
Proline Oxidation Supports Mitochondrial ATP Production When Complex I Is Inhibited

 , ,
, , 
Abstract
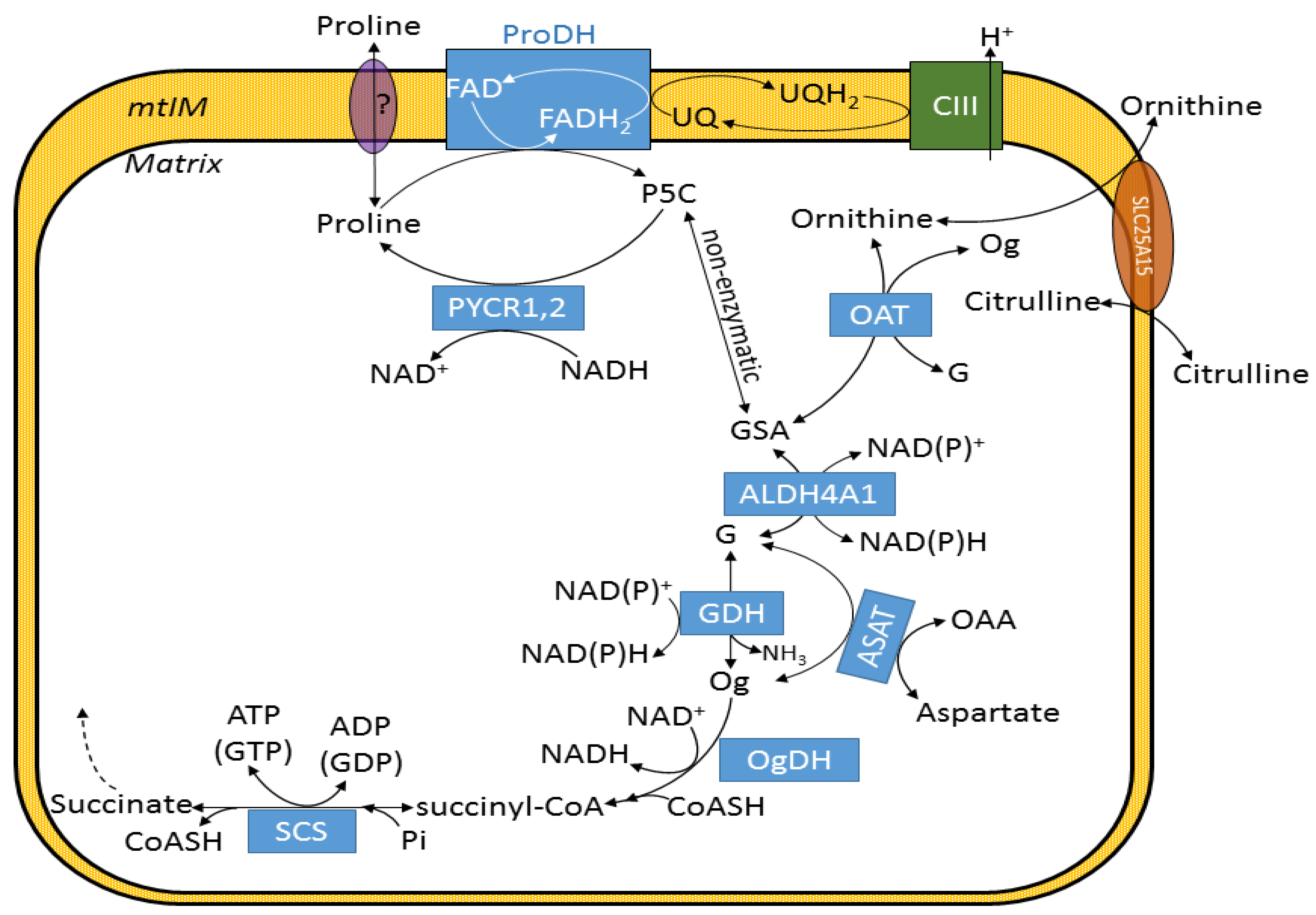
:1. Introduction
2. Results
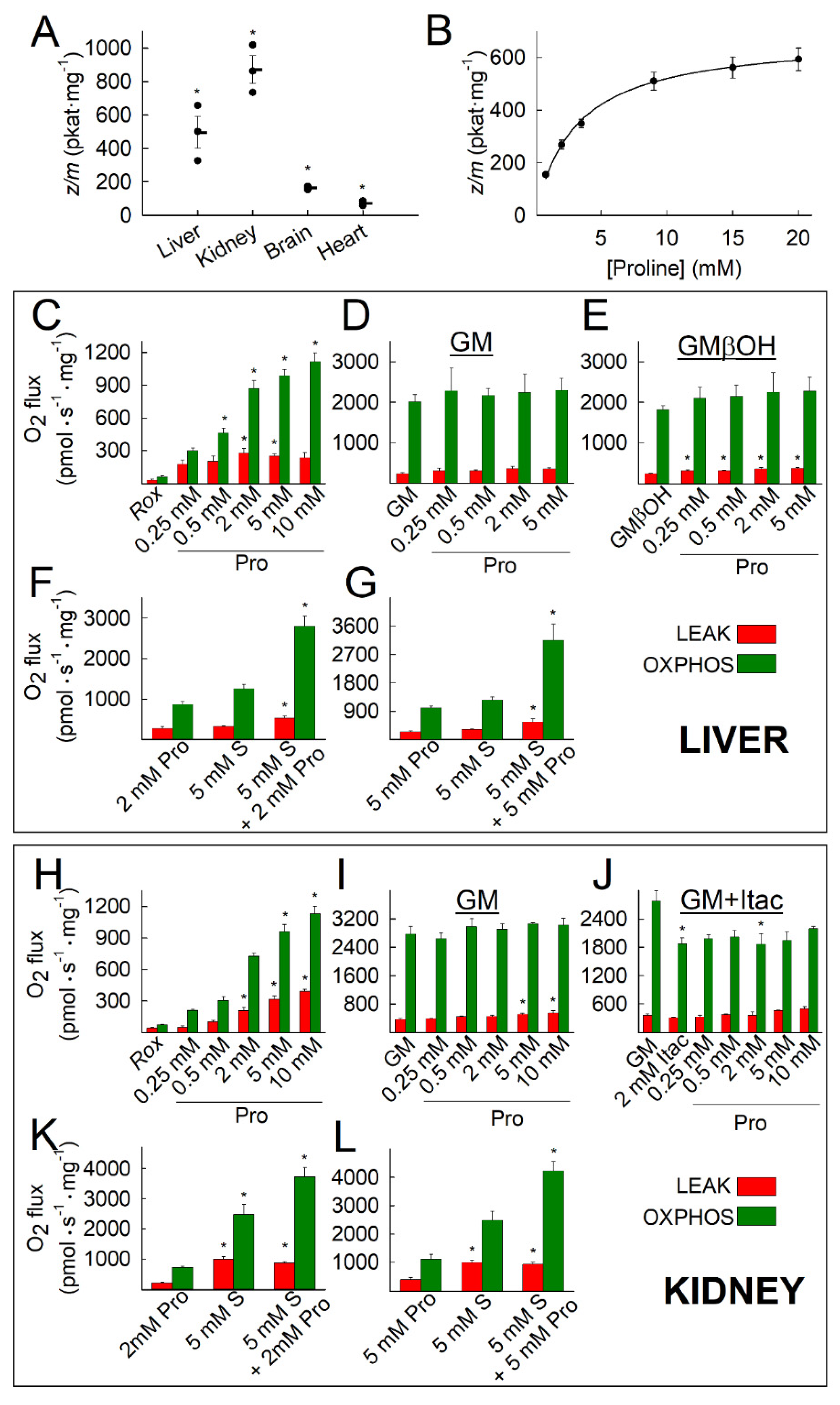
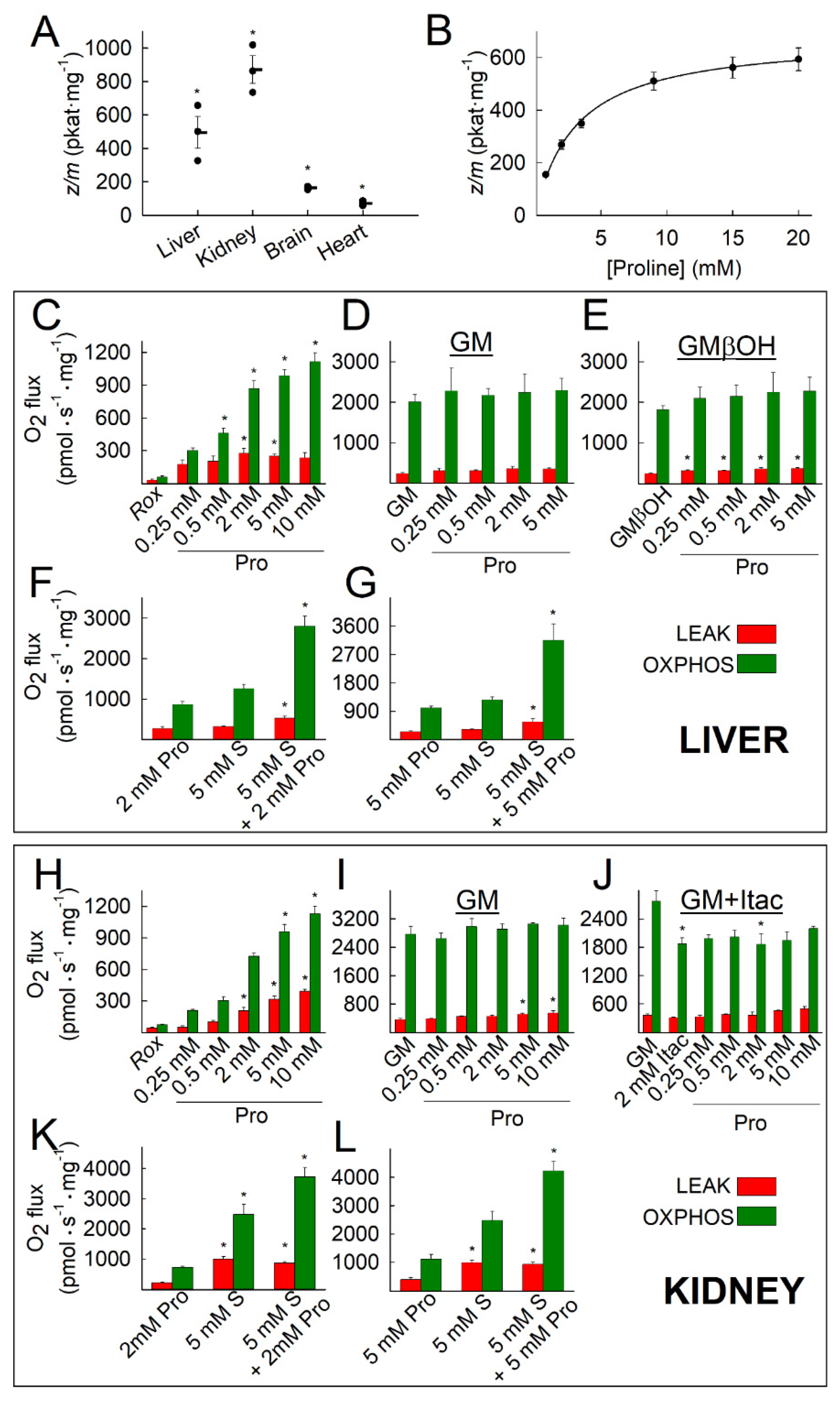
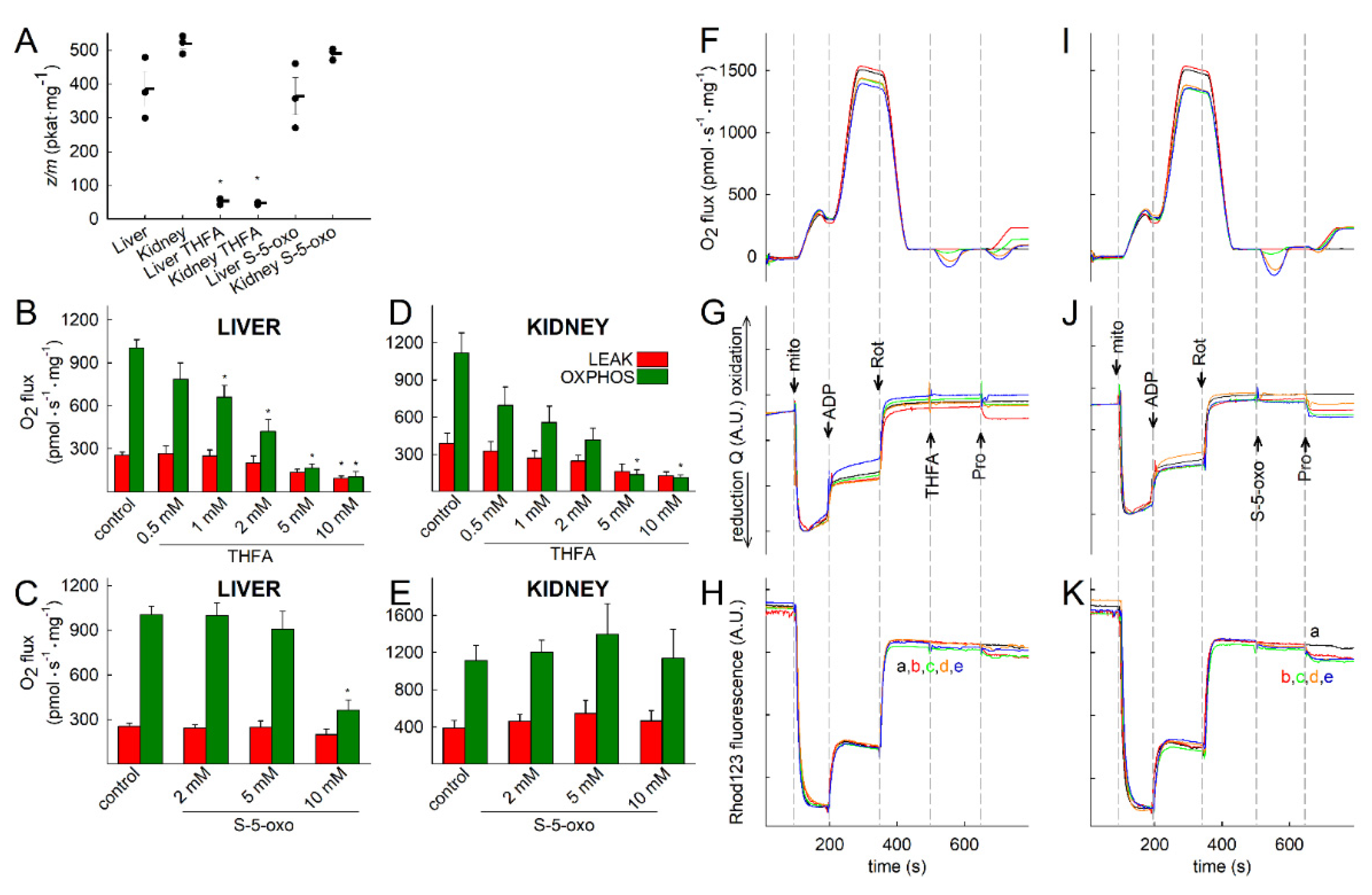
2.1. Kinetic Characterization of Proline Dehydrogenase in Mitochondria Isolated from Various Mouse Tissues
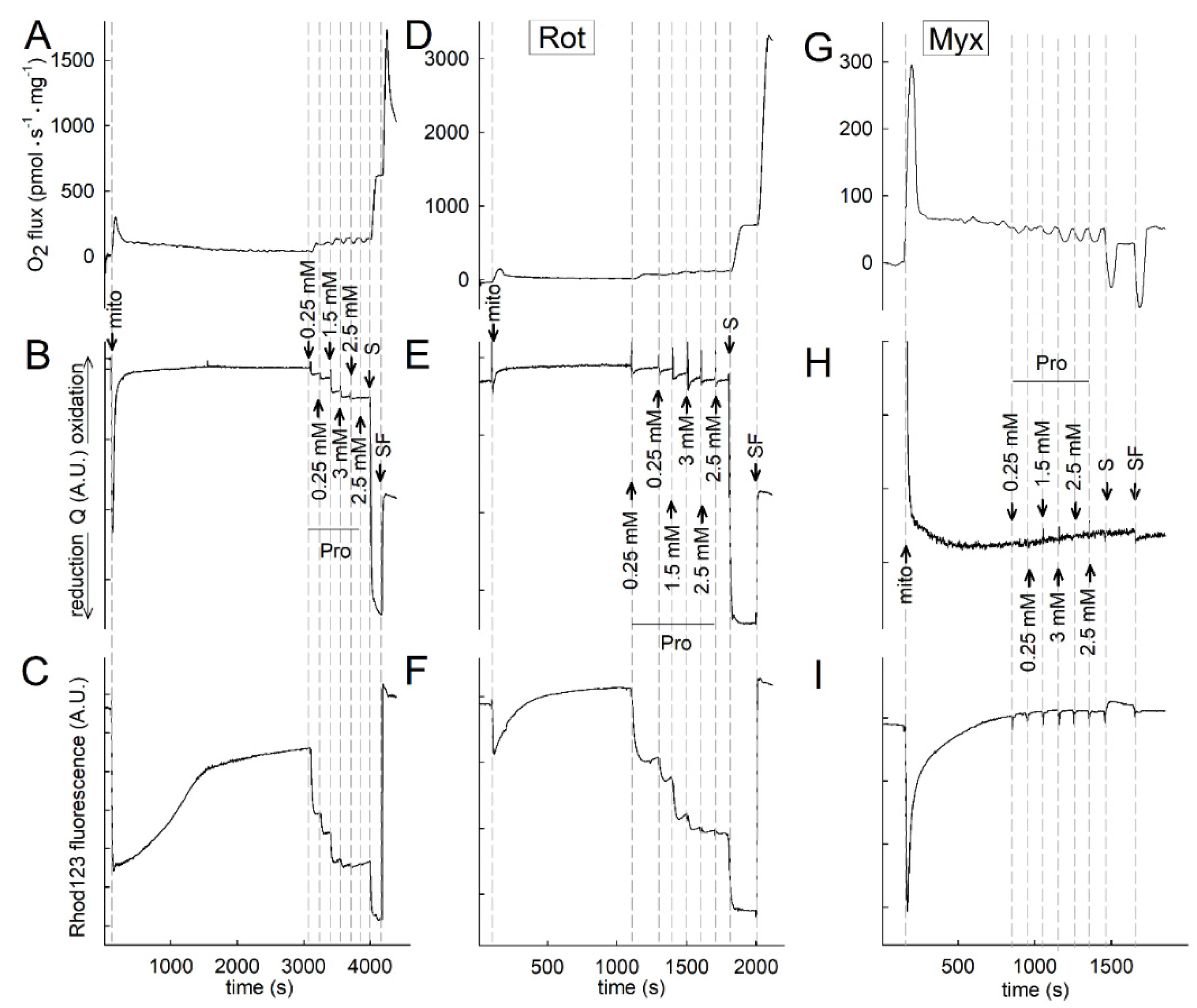
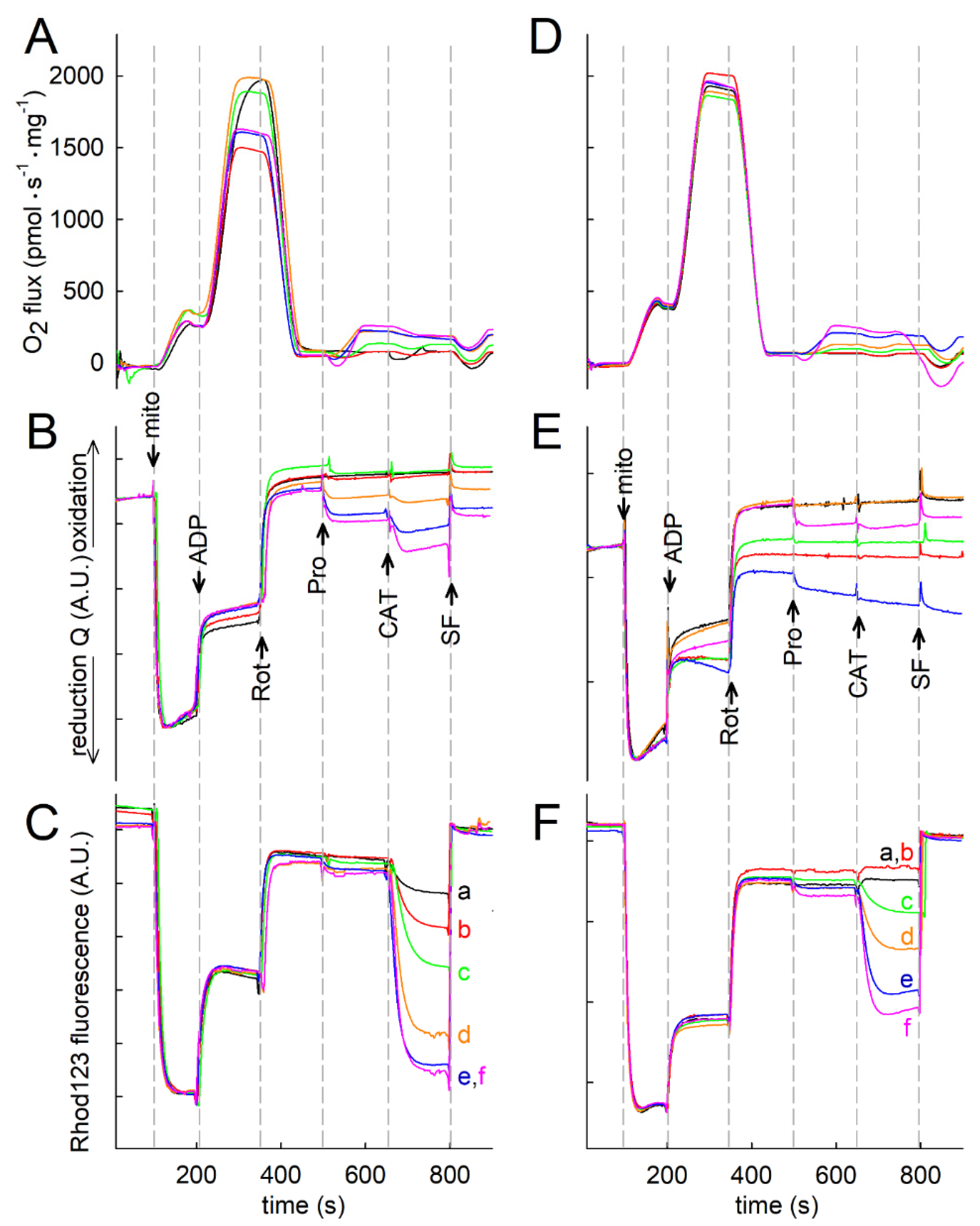
2.2. Liver and Kidney Mitochondria Respire on Proline
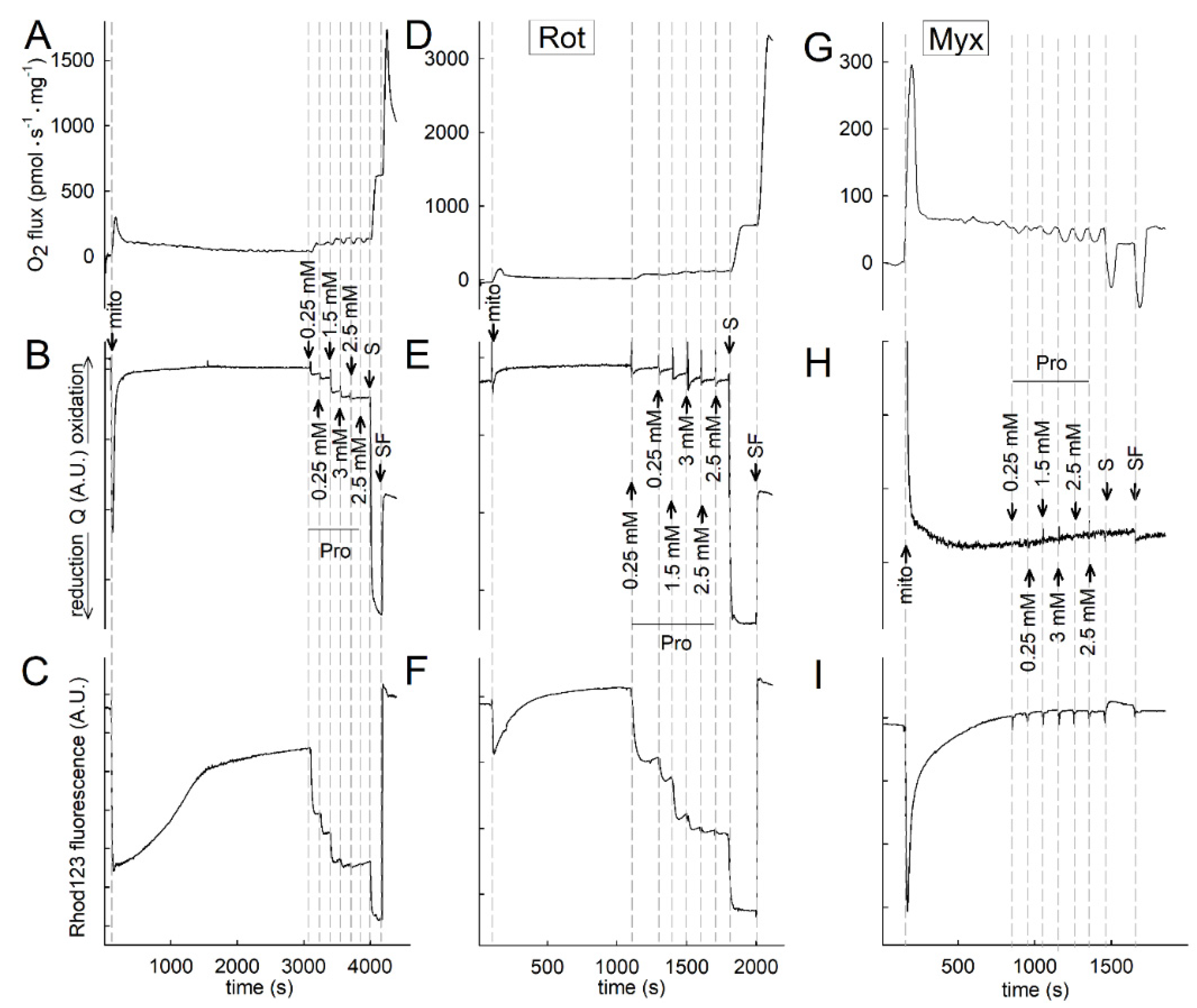
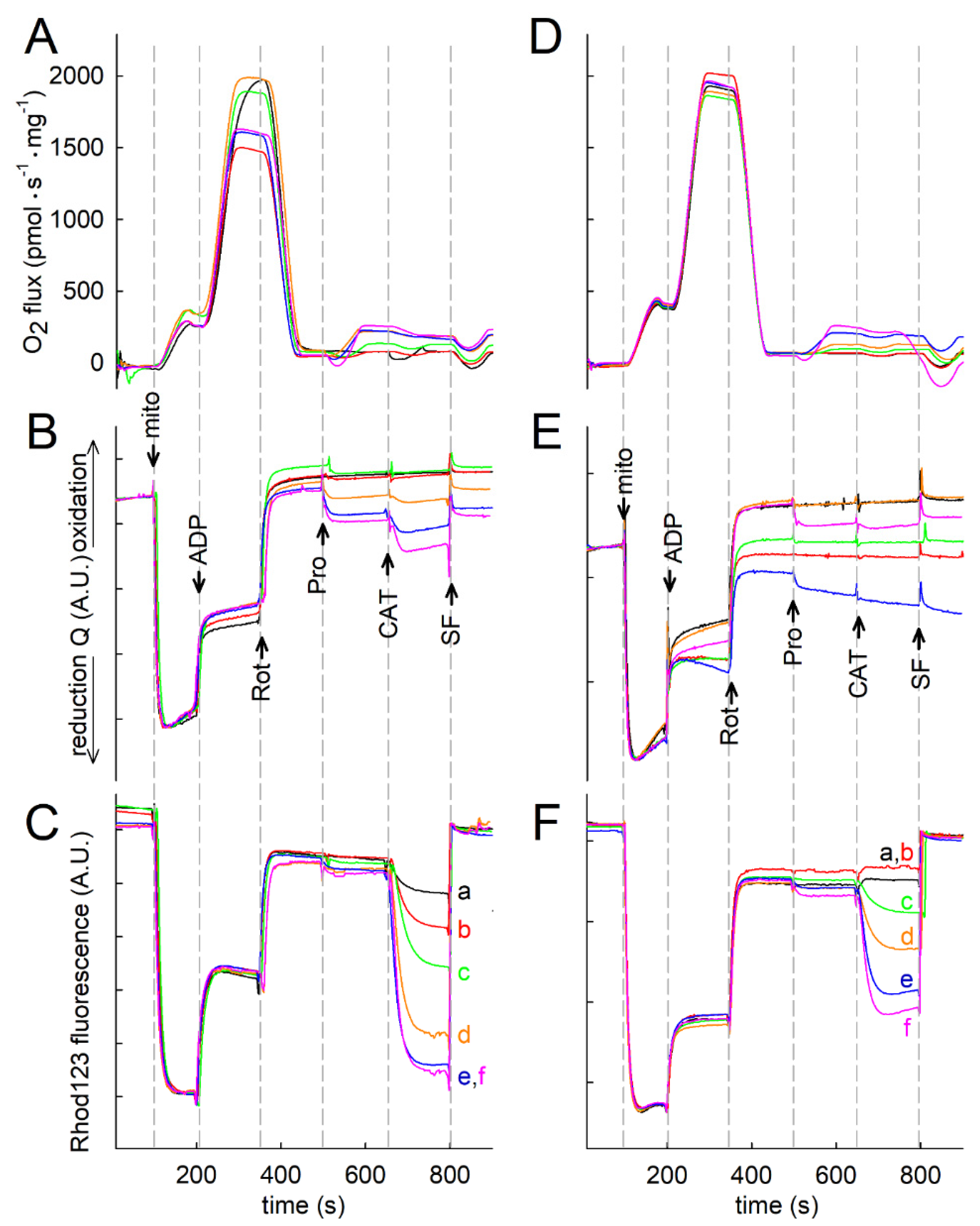
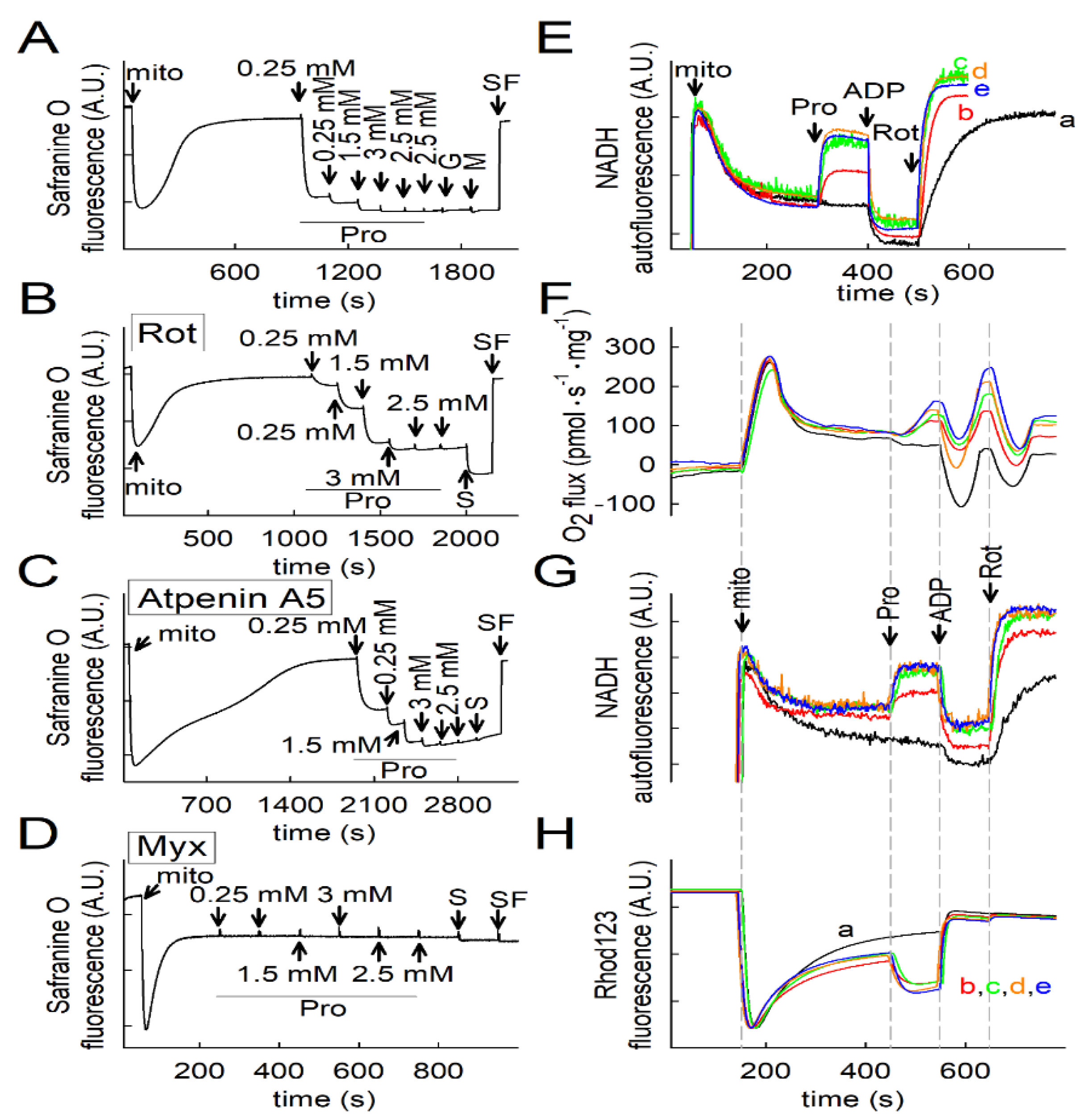
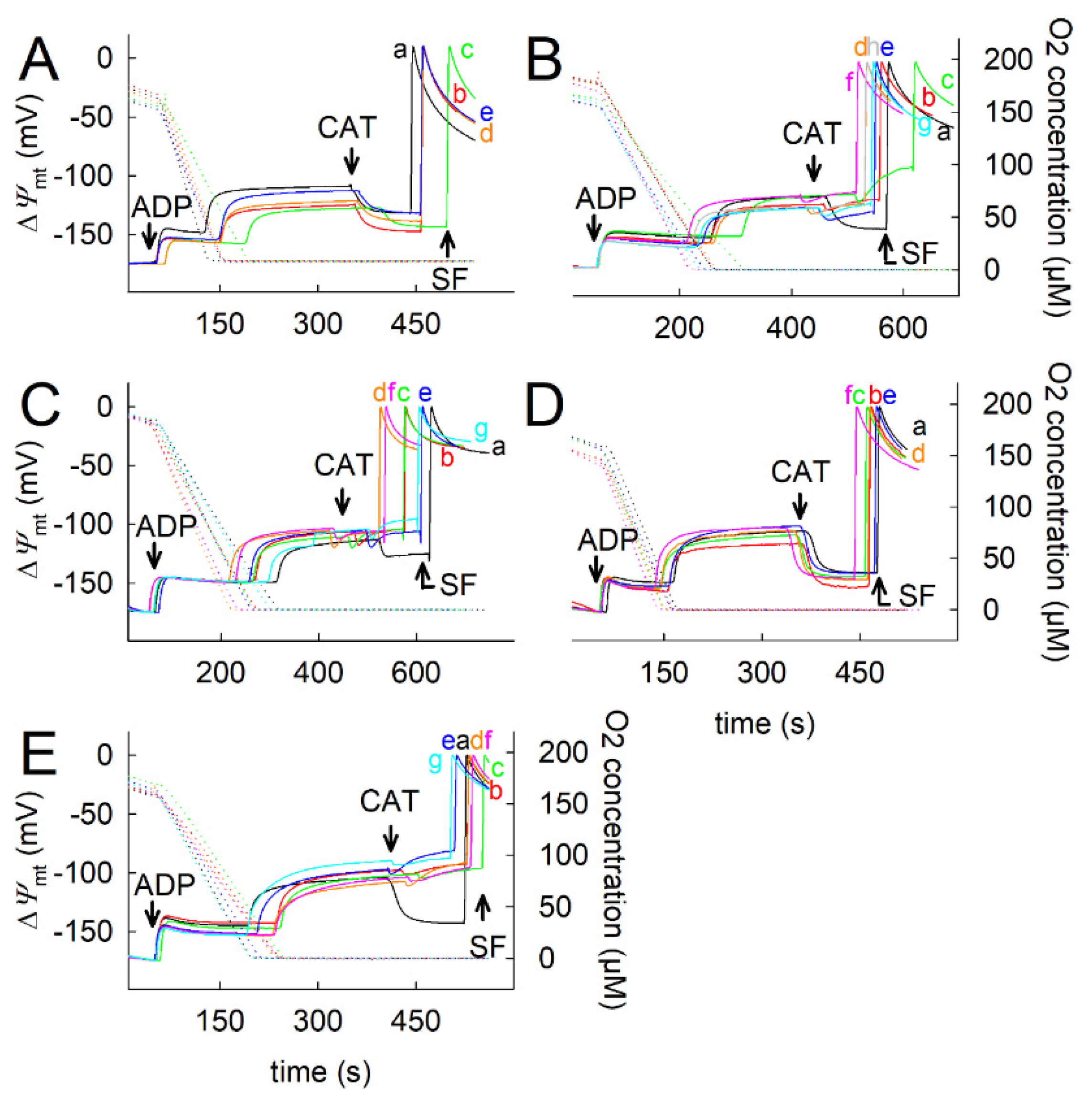
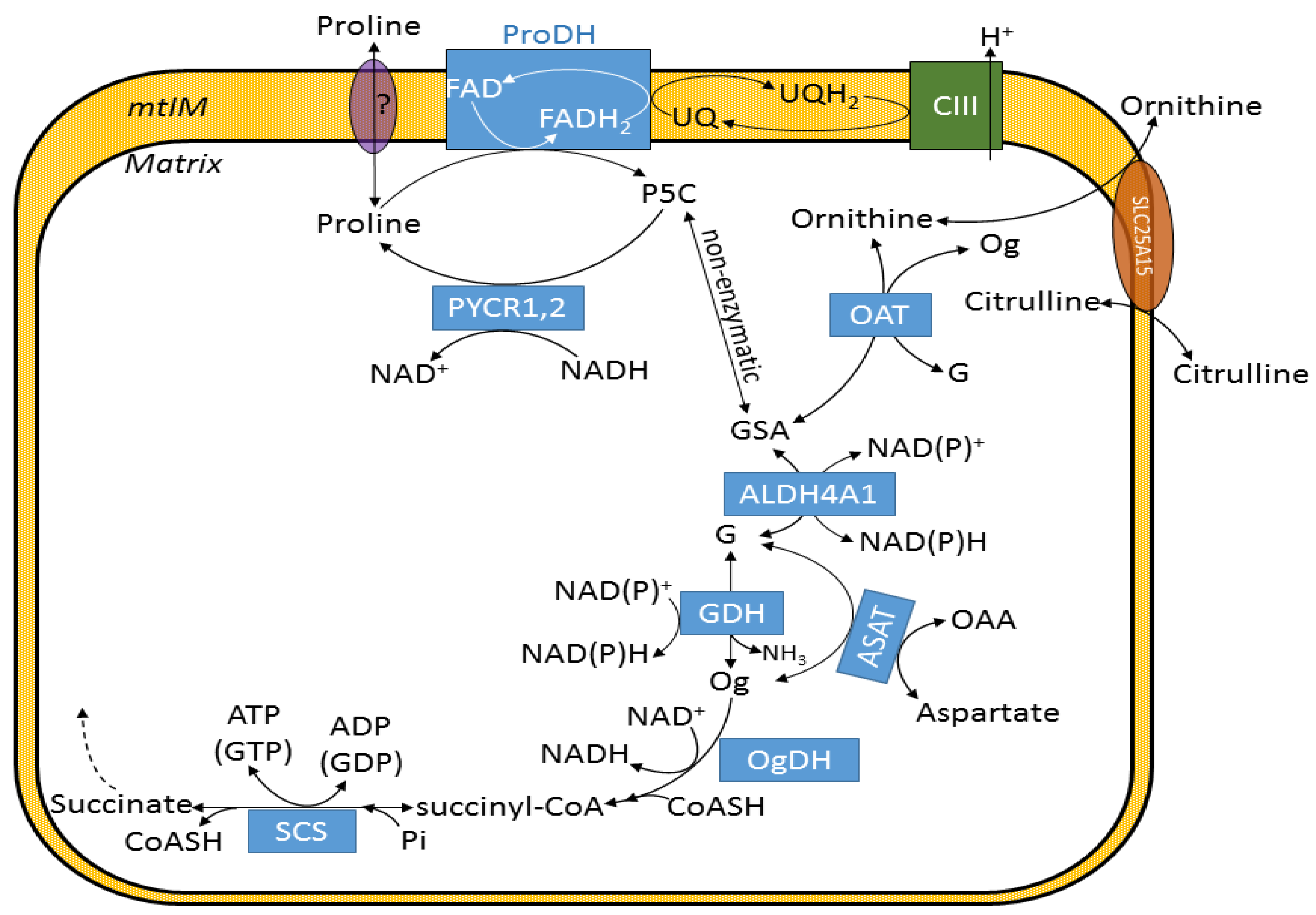
2.3. Effect of Proline on ΔΨmt, NADH Autofluorescence and Q Redox State of Isolated Mitochondria
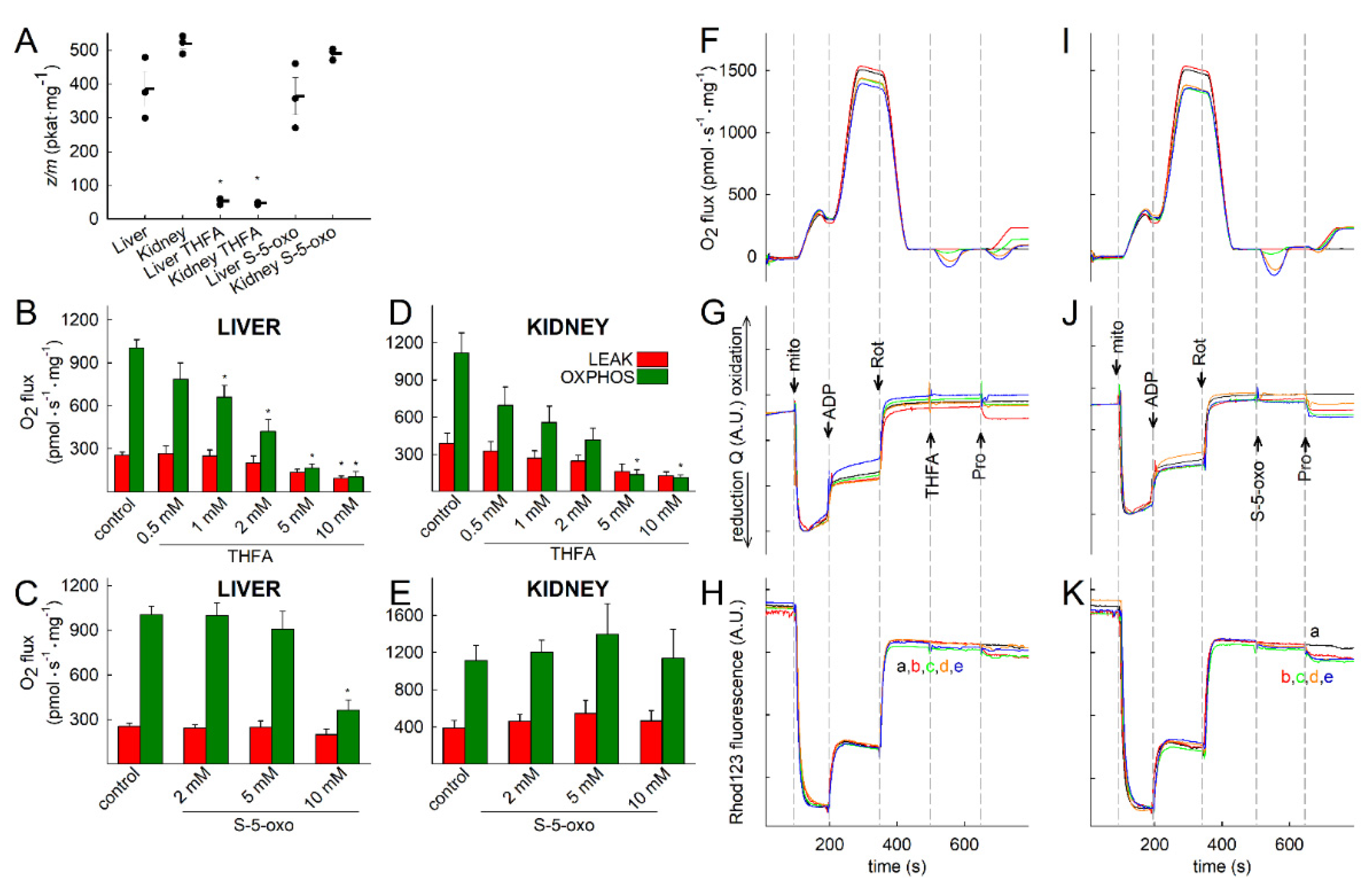
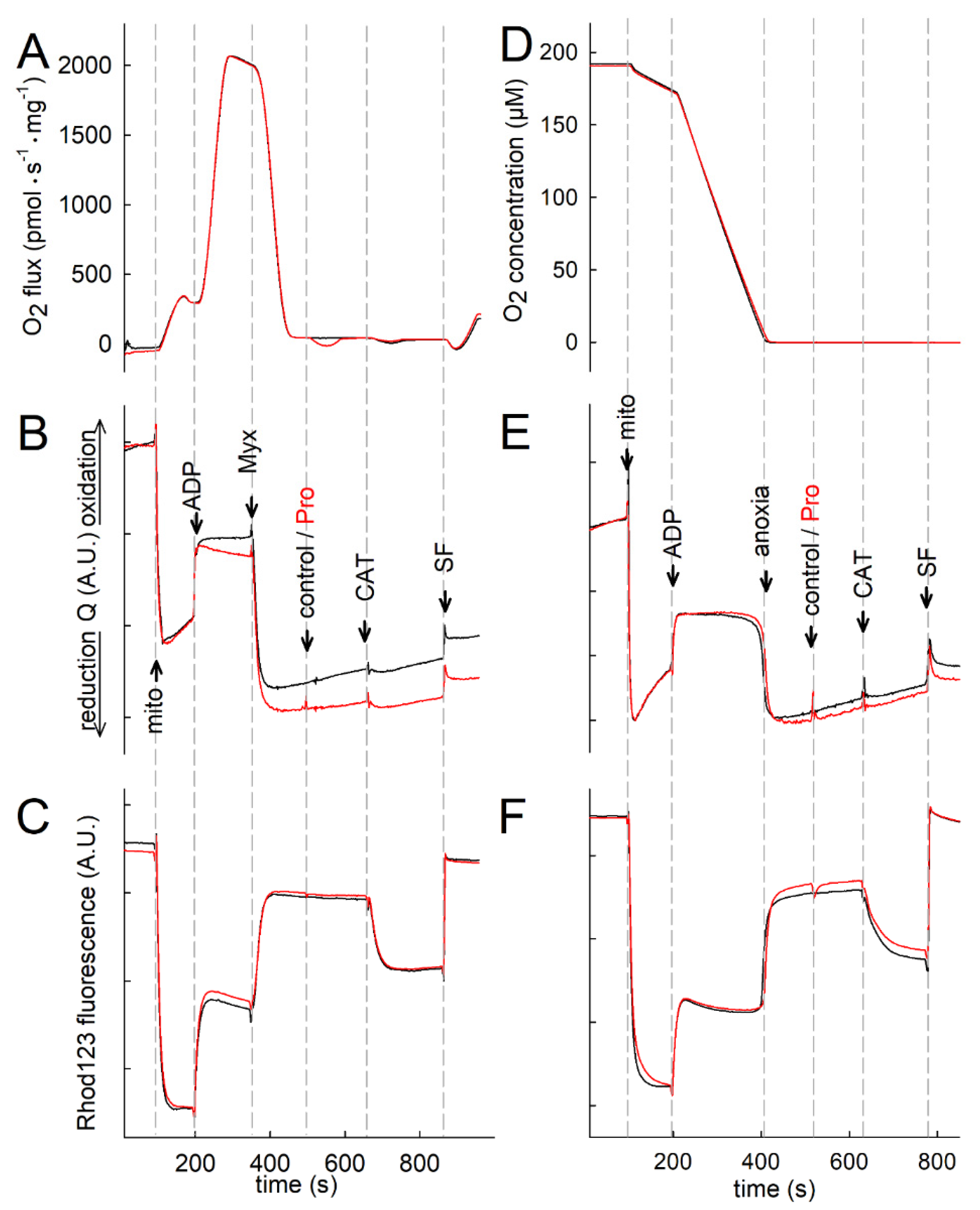
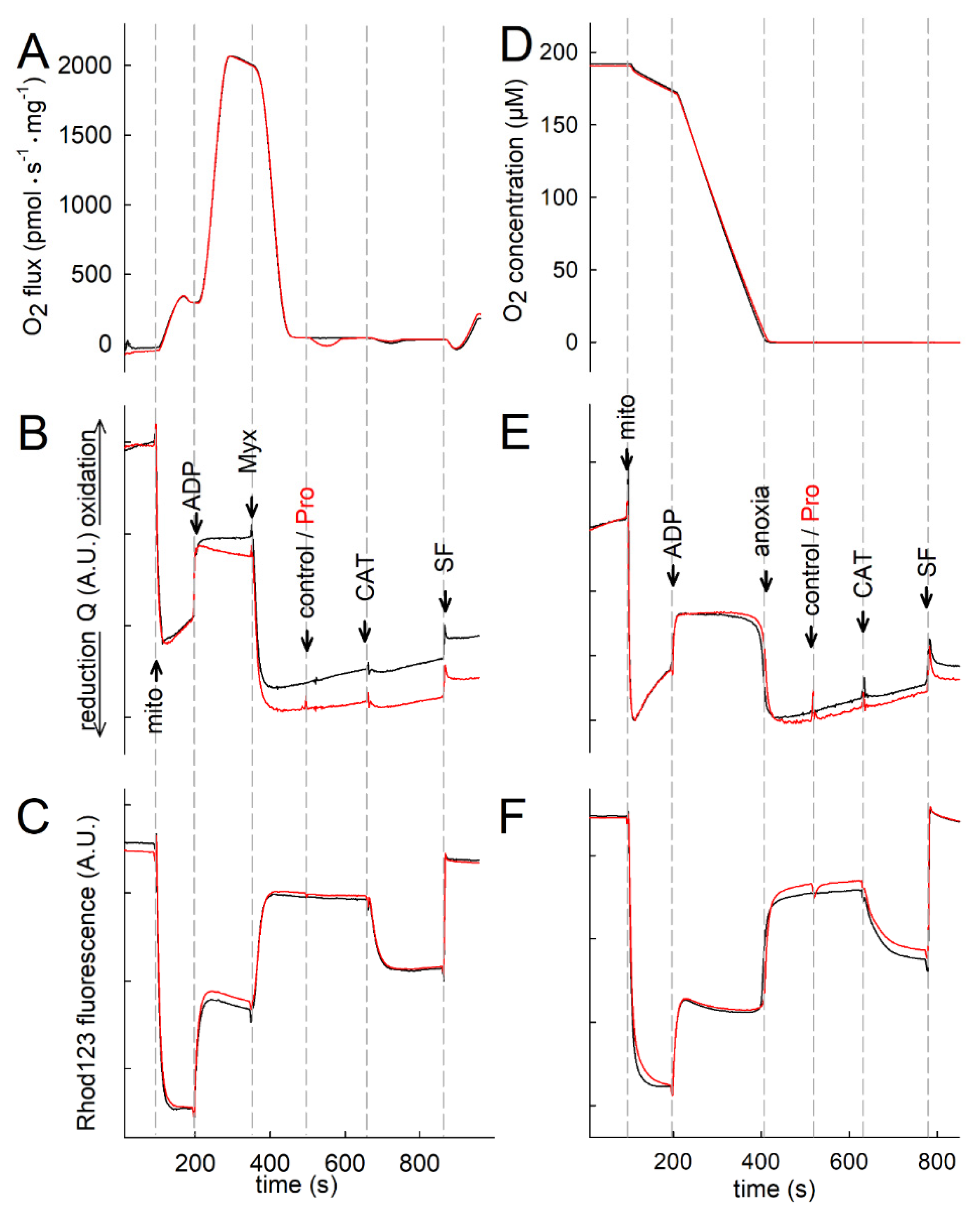
2.4. Proline Oxidation Is Sensitive to Tetrahydro-2-Furoic Acid (THFA) and S-5-Oxo-2-Tetrahydrofurancarboxylic Acid (S-5-oxo)
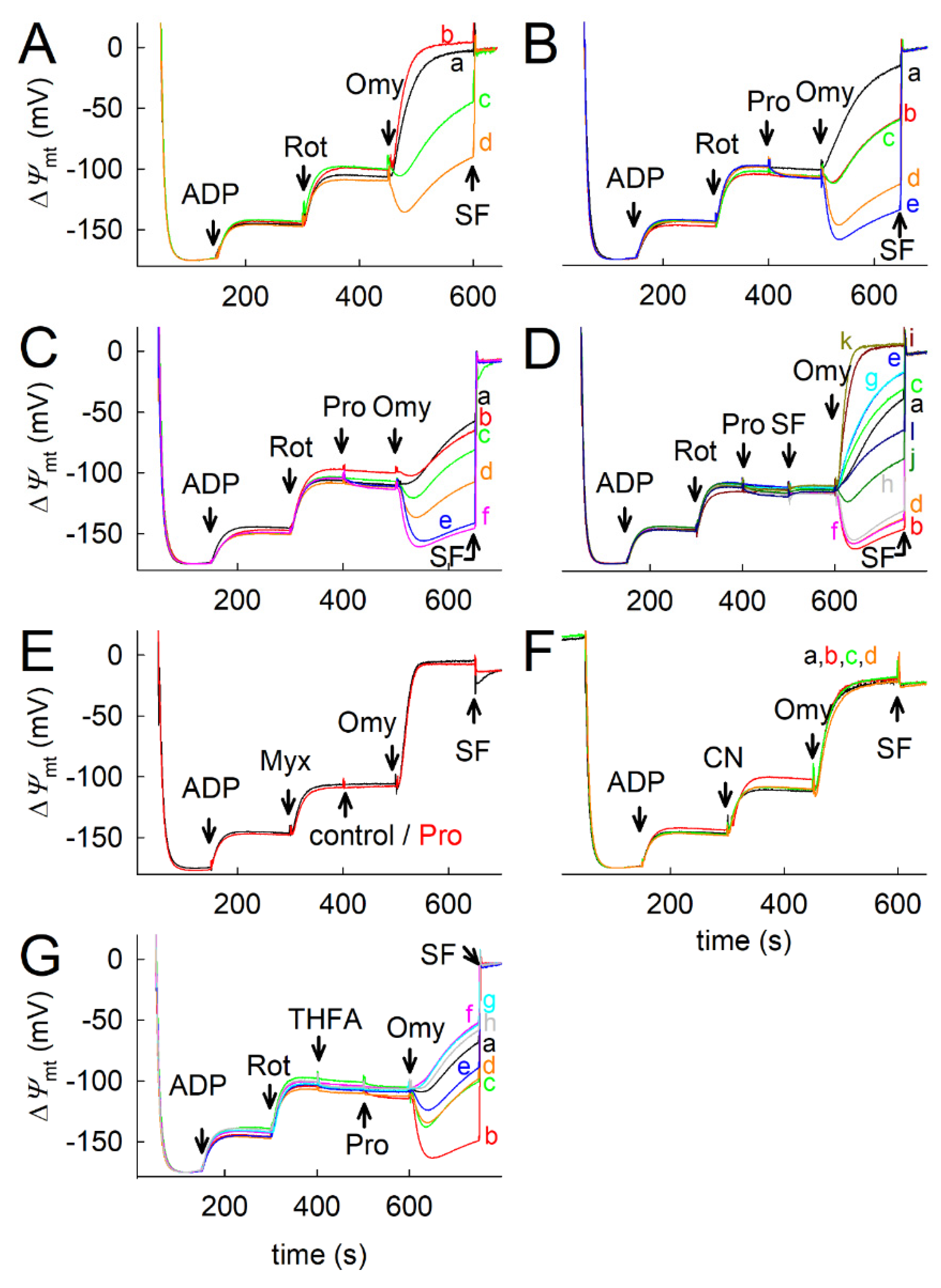
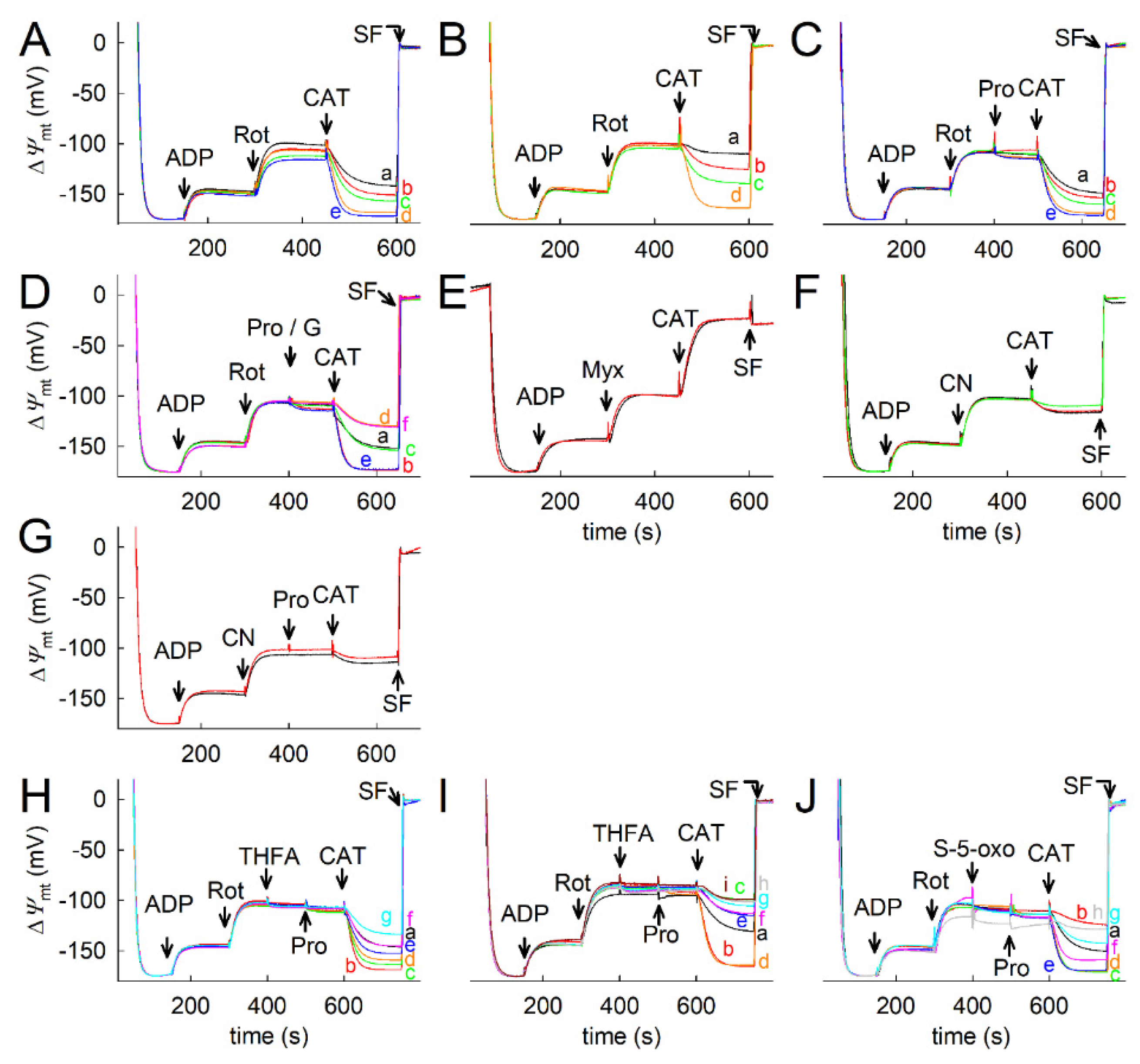
2.5. Effect of Proline Dehydrogenase Inhibitors on Proline-Induced Changes in Q Redox State and ΔΨmt
2.6. Effect of Proline on ANT Directionality
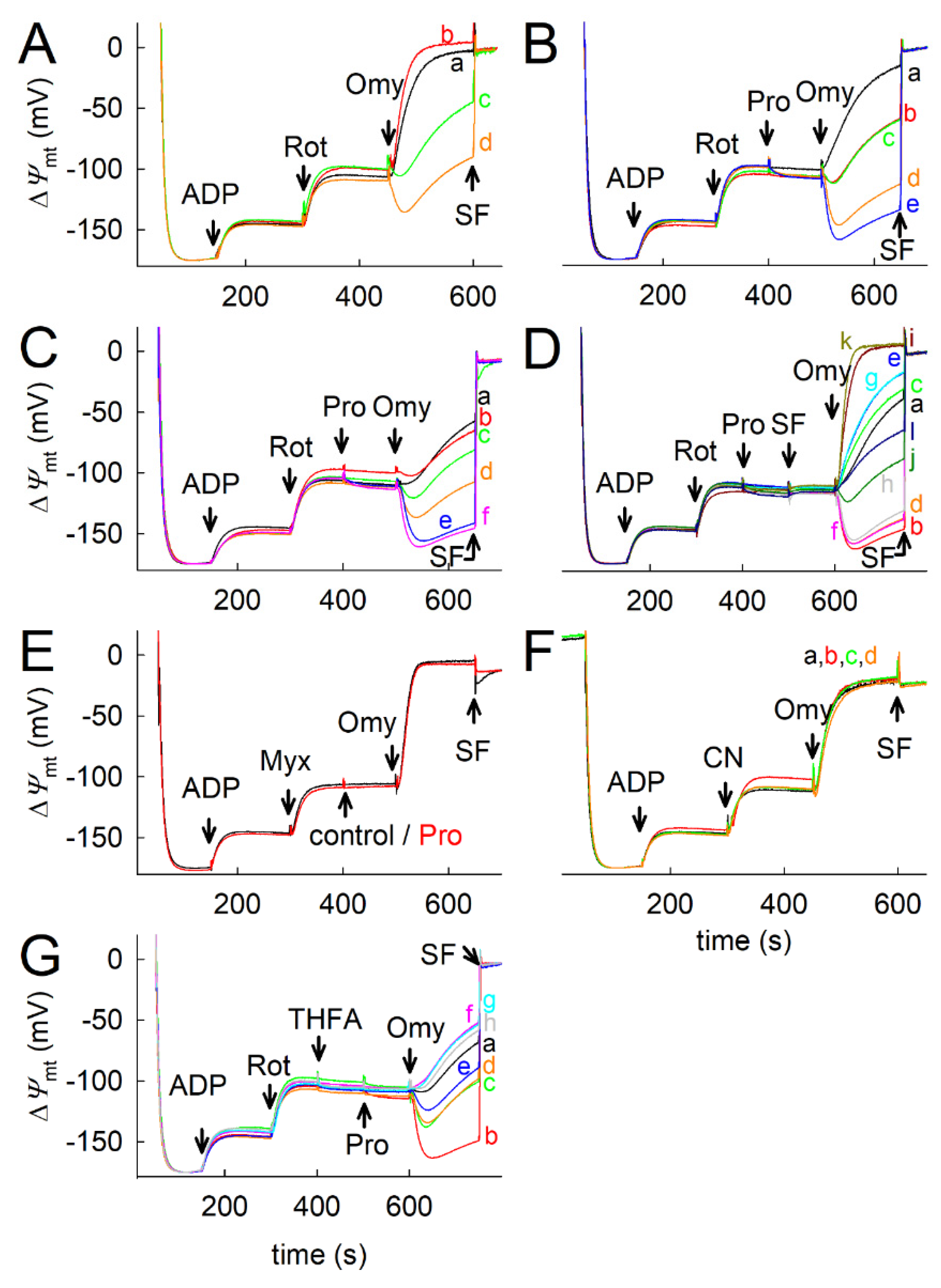
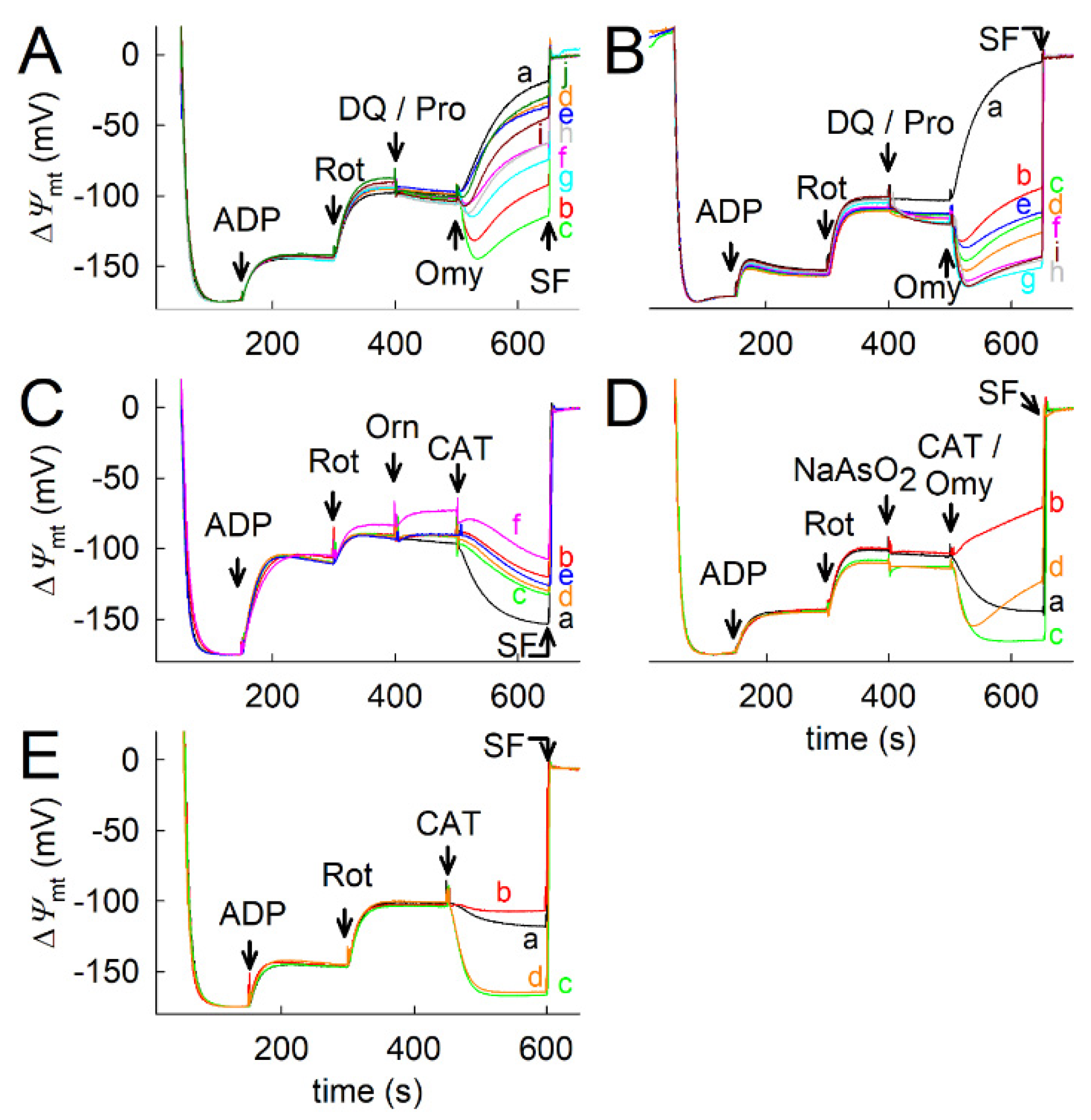
2.7. Proline Oxidation Maintains F1FO-ATPase in Forward Mode during Complex I Inhibition
2.8. Fueling Complex III with Duroquinone Only Partially Mimics the Benefits of Proline
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Isolation of Mitochondria
4.3. Determination of Membrane Potential (ΔΨmt) in Issolated Mitochondria
4.4. Mitochondrial Respiration
4.5. Determination of NADH Autofluorescence in Isolated Mitochondria
4.6. Mitochondrial Q Redox State
4.7. Determination of Proline Dehydrogenase Activity
4.8. Reagents
4.9. Figures on Time Courses
4.10. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ALDH4A1 | delta-1-pyrroline-5-carboxylate dehydrogenase |
| ANT | adenine nucleotide translocase |
