1. Introduction
Mitochondrial diseases are complex diseases resulting from mutations affecting the levels or activities of mitochondrial proteins and thereby impair the key functions of mitochondria in cells, most notably the provision of ATP by oxidative phosphorylation. Mutational reduction or loss of these essential biochemical functions of mitochondria cause mitochondrial disease, a complex array of diseases that can and do affect multiple cell types, tissues and organ systems (Schlieben et al., 2020). Nonetheless, the high energy demands of the neuromuscular and central nervous systems mean that mitochondrial disease often produces muscular and neurological deficits, including neurodegeneration. In this paper we focus on the cytopathological consequences of knocking down the expression of one particular mitochondrial protein complex, respiratory complex II or succinate dehydrogenase (Sdh). This is of particular interest because, as described below, the patterns of disease caused by mutations in different Sdh subunits are surprisingly very different and the reasons are not understood.
Sdh is a heterotetramer composed of four subunits named SdhA-D. The subunits are encoded on different chromosomes, sdhA on chromosome 5, sdhB and sdhC on chromosome 1 and sdhD on chromosome 11 [
1]. It has the smallest number of subunits of all the respiratory complexes and is unique in that all of the subunits are nuclear-encoded. It is also the only complex which does not pump protons across the inner mitochondrial membrane. Another unique property of Complex II is that it participates not only in the oxidative phosphorylation pathway but also in the TCA cycle and thus has dual functions in carbon metabolism and mitochondrial respiration [
2].
The four core subunits of Complex II are assembled with the assistance of four assembly factors and cofactors. The catalytic subunit, SdhA, is imported into the mitochondrial matrix and folded before FAD is covalently attached. Likewise, SdhB is also matured in the matrix by the addition of three Fe-S clusters. The flavinylated and mature SdhA subunit then forms a subcomplex by binding to the mature SdhB subunit. The SdhA/B subcomplex then binds irreversibly to the SdhC/D subunits which reside in the IMM to form the holo-enzyme [
3]. SdhA is responsible for oxidising succinate to fumarate in the TCA cycle and passes the electrons to the Fe-S clusters in subunit B. SdhC and D form the membrane-bound components and together with subunit B they form a ubiquinone binding site. Electrons produced from the oxidation of succinate are then transferred to ubiquinone residing in the ubiquinone binding site and used to catalyse the reduction of ubiquinone to ubiquinol. The electrons are subsequently passed to Complex III and then Complex IV, thus contributing to oxidative phosphorylation [
3].
Complex II is thus an essential component in energy production and mutations which affect its function are rare [
4]. Interestingly mutations in different subunits result in very different patterns of clinical presentation and disease which can be grouped into two main categories: cancer or mitochondrial Complex II deficiency (presenting as neurodegeneration or cardiomyopathy). Mutations affecting SdhA most commonly result in a mitochondrial respiratory defect, often presenting as a progressive neurodegenerative disorder called Leigh syndrome [
5] although cardiomyopathy or other neurodegenerative disorders have also been reported including late-onset optic atrophy, ataxia and leukodystrophy [
6,
7]. In a recent, comprehensive review of the literature, Fullerton et al. (2020) found that the vast majority of isolated Complex II deficiencies with mitochondrial phenotypes involved either homozygous or complex heterozygous alleles of sdhA. Only 5 autosomal recessive variants of sdhB and 2 variants of sdhD were found associated with mitochondrial Complex II deficiency. Mutations in sdhA are also sometimes associated with cancers, notably gastrointestinal stromal tumors (Nazar et al., 2019; McFarlane et al., 2020), about 7.5% of which involve complex II deficiency, mostly involving sdhA alleles (Rasheed and Tarjan, 2018).
In contrast with SdhA, mutations in the other subunits SdhB-D are generally associated with cancers, especially paragangliomas (PGL) and phaeochromocytomas (PCC), but also with other cancers including renal carcinoma, thyroid cancer, ovarian cancer, neuroblastoma and gastrointestinal stromal tumor [
8]. There have been a small number of reports, restricted to only a few patients, of mutations to SdhB and SdhD that are associated with leukodystrophy (Fullerton et al., 2020), but generally mutations to subunits B, C and D result in neoplasty. Mutations in all subunits result in impaired Sdh activity, so it is not clear why there is such a distinction in the pattern of clinical phenotypes from the different subunit mutations.
Complex II has been well conserved throughout evolution and simple systems have been used to study its structure and function. Its crystal structure was solved in
E.coli [
9]; its assembly was aided by studies in yeast [
2] and functional studies have been done in animal models [
10,
11].
Dictyostelium discoideum is a simple eukaryotic model organism which belongs to the Amoebozoa, a sister lineage to the animals and fungi that diverged from them after the plant kingdom but before the separation of the animal and fungal kingdoms [
12]. The
D. discoideum genome encodes subunits of all five mitochondrial respiratory complexes including Complex I, in contrast to some fungi, including
Saccharomyces cerevisae. Yet, unlike animals but like most other eukaryotic lineages, it also contains an additional enzyme of oxidative phosphorylation called Alternative Oxidase (AOX). AOX is generally associated with maintaining respiration when the organisms are exposed to various stress conditions and can transfer electrons from Complex I or II via ubiquinone to oxygen.
