Monohydroxy Alcohols: Evidence for a Short-Chain-Polymer Rheological Response. Phys. Rev. Lett. 2014, 112, 098301. [Google Scholar] [CrossRef]
- Hecksher, T.; Jakobsen, B. Communication: Supramolecular structures in monohydroxy alcohols: Insights from shear-mechanical studies of a systematic series of octanol structural isomers. J. Chem. Phys. 2014, 141, 101104. [Google Scholar] [CrossRef]
- Iwahashi, M.; Ohbu, Y.; Kato, T.; Suzuki, Y.; Yamauchi, K.; Yamaguchi, Y.; Muramatsu, M. The dynamical structure of normal alcohols in their liquids as determined by the viscosity and self-diffusion measurements. Bull. Chem. Soc. Jpn. 1986, 59, 3771–3774. [Google Scholar] [CrossRef]
- Rymdén, R.; Carlfords, J.; Stilbs, P. Substrate binding to cyclodextrins in aqueous solution: A multicomponent self-diffusion study. J. Inclusion Phenomena 1983, 1, 159–167. [Google Scholar] [CrossRef]
- Alcoutlabi, M.; McKenna, G.B. Effects of confinement on material behaviour at the nanometre size scale. J. Phys. Condens. Matter 2005, 17, R461. [Google Scholar] [CrossRef]
- Richert, R. Dynamics of nanoconfined supercooled liquids. Annu. Rev. Phys. Chem. 2011, 62, 65–84. [Google Scholar] [CrossRef] [PubMed]
- Cerveny, S.; Mallamace, F.; Swenson, J.; Vogel, M.; Xu, L. Confined Water as Model of Supercooled Water. Chem. Rev. 2016, 116, 7608–7625. [Google Scholar] [CrossRef] [PubMed]
- Buntkowsky, G.; Vogel, M. Small Molecules, Non-Covalent Interactions, and Confinement. Molecules 2020, 25, 3311. [Google Scholar] [CrossRef] [PubMed]
- Gradmann, S.; Medick, P.; Rössler, E.A. Glassy Dynamics in Nanoconfinement as Revealed by 31P NMR. J. Phys. Chem. B 2009, 113, 8443–8445. [Google Scholar] [CrossRef]
- Vogel, M. NMR studies on simple liquids in confinement. Eur. Phys. J. Special Topics 2010, 189, 47–64. [Google Scholar] [CrossRef]
- Demuth, D.; Sattig, M.; Steinrücken, E.; Weigler, M.; Vogel, M. 2H NMR studies on the dynamics of pure and mixed hydrogen-bonded liquids in confinement. Z. Phys. Chem. 2018, 232, 1059–1087. [Google Scholar] [CrossRef]
- Geske, J.; Harrach, M.; Heckmann, L.; Horstmann, R.; Klameth, F.; Müller, N.; Pafong, E.; Wohlfromm, T.; Drossel, B.; Vogel, M. Molecular dynamics simulations of water, silica, and aqueous mixtures in bulk and confinement. Z. Phys. Chem. 2018, 232, 1187–1225. [Google Scholar] [CrossRef]
- Klameth, F.; Henritzi, P.; Vogel, M. Static and dynamic length scales in supercooled liquids: Insights from molecular dynamics simulations of water and tri-propylene oxide. J. Chem. Phys. 2014, 140, 144501. [Google Scholar] [CrossRef]
- Horstmann, R.; Sanjon, E.P.; Drossel, B.; Vogel, M. Effects of confinement on supercooled tetrahedral liquids. J. Chem. Phys. 2019, 150, 214704. [Google Scholar] [CrossRef]
- Weigler, M.; Brodrecht, M.; Buntkowsky, G.; Vogel, M. Reorientation of deeply cooled water in mesoporous silica: NMR studies of the pore-size dependence. J. Phys. Chem. B 2019, 123, 2123–2134. [Google Scholar] [CrossRef] [PubMed]
- Weigler, M.; Winter, E.; Kresse, B.; Brodrecht, M.; Buntkowsky, G.; Vogel, M. Static field gradient NMR studies of water diffusion in mesoporous silica. Phys. Chem. Chem. Phys. 2020, 22, 13989–13998. [Google Scholar] [CrossRef] [PubMed]
- Reuhl, M.; Weigler, M.; Brodrecht, M.; Buntkowsky, G.; Vogel, M. Nuclear Magnetic Resonance and Broadband Dielectric Spectroscopy Studies on the Dynamics of Ethylene Glycol in Mesoporous Silica. J. Phys. Chem. C 2020, 124, 20998–21012. [Google Scholar] [CrossRef]