| ASAT | aspartate aminotransferase |
| βOH | β-hydroxybutyrate |
| CAT | carboxyatractyloside |
| CI, CII, CIII, CIV | respiratory Complexes I to IV |
| CN | cyanide (NaCN) |
| DQ | duroquinone |
| ET | electron transfer |
| ETS | electron transfer system |
| ΔΨmt | mitochondrial membrane potential |
| G | glutamate |
| GDH | glutamate dehydrogenase |
| GM | glutamate & malate |
| GSA | glutamate semi-aldehyde |
| Itac | itaconate |
| LEAK respiration | resting respiration in the absence of ADP |
| M | malate |
| mtIM | mitochondrial inner membrane |
| Myx | myxothiazol |
| OAT | ornithine aminotransferase |
| Og | 2-oxoglutarate (α-ketoglutarate) |
| OgDH | oxoglutarate dehydrogenase |
| Omy | oligomycin |
| OXPHOS capacity | respiration at kinetically saturating [ADP] |
| Pro | proline |
| ProDH | proline dehydrogenase |
| P5C | pyrroline-5-carboxylate |
| Q | mitochondrial ETS-reactive coenzyme Q |
| Rot | rotenone |
| Rox | residual oxygen consumption |
| S | succinate |
| SCS | succinate-CoA ligase (succinyl-CoA synthetase) |
| SF | uncoupler SF6847 |
| S-5-oxo | S-5-oxo-2-tetrahydrofurancarboxylic acid |
| THFA | tetrahydro-2-furoic acid |
| UQ | ubiquinone, oxidized Q |
| UQH2 | ubiquinol, reduced Q |
References
- Taggart, J.V.; Krakaur, R.B. Studies on the cyclophorase system; the oxidation of proline and hydroxyproline. J. Biol. Chem. 1949, 177, 641–653. [Google Scholar] [CrossRef]
- Johnson, A.B.; Strecker, H.J. The interconversion of glutamic acid and proline. IV. The oxidation of proline by rat liver mito-chondria. J. Biol. Chem. 1962, 237, 1876–1882. [Google Scholar] [CrossRef]
- McKnight, J.A.; Hird, F.J. The oxidation of proline by mitochondrial preparations. Comp. Biochem. Physiol. Part B Comp. Biochem. 1986, 85, 289–294. [Google Scholar] [CrossRef]
- Scaraffia, P.; Wells, M. Proline can be utilized as an energy substrate during flight of Aedes aegypti females. J. Insect Physiol. 2003, 49, 591–601. [Google Scholar] [CrossRef]