The
D. discoideum nuclear and mitochondrial genomes have been fully sequenced [
13,
14]. As in humans, the nuclear genome encodes homologues of all four Sdh subunits and several Complex II assembly factors. The organism is amenable to genetic manipulation, and mitochondrial disease has been created in this organism via gene knockouts of nuclear and mitochondrially encoded genes, knockdown of expression of mRNA transcripts encoding mitochondrial chaperonin 60 and treatment with ethidium bromide [
15,
16]. Analysis of the effects of gene mutations is facilitated in
D.discoideum by its unique life cycle featuring unicellular and multicellular stages which provide a wide variety of phenotypes to study as readouts of the underlying signalling pathways. The growth of the unicellular amoebae is halted upon removal of nutrients which initiates a starvation-induced cascade of differentiation events beginning with the acquisition of the ability to secrete and respond chemotactically to cAMP. This results in the aggregation of approximately 100,000 cells which then proceed to form a sequence of multicellular structures including a motile slug and ultimately a fruiting body with a long slender stalk supporting a sorus, which is a droplet of spores. In
D. discoideum, impairing mitochondrial respiration by manipulating genes encoding mitochondrial proteins results in the chronic activation of the energy-sensing protein AMP-activated protein kinase (AMPK) [
17]. This chronic activation of AMPK results in a clear set of defective phenotypes namely defective slug phototaxis, altered fruiting body morphology and decreased growth rates independent of endocytosis.
AMPK is a highly conserved protein kinase that is ubiquitous amongst eukaryotic organisms. It can very sensitively detect energy stress, as it detects altered levels of AMP which rise when ATP levels drop even by a modest amount. AMPK is composed of three subunits—a catalytic α subunit, a scaffolding β subunit and a γ regulatory subunit. During energy stress AMP can bind to the γ subunit causing a conformational change of the complex, allowing phosphorylation by upstream kinases [
18]. Phosphorylation and activation of AMPK can also occur independently of AMP binding in response to oxidative stress or elevated calcium levels, mediated respectively by the upstream kinases TAK1 and Ca
2+/calmodulin-dependent protein kinase β (CaMMKβ) [
19,
20]. Once activated, AMPK works to restore energy levels by inhibiting energy-consuming anabolic pathways and activating energy-producing catabolic ones. One such energy-consuming process is the translation of proteins which is largely governed by a key regulator of nutrient stress called Target of Rapamycin Complex I (TORC1). Activated AMPK can phosphorylate and thereby inhibit TORC1.
Here we have used the model organism D. discoideum and deployed antisense inhibition to individually knockdown expression of three of the Sdh subunits (SdhA, B and C). This allowed us to investigate if the reduction or loss of function of these subunits produces different cytopathological outcomes and could potentially shed light on why mutations affecting these subunits are associated with such different clinical outcomes in humans.
3. Discussion
An abiding mystery in understanding the pathological outcomes of loss of function of individual Sdh subunits has been the distinct patterns of disease outcomes associated with mutations in the different subunits. While either cancer phenotypes or typical mitochondrial disease phenotypes (neurodegeneration and/or myopathies) can be caused by loss of function mutations to any of the subunits, the relative frequencies of these outcomes differ for the different subunits (Fullerton et al., 2020). SdhA mutations mostly cause clinical outcomes reminiscent of other mitochondrial diseases, while loss of function of any of the other subunits mostly causes a cancer phenotype [
4]. This is the case even though all subunits are necessary for and contribute to the efficiency of the complex as a whole. In this work, we studied the outcomes of knocking down the expression of three of the four Sdh subunits in the established mitochondrial disease model,
Dictyostelium discoideum. Our results showed that in
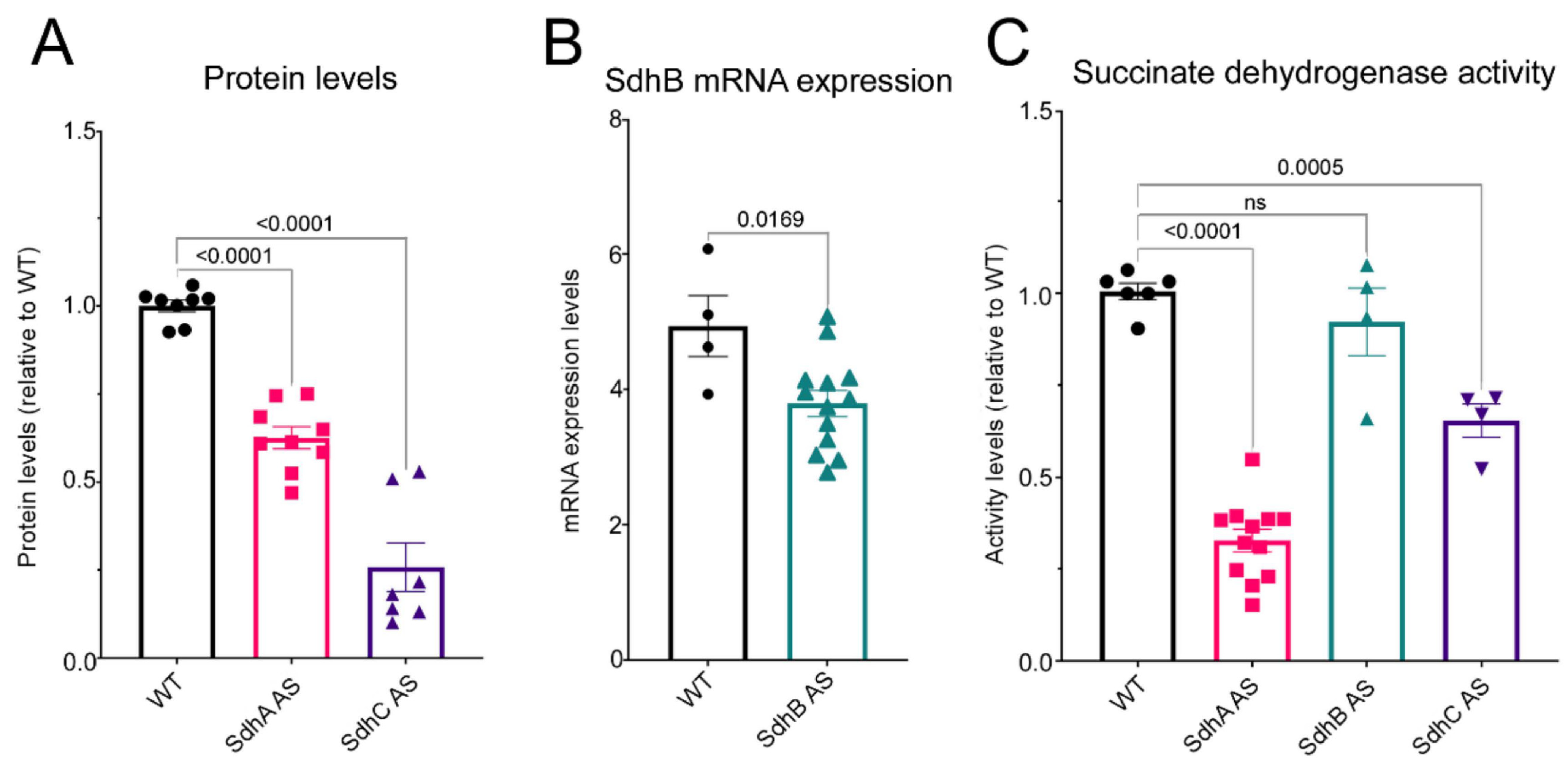
D. discoideum a reduction in SdhA, B or C expression resulted in decreases in succinate dehydrogenase activity as in mammalian cells, although this reduction did not reach significance in our SdhB antisense-inhibited transformants.