- Huwe, A.; Kremer, F.; Behrens, P.; Schwieger, W. Molecular dynamics in confining space: From the single molecule to the liquid state. Phys. Rev. Lett. 1999, 82, 2338. [Google Scholar] [CrossRef]
- Lusceac, S.; Rosenstihl, M.; Vogel, M.; Gainaru, C.; Fillmer, A.; Böhmer, R. NMR and dielectric studies of hydrated collagen and elastin: Evidence for a delocalized secondary relaxation. J. Non-Cryst. Solids 2011, 357, 655–663. [Google Scholar] [CrossRef][Green Version]
- Blochowicz, T.; Schramm, S.; Lusceac, S.; Vogel, M.; Stühn, B.; Gutfreund, P.; Frick, B. Signature of a Type-A Glass Transition and Intrinsic Confinement Effects in a Binary Glass-Forming System. Phys. Rev. Lett. 2012, 109, 035702. [Google Scholar] [CrossRef]
- Reuhl, M. Untersuchung der Dynamik Wasserstoffbrückenbildender Flüssigkeiten in Eingeschränkten Geometrien. Ph.D. Thesis, TU Darmstadt, Darmstadt, Germany, 2022. [Google Scholar]
- Kahlau, R.; Dörfler, T.; Rössler, E.A. Secondary relaxations in a series of organic phosphate glasses revealed by dielectric Spectroscopy. J. Chem. Phys. 2013, 139, 134504. [Google Scholar] [CrossRef]
- Wu, T.; Jin, X.; Saini, M.K.; Liu, Y.D.; Ngai, K.L.; Wang, L.-M. Presence of global and local α-relaxations in an alkyl phosphate glass former. J. Chem. Phys. 2017, 147, 34501. [Google Scholar] [CrossRef]
- Carignani, E.; Flämig, M.; Calucci, L.; Rössler, E.R. Dynamics in the plastic crystalline phase of cyanocyclohexane and isocyanocyclohexane probed by 1H field cycling NMR relaxometry. J. Chem. Phys. 2021, 154, 234506. [Google Scholar] [CrossRef]
- Lebon, M.J.; Dreyfus, C.; Guissani, Y.; Pick, R.M.; Cummins, H.Z. Light scattering and dielectric susceptibility spectra of glassforming liquids. Z. Phys. B 1997, 103, 433–439. [Google Scholar] [CrossRef]
- Blochowicz, T.; Kudlik, A.; Benkhof, S.; Senker, J.; Rössler, E.; Hinze, G. The spectral density in simple organic glass formers: Comparison of dielectric and spin-lattice relaxation. J. Chem. Phys. 1999, 110, 12011–12022. [Google Scholar] [CrossRef]
- Galitskaya, E.; Privalov, A.F.; Weigler, M.; Vogel, M.; Kashin, A.; Ryzhkin, M.; Sinitsyn, V. NMR studies of proton exchange membranes in wide temperature range. J. Membrane Sci. 2020, 596, 117691. [Google Scholar] [CrossRef]














Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Becher, M.; Lichtinger, A.; Minikejew, R.; Vogel, M.; Rössler, E.A. NMR Relaxometry Accessing the Relaxation Spectrum in Molecular Glass Formers. Int. J. Mol. Sci. 2022, 23, 5118. https://doi.org/10.3390/ijms23095118
Becher M, Lichtinger A, Minikejew R, Vogel M, Rössler EA. NMR Relaxometry Accessing the Relaxation Spectrum in Molecular Glass Formers. International Journal of Molecular Sciences. 2022; 23(9):5118. https://doi.org/10.3390/ijms23095118
Chicago/Turabian StyleBecher, Manuel, Anne Lichtinger, Rafael Minikejew, Michael Vogel, and Ernst A. Rössler. 2022. "NMR Relaxometry Accessing the Relaxation Spectrum in Molecular Glass Formers" International Journal of Molecular Sciences 23, no. 9: 5118. https://doi.org/10.3390/ijms23095118
APA StyleBecher, M., Lichtinger, A., Minikejew, R., Vogel, M., & Rössler, E. A. (2022). NMR Relaxometry Accessing the Relaxation Spectrum in Molecular Glass Formers. International Journal of Molecular Sciences, 23(9), 5118. https://doi.org/10.3390/ijms23095118