- McDonald, A.E.; Pichaud, N.; Darveau, C.-A. “Alternative” fuels contributing to mitochondrial electron transport: Importance of non-classical pathways in the diversity of animal metabolism. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2018, 224, 185–194. [Google Scholar] [CrossRef]
- Gäde, G.; Auerswald, L. Beetles’ choice—proline for energy output: Control by AKHs. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2002, 132, 117–129. [Google Scholar] [CrossRef]
- D’Aniello, C.; Patriarca, E.J.; Phang, J.M.; Minchiotti, G. Proline Metabolism in Tumor Growth and Metastatic Progression. Front. Oncol. 2020, 10, 776. [Google Scholar] [CrossRef]
- Phang, J.M. Proline Metabolism in Cell Regulation and Cancer Biology: Recent Advances and Hypotheses. Antioxidants Redox Signal. 2019, 30, 635–649. [Google Scholar] [CrossRef] [Green Version]
- Hancock, C.N.; Liu, W.; Alvord, W.G.; Phang, J.M. Co-regulation of mitochondrial respiration by proline dehydrogenase/oxidase and succinate. Amino Acids 2016, 48, 859–872. [Google Scholar] [CrossRef] [Green Version]
- Vettore, L.A.; Westbrook, R.L.; Tennant, D.A. Proline metabolism and redox; maintaining a balance in health and disease. Amino Acids 2021, 53, 1779–1788. [Google Scholar] [CrossRef]
- Adams, E.; Frank, L. Metabolism of Proline and the Hydroxyprolines. Annu. Rev. Biochem. 1980, 49, 1005–1061. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Zhu, S.; Lim, R.R.; Chao, J.R. Proline metabolism and transport in retinal health and disease. Amino Acids 2021, 53, 1789–1806. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J. Proline transport in rat liver mitochondria. Arch. Biochem. Biophys. 1977, 178, 387–395. [Google Scholar] [CrossRef]
- Atlante, A.; Passarella, S.; Pierro, P.; Quagliariello, E. Proline Transport in Rat Kidney Mitochondria. Arch. Biochem. Biophys. 1994, 309, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Summitt, C.B.; Johnson, L.C.; Jönsson, T.J.; Parsonage, D.; Holmes, R.P.; Lowther, W.T. Proline dehydrogenase 2 (PRODH2) is a hydroxyproline dehydrogenase (HYPDH) and molecular target for treating primary hyperoxaluria. Biochem. J. 2015, 466, 273–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandhare, J.; Donald, S.P.; Cooper, S.K.; Phang, J.M. Regulation and function of proline oxidase under nutrient stress. J. Cell. Biochem. 