It has been shown that SdhA activity assayed in vitro is dependent on its binding to the other subunits and mature free SdhA unbound to other subunits has very little catalytic activity [
24]. In our semiquantitative western blots and quantitative in vitro activity assays, the subunit levels and associated Sdh activities were significantly reduced in both the SdhA and SdhC antisense strains. The lack of a significant decrease of Sdh activity in the SdhB transformants may be because the reduction of SdhB expression was not low enough. Unfortunately, this could not be verified because we did not have an antibody suitable for measuring SdhB protein expression levels. However, the reduced SdhB mRNA levels in these knockdown strains were accompanied by phenotypic abnormalities, the simplest explanation of which is that they were caused by reduced expression of the protein itself.
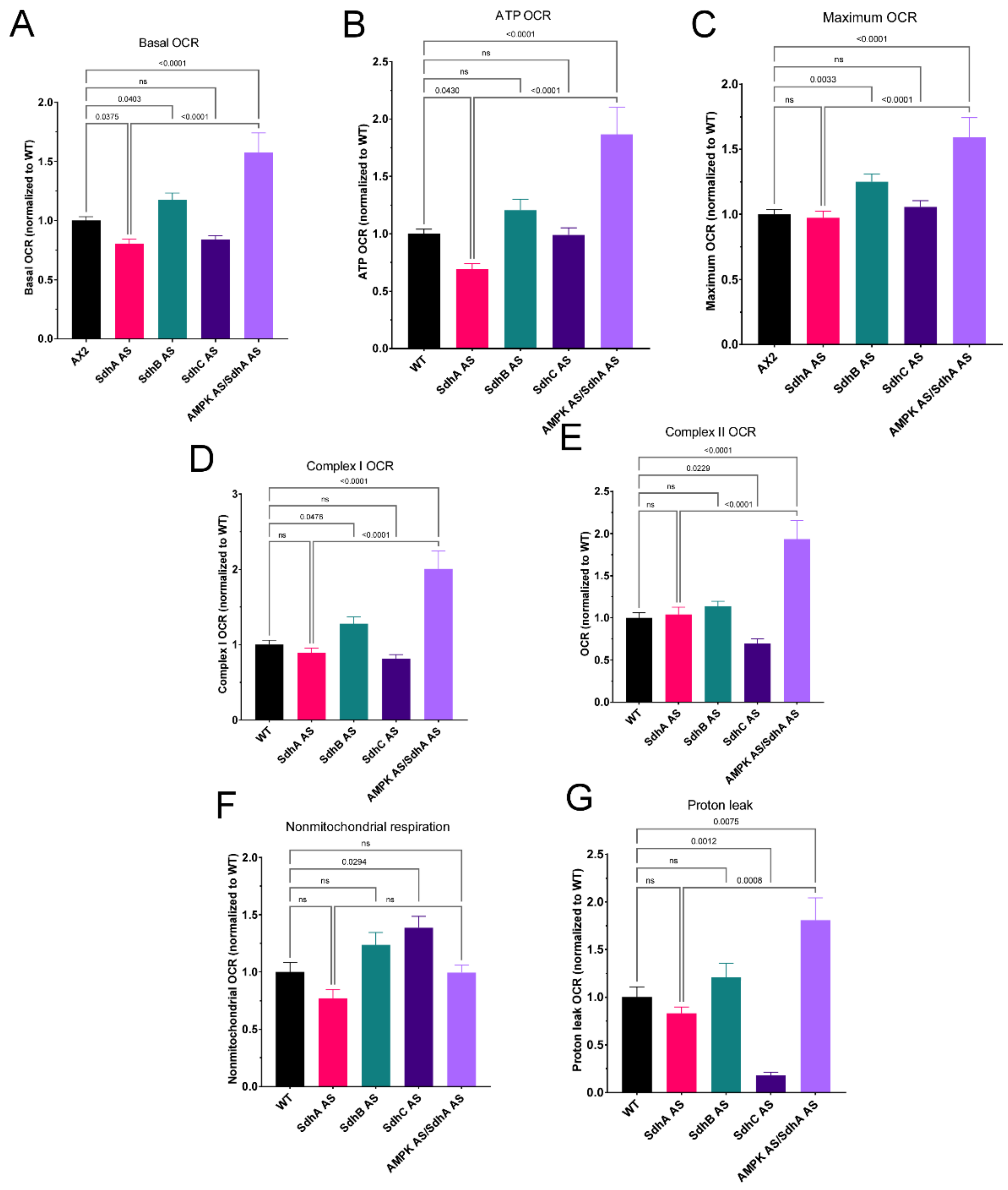
Despite the demonstrably lower Sdh enzyme activities in both the SdhA and SdhC knockdowns (
Figure 1), Seahorse respirometry detected reduced Complex II activity in intact cells only in the latter (
Figure 6E). This may be due to the difference in how Complex II activity is measured in the two methods. The in vitro Sdh activity assay measures, in crude cell lysates, the donation of electrons from experimentally provided substrate succinate, via FADH and the iron-sulphur cluster of SdhB to a colour-forming ubiquinone analogue (via Complex III/IV or AOX). This measure is thus independent of the cellular context, including the activity of the TCA cycle. In the Seahorse assay, the contribution of Complex II to maximum, uncoupled O
2 consumption is measured in intact cells provided with excess TCA cycle substrates in the form of pyruvate and malate. The Seahorse assay may thus be more physiologically relevant, depending as it does on the cellular context, which determines the rate of provision of succinate to complex II by the TCA cycle and is subject to multiple layers of regulation, both at the protein expression and metabolic activity levels.