2009, 107, 759–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erecińska, M. Ubiquinone in proline oxidation. Arch. Int. Pharmacodyn. Ther. 1965, 158, 209–215. [Google Scholar]
- Chinopoulos, C. The “B space” of mitochondrial phosphorylation. J. Neurosci. Res. 2011, 89, 1897–1904. [Google Scholar] [CrossRef]
- Kowaloff, E.; Granger, A.; Phang, J. Alterations in proline metabolic enzymes with mammalian development. Metabolism 1976, 25, 1087–1094. [Google Scholar] [CrossRef]
- Schmidt, J.A.; Rinaldi, S.; Scalbert, A.; Ferrari, P.; Achaintre, D.; Gunter, M.J.; Appleby, P.N.; Key, T.J.; Travis, R.C. Plasma concentrations and intakes of amino acids in male meat-eaters, fish-eaters, vegetarians and vegans: A cross-sectional analysis in the EPIC-Oxford cohort. Eur. J. Clin. Nutr. 2016, 70, 306–312. [Google Scholar] [CrossRef] [Green Version]
- McMenamy, R.H.; Lund, C.C.; Oncley, J.L. Unbound Amino Acid Concentrations in Human Blood Plasmas. J. Clin. Investig. 1957, 36, 1672–1679. [Google Scholar] [CrossRef] [PubMed]
- Frame, E.G. The Levels of Individual Free Amino Acids in the Plasma of Normal Man at Various Intervals After a High-Protein Meal1. J. Clin. Investig. 1958, 37, 1710–1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Banaclocha, M.; Frank, A.; Diez-Tejedor, E.; Hernanz, A. Amino acid concentrations in cerebrospinal fluid and serum in Alzheimer’s disease and vascular dementia. J. Neural Transm. 1993, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Jeppesen, P.B.; Gregersen, S.; Larsen, L.B.; Hermansen, K. Chronic Exposure to Proline Causes Aminoacidotoxicity and Impaired Beta-Cell Function: Studies In Vitro. Rev. Diabet. Stud. 2016, 13, 66–78. [Google Scholar] [CrossRef] [Green Version]
- Newton, H.; Wang, Y.-F.; Camplese, L.; Mokochinski, J.B.; Kramer, H.B.; Brown, A.E.X.; Fets, L.; Hirabayashi, S. Systemic muscle wasting and coordinated tumour response drive tumourigenesis. Nat. Commun. 2020, 11, 4653. [Google Scholar] [CrossRef]
- Scharff, R.; Wool, I.G. Effect of diabetes on the concentration of amino acids in plasma and heart muscle of rats. Biochem. J. 1966, 99, 173–178. [Google Scholar] [CrossRef] [Green Version]
- Németh, B.; Doczi, J.; Csete, D.; Kacso, G.; Ravasz, D.; Adams, D.; Kiss, G.; Nagy, A.M.; Horvath, G.; Tretter, L.; et al. Abolition of mitochondrial substrate-level phosphorylation by itaconic acid produced by LPS-induced Irg1 expression in cells of murine macrophage lineage. FASEB J. 2016, 30, 286–300. [Google Scholar] [CrossRef] [Green Version]
- Gnaiger, E. Mitochondrial Pathways and Respiratory Control. An introduction to OXPHOS analysis 5th ed. Bioenerg. Commun. 2020, 2020, 2. [Google Scholar] [CrossRef]
- Gnaiger, E.; MitoEAGLE Task Group. Mitochondrial physiology. Bioenerg. Commun. 2020, 2020, 1–44. [Google Scholar] [CrossRef]
- Luo, M.; Arentson, B.W.; Srivastava, D.; Becker, D.F.; Tanner, J.J. Crystal Structures and Kinetics of Monofunctional Proline Dehydrogenase Provide Insight into Substrate Recognition and Conformational Changes Associated with Flavin Reduction and Product Release. Biochemistry 2012, 51, 10099–10108. [Google Scholar] [CrossRef]
- Scott, G.K.; Yau, C.; Becker, B.C.; Khateeb, S.; Mahoney, S.; Jensen, M.B.; Hann, B.; Cowen, B.J.; Pegan, S.D.; Benz, C.C. Targeting Mitochondrial Proline Dehydrogenase with a Suicide Inhibitor to Exploit Synthetic Lethal Interactions with p53 Upregulation and Glutaminase Inhibition. Mol. Cancer Ther. 2019, 18, 1374–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinopoulos, C.; Gerencser, A.A.; Mandi, M.; Mathe, K.; Töröcsik, B.; Doczi, J.; Turiak, L.; Kiss, G.; Konràd, C.; Vajda, S.; et al. Forward operation of adenine nucleotide translocase during F0F1-ATPase reversal: Critical role of matrix substrate-level phosphorylation. FASEB J. 2010, 24, 2405–2416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinopoulos, C. Mitochondrial consumption of cytosolic ATP: Not so fast. FEBS Lett. 2011, 585, 1255–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klingenberg, M. The ADP and ATP transport in mitochondria and its carrier. Biochim. Biophys. Acta (BBA)—Biomembr. 