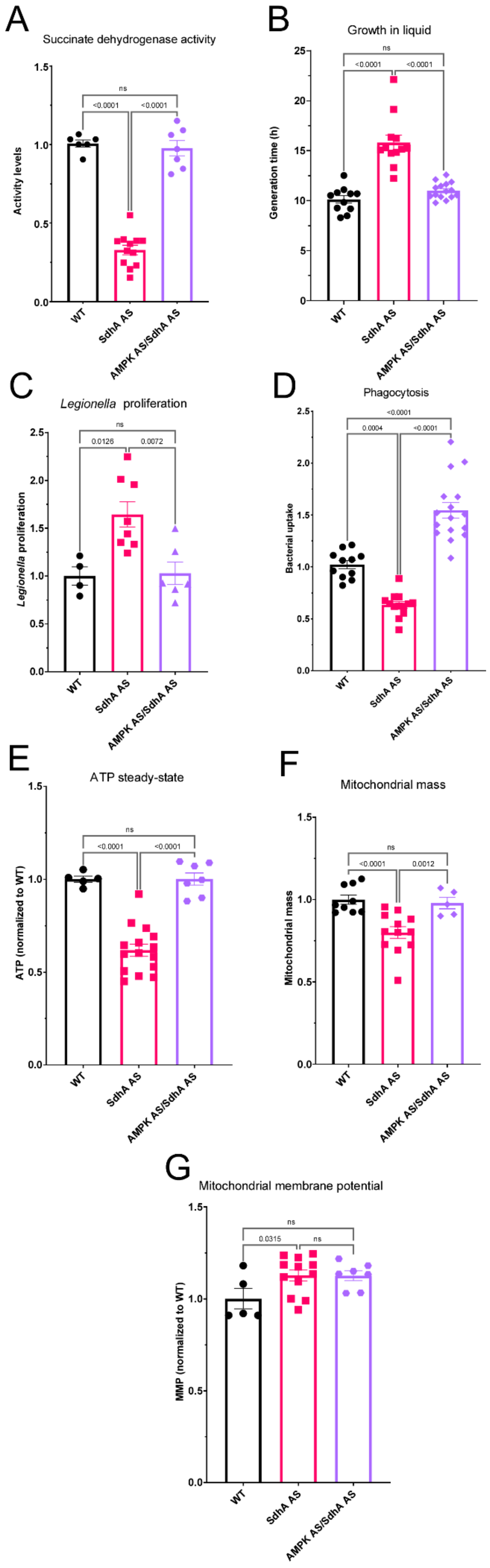
Our Seahorse results suggest a model for the consequences of SdhA knockdown in intact cells, according to which the reduced availability of the SdhA subunit is not the rate-limiting factor for maximal complex II activity. Instead, it rate limits and diminishes the flux of carbon through the TCA cycle, reducing both the rate of supply of TCA cycle intermediates for biosynthetic reactions and of electrons for biosynthesis and oxidative phosphorylation. The result is a new steady-state in which the lower ATP synthesis rates are accompanied by lower ATP steady-state levels (
Figure 5E) and chronic activation of AMPK which homeostatically reduces cellular ATP demand by inhibiting biosynthetic processes including cell growth (
Figure 5B) and mitochondrial biogenesis (
Figure 5F). The mitochondrial membrane potential is higher (
Figure 5G) because its use by complex V to drive ATP synthesis is reduced.
Chronic activation of AMPK has been reported in many pathological conditions such as polycystic kidney disease [
25], Alzheimer’s disease [
26], ischemia and aging [
27,
28]. One of the phenotypic consequences of chronic AMPK hyperactivity in the
Dictyostelium mitochondrial disease model is the greater intracellular proliferation of the intracellular bacterial pathogen
Legionella pneumophila [
22]. We showed here that this is also the case in the SdhA knockdowns (
Figure 5C), supporting the view that chronically hyperactive AMPK is responsible for many of the phenotypic outcomes of SdhA knockdown. If this is so, then AMPK knockdown in the same cells should reverse many of these outcomes. Our results here showed that when AMPK and SdhA were both knocked down in the same cells, the phenotypic consequences of SdhA knockdown were all reversed. In fact, in some cases (impaired phagocytosis and mitochondrial respiratory function) the reversal actually surpassed what was needed to restore wild type levels.
A traditional advantage of working with clonally grown model organisms, such as Dictyostelium discoideum in this work, is that independent, clonal mutants altered in a single target gene can be readily created and studied. This allows clear cause-effect relationships to be established experimentally without the unavoidable complications in clinical observational studies of multiple, unknown genetic modifiers elsewhere in the genome. On the other hand, those genetic modifiers elsewhere may play important roles in the penetrance and disease outcomes in humans and such effects would not be apparent unless explicitly tested. In Dictyostelium, it is easy to explicitly test the effect of potential modifiers as we have done here in the case of AMPK. Our results suggest that differences in AMPK expression or activity could be one such modifier in SdhA-deficient cells, with the phenotype being ameliorated by reduced AMPK activity or exacerbated by higher AMPK activity.