2008, 1778, 1978–2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandre, A.; Reynafarje, B.; Lehninger, A.L. Stoichiometry of vectorial H + movements coupled to electron transport and to ATP synthesis in mitochondria. Proc. Natl. Acad. Sci. USA 1978, 75, 5296–5300. [Google Scholar] [CrossRef] [Green Version]
- Kiss, G.; Konrad, C.; Doczi, J.; Starkov, A.A.; Kawamata, H.; Manfredi, G.; Zhang, S.F.; Gibson, G.E.; Beal, M.F.; Adam-Vizi, V.; et al. The negative impact of α-ketoglutarate dehydrogenase complex deficiency on matrix substrate-level phosphorylation. FASEB J. 2013, 27, 2392–2406. [Google Scholar] [CrossRef] [Green Version]
- Bui, D.; Ravasz, D.; Chinopoulos, C. The Effect of 2-Ketobutyrate on Mitochondrial Substrate-Level Phosphorylation. Neurochem. Res. 2019, 44, 2301–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinopoulos, C.; Tretter, L.; Adam-Vizi, V. Depolarization of in Situ Mitochondria Due to Hydrogen Peroxide-Induced Oxidative Stress in Nerve Terminals: Inhibition of alpha-ketoglutarate dehydrogenase. J. Neurochem. 2002, 73, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Reyes, I.; Diebold, L.P.; Kong, H.; Schieber, M.; Huang, H.; Hensley, C.T.; Mehta, M.; Wang, T.; Santos, J.H.; Woychik, R.; et al. TCA Cycle and Mitochondrial Membrane Potential Are Necessary for Diverse Biological Functions. Mol. Cell 2016, 61, 199–209. [Google Scholar] [CrossRef] [Green Version]
- Vafai, S.B.; Mevers, E.; Higgins, K.W.; Fomina, Y.; Zhang, J.; Mandinova, A.; Newman, D.; Shaw, S.Y.; Clardy, J.; Mootha, V.K. Natural Product Screening Reveals Naphthoquinone Complex I Bypass Factors. PLoS ONE 2016, 11, e0162686. [Google Scholar] [CrossRef]
- Ravasz, D.; Kacso, G.; Fodor, V.; Horvath, K.; Adam-Vizi, V.; Chinopoulos, C. Reduction of 2-methoxy-1,4-naphtoquinone by mitochondrially-localized Nqo1 yielding NAD+ supports substrate-level phosphorylation during respiratory inhibition. Biochim. Biophys. Acta 2018, 1859, 909–924. [Google Scholar] [CrossRef] [PubMed]
- Zieliński, Ł.P.; Smith, A.C.; Smith, A.G.; Robinson, A.J. Metabolic flexibility of mitochondrial respiratory chain disorders predicted by computer modelling. Mitochondrion 2016, 31, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Fato, R.; Bergamini, C.; Leoni, S.; Lenaz, G. Mitochondrial production of reactive oxygen species: Role of Complex I and quinone analogues. BioFactors 2008, 32, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Tanner, J.J.; Fendt, S.-M.; Becker, D.F. The Proline Cycle As a Potential Cancer Therapy Target. Biochemistry 2018, 57, 3433–3444. [Google Scholar] [CrossRef]
- Chinopoulos, C.; Vajda, S.; Csanády, L.; Mándi, M.; Mathe, K.; Adam-Vizi, V. A Novel Kinetic Assay of Mitochondrial ATP-ADP Exchange Rate Mediated by the ANT. Biophys. J. 2009, 96, 2490–2504. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Åkerman, K.E.; Wikström, M.K. Safranine as a probe of the mitochondrial membrane potential. FEBS Lett. 1976, 68, 191–197. [Google Scholar] [CrossRef] [Green Version]
- Emaus, R.K.; Grunwald, R.; Lemasters, J.J. Rhodamine 123 as a probe of transmembrane potential in isolated rat-liver mitochondria: Spectral and metabolic properties. Biochim. Biophys. Acta 1986, 850, 436–448. [Google Scholar] [CrossRef]
- Valle, V.G.; Pereira-Da-Silva, L.; Vercesi, A.E. Undesirable feature of safranine as a probe for mitochondrial membrane potential. Biochem. Biophys. Res. Commun. 1986, 135, 189–195. [Google Scholar] [CrossRef]
- Chinopoulos, C.; Adam-Vizi, V. Mitochondrial Ca2+ sequestration and precipitation revisited. FEBS J. 2010, 277, 3637–3651. [Google Scholar] [CrossRef]
- Krumschnabel, G.; Eigentler, A.; Fasching, M.; Gnaiger, E. Use of Safranin for the Assessment of Mitochondrial Membrane Potential by High-Resolution Respirometry and Fluorometry. Methods Enzymol. 2014, 542, 163–181. [Google Scholar] [CrossRef] [PubMed]
- Komlódi, T.C.L.; Doerrier, C.; Moore, A.L.; Rich, P.R.; Gnaiger, E. Coupling and pathway control of coenzyme Q redox state and respiration in isolated mitochondria. Bioenerg. Commun. 2021, 2021. [Google Scholar] [CrossRef]