Not only were the phenotypic consequences of SdhA knockdown reversed by AMPK antisense inhibition, but the activity of SdhA itself was restored to normal. Since AMPK is known to activate the transcription of nuclear-encoded mitochondrial proteins, this result was unexpected. The simplest explanation is that the lower levels of SdhA mRNA in the knockdowns are translated at higher rates when AMPK is also knocked down. The canonical mechanism by which AMPK inhibits translation is by inhibiting TORC1, a known translational activator of mitochondrial biogenesis, including the proteins involved in oxidative phosphorylation. It would be valuable in future work to test this hypothesis that AMPK-mediated inhibition of TORC1 is responsible for many of the phenotypic outcomes of reduced SdhA activity.
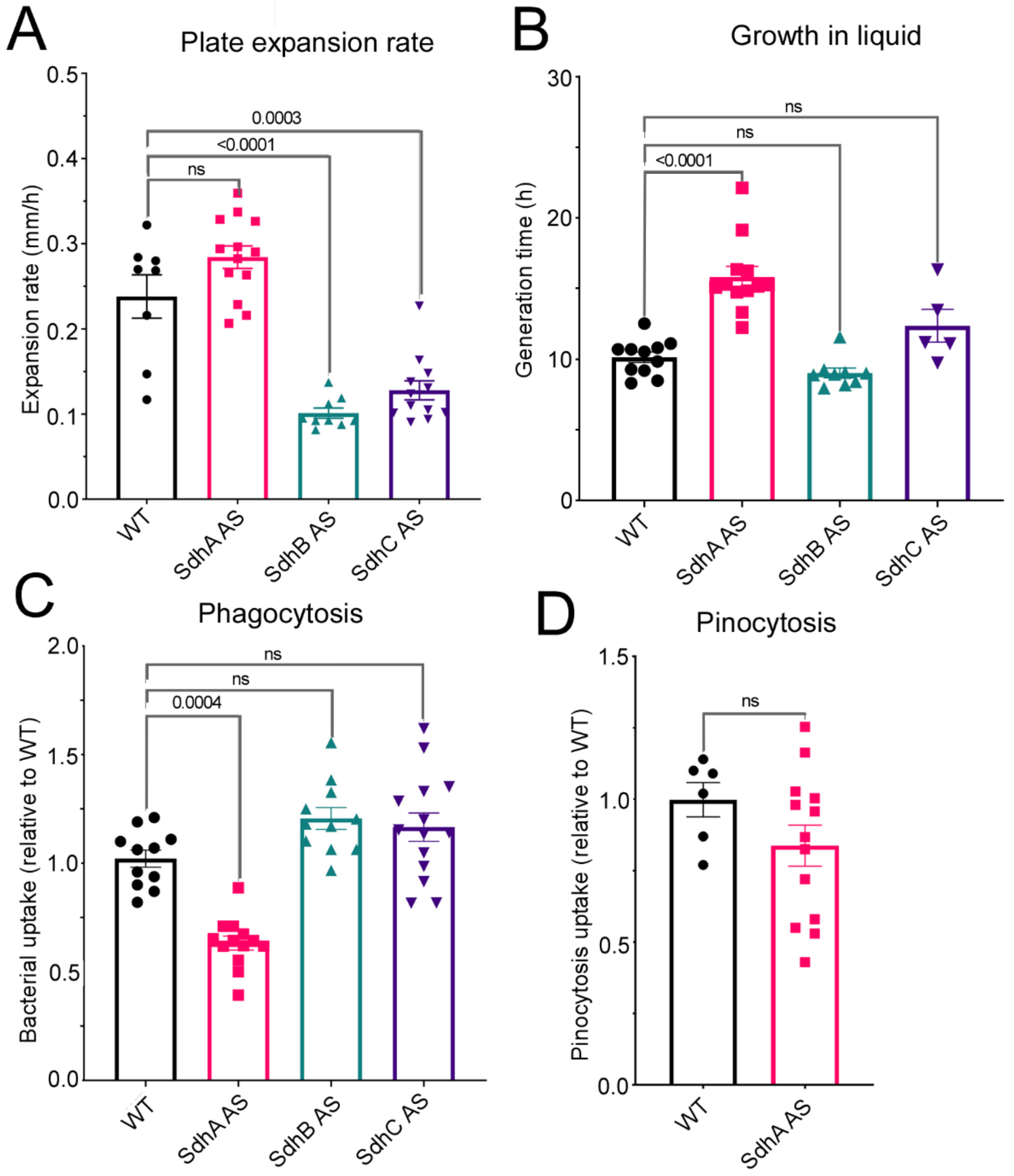
Unlike in other
D. discoideum cells with mitochondrial dysfunction, SdhA antisense-inhibited transformants exhibited decreased phagocytosis and no significant defect in plaque expansion rates on bacterial lawns. The phagocytosis defect was mediated by AMPK, as evidenced by the fact that it was reversed by AMPK knockdown. This finding suggests that under some circumstances AMPK hyperactivity can inhibit phagocytosis. What might those circumstances be? Recent data from our laboratory has shown that in oxidatively stressed cells, phagocytosis becomes sensitive to and inhibited by AMPK signaling [
29]. Whilst ROS levels were slightly elevated in our Sdh knockdowns, they did not reach statistically significant levels (
Figure 4D). However, a significant increase in the mitochondrial membrane potential was observed and it has been established by others that an increased MMP results in increased ROS production [
30]. Reduced Complex II activity can also lead to increased ROS production [
31,
32].