- Kawabata, Y.; Katunuma, N.; Sanada, Y. Characteristics of Proline Oxidase in Rat Tissues. J. Biochem. 1980, 88, 281–283. [Google Scholar] [CrossRef] [PubMed]
- Mezl, V.A.; Knox, W.E. Properties and analysis of a stable derivative of pyrroline-5-carboxylic acid for use in metabolic studies. Anal. Biochem. 1976, 74, 430–440. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proline | Succinate | JPro/JSPro | JS/JSPro | Additivity | |
|---|---|---|---|---|---|
| [mM] | [mM] | ||||
| Liver OXPHOS | 2 | 5 | 0.30 | 0.40 | 2.04 |
| 5 | 5 | 0.30 | 0.41 | 1.95 | |
| Liver LEAK | 2 | 5 | 0.55 | 0.67 | 0.59 |
| 5 | 5 | 0.40 | 0.59 | 1.03 | |
| Kidney OXPHOS | 2 | 5 | 0.19 | 0.60 | 2.11 |
| 5 | 5 | 0.26 | 0.58 | 1.62 | |
| Kidney LEAK | 2 | 5 | 0.25 | 1.10 | −0.39 |
| 5 | 5 | 0.39 | 1.08 | −0.20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pallag, G.; Nazarian, S.; Ravasz, D.; Bui, D.; Komlódi, T.; Doerrier, C.; Gnaiger, E.; Seyfried, T.N.; Chinopoulos, C. Proline Oxidation Supports Mitochondrial ATP Production When Complex I Is Inhibited. Int. J. Mol. Sci. 2022, 23, 5111. https://doi.org/10.3390/ijms23095111
Pallag G, Nazarian S, Ravasz D, Bui D, Komlódi T, Doerrier C, Gnaiger E, Seyfried TN, Chinopoulos C. Proline Oxidation Supports Mitochondrial ATP Production When Complex I Is Inhibited. International Journal of Molecular Sciences. 2022; 23(9):5111. https://doi.org/10.3390/ijms23095111
Chicago/Turabian StylePallag, Gergely, Sara Nazarian, Dora Ravasz, David Bui, Timea Komlódi, Carolina Doerrier, Erich Gnaiger, Thomas N. Seyfried, and Christos Chinopoulos. 2022. "Proline Oxidation Supports Mitochondrial ATP Production When Complex I Is Inhibited" International Journal of Molecular Sciences 23, no. 9: 5111. https://doi.org/10.3390/ijms23095111
APA StylePallag, G., Nazarian, S., Ravasz, D., Bui, D., Komlódi, T., Doerrier, C., Gnaiger, E., Seyfried, T. N., & Chinopoulos, C. (2022). Proline Oxidation Supports Mitochondrial ATP Production When Complex I Is Inhibited. International Journal of Molecular Sciences, 23(9), 5111. https://doi.org/10.3390/ijms23095111