As discussed above, the cytopathological consequences of SdhA knockdown can, in principle, all be traced to reduced supply of electrons both for biosynthesis and oxidative phosphorylation by the TCA cycle. Why would these same consequences not be apparent when the supply of other Sdh subunits is reduced? Our results do not provide an explanation, but they do confirm that in the Dictyostelium model the cytopathological consequences of reducing or losing SdhB or C function are unlike those caused by loss of SdhA. Unlike knocking down SdhA, knocking down either SdhB or SdhC resulted in defects in growth on bacterial lawns without any corresponding impairment of phagocytosis. This was unaccompanied by any impairment of mitochondrial biogenesis, ATP synthesis rates or ATP steady state levels. The SdhC knockdowns exhibited reductions in the mitochondrial components of basal respiration (statistically significant only in the case of the proton leak) and the mitochondrial components of the maximum respiratory capacity (significant only in the case of complex II activity). This was accompanied by elevated rates of nonmitochondrial O2 consumption, which is driven by cellular oxygenases that directly use molecular O2. These results suggest a dysregulation of cellular metabolism that is distinct from that caused by SdhA knockdown. On the other hand, the SdhB knockdowns exhibited an increase in respiratory function that reached statistical significance only in the case of the basal respiration rate, the maximum respiratory capacity and its major component, complex I activity.
Whatever the explanation of these distinct patterns of cytopathological outcomes is, they suggest that the catalytic subunit, SdhA, and its heterodimeric partner SdhB, may exist in the cell in more than one pool, perhaps in association with other proteins or alternative acceptors of the electrons from succinate. This would help explain differences in the phenotypic consequences of knocking down the different Sdh subunits. By way of a speculative example, if the SdhC/D heterodimer in the inner mitochondrial membrane is the preferred assembly partner for SdhA/B, then reducing its availability (eg., by knocking down or mutating SdhC) could cause accumulation of SdhA and/or SdhB in an alternative pool. Knocking down or mutating SdhA activity on the other hand would preferentially reduce its levels in the alternative, non-complex II pool. Another key TCA cycle enzyme, fumarate hydratase, whose mutation can cause cancer or a neurological phenotype, is known to be present in two pools, one in the mitochondria and one in the cytosol (Schmidt et al., 2020). A study by Raimundo et al. (2008) showed that a patient with a fumarase mutation and presentation of cancer had normal levels of mitochondrial fumarase but no cytosolic fumarase, suggesting different roles of the differentially localised fumarase.
The possibility of alternative pools and binding partners for SdhA has been little investigated. However, it has recently been suggested that when energy is low or when SdhB expression is reduced, an alternative assembly of Complex II is formed so that SdhA forms a complex with its two accessory proteins and is not bound by other subunits [
33]. The authors termed this alternative assembly CII
low and noted that when SdhB was knocked out, the cells differentially regulated a number of proteins including upregulation of catabolic and salvage pathways. In these cells, this resulted in a decrease in mitochondrial respiration [
33] rather than the increase we observed in this work. In our case, we are using a knockdown rather than a knockout strain and a simpler organism, but the premise remains that depletion of SdhB could result in the activation of compensatory pathways to restore the energy and redox imbalance. In
D. discoideum this may result in the upregulation of these pathways so effectively in the SdhB case that respiration is actually increased beyond wild type levels.
In conclusion, our results reveal differences in the cytopathological outcomes of knocking down different Sdh subunits in Dictyostelium. Partial loss of SdhA resulted in mitochondrial dysfunction and AMPK-mediated phenotypic outcomes similar to those in previously studied D. discoideum mitochondrial disease models. Partial loss of SdhB or SdhC however resulted in defects in growth on bacterial lawns and unique effects on mitochondrial respiration.
4. Materials and Methods
4.1. Dicytostelium Discoideum Strains, Culture Conditions and Multicellular Development
All experiments were conducted using
D. discoideum parental strain AX2 and transformants derived from it. For bacterial growth, the cultures were grown on lawns of
Enterobacter aerogenes on SM agar (Formedium, Hunstanton, Norfolk, UK) at 21 °C as previously described [
34]. Cells were also grown axenically in HL-5 (Formedium, Hunstanton, Norfolk, UK) liquid medium with shaking (150 rpm) at 21 °C. As a selective marker, G418 (ThermoFisher Scientific, Waltham, MA, USA) at 25 µg/mL was added to the growth media during subculturing for all the transformants. However, for all the phenotypic investigations, all antibiotics were excluded from the growth medium in order to exclude any possible antibiotic-associated effects. The transformants contained the constructs described below for antisense inhibition of expression of SdhA (pPROF227), SdhB (pPROF765) or SdhC (pPROF756).
The fruiting body morphology of AX2 and transformants was examined after growth on KK2 (20 mM potassium phosphate buffer, pH 6.4, 1.2% agar) agar plates for several days at 21 °C. Aerial view images were taken of the fruiting bodies using an Olympus S761TM dissecting microscope equipped with a Mitocam 2300TM camera.
4.2. Plasmid Construction
For the construction of the Sdh antisense plasmids, the following gene-specific primers were used to amplify a fragment of the genes using AX2 genomic DNA as a template. For the sdhA gene, a 1493 bp fragment was PCR-amplified using the primers 5′sdha2 (5′-GTCGCAGCACAAGGTGCTATTAATAATGC-3′) and 3′sdha1 (5′-GCGAATTCGGATCCATGAGCACCACGACTTTCTTT-3′). For the sdhB gene, a fragment 338 bp in size was PCR amplified using the primers SdhBF (5′-GCGAATTCGGTACCATTACCACACATGCAT-3′) and SdhBR (5′-GCGAATTCGGATCCGATCTTCAGAGAGGAT-3′). For the sdhC, gene a 225 bp fragment was amplified via PCR using the primers SdhCAS_F 5′-GCGAATTCCTCGAGGCACCACTTGTCAGAAATTG-3′), and SdhCAS_R (5′-GCGAATTCGGATCCCATAACGGCTGGTAATGGG-3′). The PCR products were subcloned into the EcoRI site of the pDNeo2 plasmid in the antisense orientation to create the constructs named pPROF227 (SdhA), pPROF765 (SdhB) and pPROF756 (SdhC). All the constructs created for this study were verified by sequence analysis at the AGRF. We did not deploy empty vector controls in this work, because it is well established that there are no adverse phenotypic outcomes in Dictyostelium of the vectors themselves, or vectors with antisense fragments cloned in the sense orientation, out of frame, and following a stop codon, or vectors expressing nonfunctional portions of Dictyostelium proteins or vectors ectopically expressing marker proteins such as GFP or aequorin that do not interact with other proteins in the cell (Kotsifas et al. 2002; Bokko et al., 2007; Annesley et al., 2007; Nebl and Fisher, 200; Ahmed and Fisher, 2006).
4.3. D. discoideum Transformation
AX2 cells were transformed with 15 µg DNA of each of the necessary constructs using the calcium phosphate DNA precipitation method [
35]. All transformants were selected from isolated colonies on
Micrococcus luteus lawns grown on SM agar supplemented with 25 µg/mL G418 [
36] and subsequently subcultured and maintained on
E. aerogenes lawns and axenically in HL-5.
4.4. Measurement of Construct Copy Number
To measure the number of copies of the antisense inhibition constructs per genome in the transformants, quantitative PCR (qPCR) was employed using a CFX Connect real-time PCR detection system and the iQ SYBR Green Supermix (Bio-Rad, Hercules, CA, USA) according to the manufacturer’s instructions. Firstly, genomic DNA was extracted from D. discoideum WT and transformants using DNAzol (Molecular Research Center, CI, Ohio), according to the manufacturer’s instructions and the resulting DNA was resuspended in 50 µL milliQ H2O containing 100 µg/mL RNaseI. The following primer sequences were used for amplification of a portion of each gene including abpC (encoding filamin), a single copy gene which was used as a loading control:
Filamin: Forward primer 5′-CCACAGAGATATTGGAGTTGCGTACC-3′, Reverse primer 5′-CAACTCAACCAATGTGCCTGCCAA-3′.
SdhA: Forward primer 5′-TGCTCGTGGTGAAGGTGGTTATCT-3′, Reverse primer 5′- ACATCACGAGAGGCTAAATCGGCT-3′.
SdhB: Forward primer 5′-CAACACAACCAACACCCCATT-3′ Reverse primer 5′-GTGAGGTTGGTTGGTATGGAG-3′
SdhC: Forward primer 5′-CTGGTCTTGCAGGTGTTACT-3′, Reverse primer 5′-GGGTATTGAGTATGGAGGAGTTG-3′.
Two standard curves were prepared. One was used for estimation of the total DNA loaded and deployed known quantities of pure genomic DNA of the parental strain AX2 with the filamin primers. The second was for estimation of the quantity of the plasmid construct in question and deployed purified plasmid DNA and the cognate gene primers of interest. The standard curves were used in combination with the known sizes of the amplified fragment and of the D. discoideum genome to estimate the construct copy number. These raw copy number estimates were then normalised against the estimated number of copies of the target gene in the control parental strain AX2 included in the same experiment as an internal control (bringing the control copy number to the known value of 1).
4.5. Calculation of mRNA Expression
Semiquantitative real-time RT-PCR was used to measure relative expression levels of SdhA, SdhB and SdhC mRNA. Real-time PCR was performed as described previously [
37] using SensiFast SYBR and Fluorescein One-Step Kit and CFX Connect (Bio-Rad, Hercules, California, USA) real-time PCR detection system. Filamin was used as a loading control to estimate the total RNA loaded. The same filamin and Sdh primers were used as described in the previous section. RNA from WT and transformants was prepared using TriZol (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions.
4.6. Western Blotting
Whole-cell extracts were prepared by lysing 5 × 106 cells in 50 µL 2× Laemmli sample buffer (4% SDS, 20% glycerol, 0.004% bromophenol blue and 0.125 M Tris-Cl, pH 6.8). A 15 µL aliquot of cell lysate was heated at 95 °C for 10 min and then loaded onto 10% stain-free polyacrylamide gels for SDS-PAGE using a Bio-Rad MiniProtean II apparatus. The proteins thus separated were transferred to a PVDF membrane (Bio-Rad, Hercules, CA, USA) with the help of the Bio-Rad Mini Trans-Blot electrophoretic transfer cell. The membrane was probed with rabbit polyclonal anti-SdhA (1:1000, custom-made), and anti-SdhC (1:1000, Santa Cruz Biotechnology, Dallas, TX, USA) diluted in blocking buffer (1% casein, in TBST) overnight at 4 °C, followed by incubation in secondary goat anti-rabbit, HRP-conjugated (ThermoFisher Scientific, Waltham, MA, USA) for 1 h at RT. The protein bands of interest were visualized using Clarity ECL Western blotting substrate (Bio-Rad, Hercules, CA, USA) and acquired on an Amersham Imager 600 (GE Healthcare, Chicago, IL, USA) or a Chemidoc Touch (Bio-Rad, Hercules, CA, USA).
4.7. Succinate Dehydrogenase Activity Assay
Succinate dehydrogenase activity was determined following the reduction of the dye DCPIP (2,6-Dichlorophenolindophenol sodium salt hydrate) (an artificial electron acceptor) and succinate at 600 nm at 25 °C as described in [
38]. Exponentially grown
D. discoideum vegetative cells were harvested, washed, and resuspended in H
2O to a density of 1 × 10
6 /mL. The cells were lysed by quick freezing at −80 °C for 15–30 min. In a clear 96 well, flat bottom plate, 40 μL of succinate reductase assay buffer (10 mM KH
2PO
4, 2 mM EDTA, 1 mg/mL BSA, 80 μM DCPIP, 4 μM rotenone, 0.2 mM ATP, 10 mM succinate) was added, followed by 50 μL of cell lysate. The reaction was started by the injection of 10 μL of 80 μM decylubiquinone, while measuring the absorbance at 600 nm for 10–15 min using a Clariostar microplate reader and finalized by the complete inhibition of Complex II with the injection of 10 mM malonate.
4.8. Growth in Axenic Medium
D. discoideum AX2 and transformant strains were grown exponentially in HL-5 medium without antibiotics and then subcultured into fresh 50 mL HL-5 at a density of 1–2 × 10
4 cells/mL and incubated shaking at 150 rpm at 21 °C. The cell densities were determined using a haemocytometer at 6–12 h intervals over 5 days and the generation times were estimated by log-linear regression during the exponential phase of growth using the R environment for statistical computing and graphics (
http://www.R-project.org, accessed on 27 September 2021).
4.9. Growth on Bacterial Lawns
A scraping of amoebae from the edge of AX2 and transformant D. discoideum plaques was inoculated in the centre of an E. coli B2 lawn grown on Normal agar (20 g/L agar (ThermoFisher Scientific, Waltham, MA, USA), 1 g/L bactopeptone (ThermoFisher Scientific, Waltham, MA, USA), 1.1 g/L glucose, 1.9972 g/L KH2PO4, 0.356 g/L Na2HPO4*2H2O). The plates were incubated at 21 °C and the diameter of each plaque was recorded at intervals of 8–12 h for a period of 5–7 days. The results were analysed by linear regression using the R software to calculate the growth rate for each strain.
4.10. Endocytosis Assays
Phagocytosis by
D. discoideum strains was measured by determining uptake of an
E.coli strain expressing the fluorescent protein DsRed [
39] as previously described [
34]. Macropinocytosis rates were determined via measuring uptake of fluorescein isothiocyanate (FITC)-dextran (Sigma-Aldrich, average mol. Mass 70 kDa, St. Louis, MO, USA) as previously described [
34].
4.11. Phototaxis Assay
Qualitative phototaxis was done as previously described [
40]. Scrapings of amoebae from the edges of
D. discoideum plaques grown on SM agar plates with a lawn of
E. aerogenes were placed on the centre of charcoal agar plates (5% activated charcoal, 1.5% agar). The plates were incubated at 21 °C for 2 days with a single lateral source to allow slugs to form and migrate. The slug trails were examined, transferred to PVC discs and stained with Coomassie Blue and then photographed for further analysis.
4.12. Seahorse Respirometry
A Seahorse Extracellular Flux Analyser (Agilent Technologies, Santa Clara, California) was used for mitochondrial respiration assays as described previously [
41].
D. discoideum AX2 and transformed cells were grown axenically in HL-5 medium to exponential phase, washed and resuspended in SIH (Formedium, Hunstanton, Norfolk, UK) media supplemented with 20 mM pyruvate and 5 mM malate, pH7.4. Each strain was inoculated onto 8 wells for transformants or 4 wells for AX2 of a 24-well cell culture plate, pre-treated with Matrigel, to ensure cell attachment during the assay. Basal Oxygen Consumption Rates (OCR) were measured, followed by the injection of a series of mitochondrial inhibitors and reagents: [10 μM DCCD (N,N0-dicyclohexylcarbodimide, an ATP synthase inhibitor (Sigma-Aldrich, St. Louis, MI, USA), 10 μM CCCP (carbonyl cyanide 3-chlorophenol hydrazone, a protonophore (Sigma-Aldrich, St. Louis, MI, USA)], 20 μM rotenone [Complex I inhibitor (Sigma-Aldrich, St. Louis, MI, USA)] and either 10 μM antimycin A [Complex III inhibitor (Sigma-Aldrich, St. Louis, MI, USA)] or 1.5 mM BHAM [benzohydroxamic acid, alternative oxidase (AOX) inhibitor (Sigma-Aldrich, St. Louis, MI, USA)]. Measurement cycles consisting of 3 min of mixing, 2 min wait, and 3 min measurement time were completed before and after each sequentially added drug, at least two cycles per condition. The measurements could then be used to calculate the contribution of different complexes and components to mitochondrial respiration.
4.13. Mitochondrial Mass and Mitochondrial Membrane Potential
Measurements of mitochondrial mass and mitochondrial membrane potential were performed simultaneously using the mitochondrial stains Mitotracker Red CMXRos and Mitotracker Green FM (ThermoFisher Scientific, Waltham, MA, USA). D. discoideum AX2 and transformed cells were harvested, resuspended in LoFlo HL5 to a density of 1 × 106 cells/mL and 100 μL of each strain was seeded into 6 wells of a 96-well black plate and allowed to equilibrate for 40 min. The mitochondrial stains (Mitotracker Red 200 nM and Mitotracker green, 400 nM) diluted in LoFlo HL5 medium were added to 2 wells each and further incubated for 1 h. For each strain, duplicate wells of unstained cells were used as blank. The fluorescence measurements were performed using a Clariostar microplate reader (BMG) with excitation and emission set at 470 nm 515 nm respectively for Mitotracker Green and 570 nm and 620 nm for Mitotracker Red. The mitochondrial mass was calculated by subtracting the fluorescence of the unstained well from that of the Mitotracker Green-stained wells, while the mitochondrial membrane potential was the background-subtracted Mitotracker Red fluorescence divided by the background-subtracted Mitotracker Green fluorescence. The parental AX2 strain was used for assay normalization.
4.14. ATP Steady-State Levels
The ATP determination kit (ThermoFisher Scientific, Waltham, MA, USA) was used to determine the ATP content for the D. discoideum AX2 and transformant strains according to the manufacturer’s instructions. D. discoideum strains were grown to exponential phase 1–3 × 106 /mL, harvested by centrifugation and 1 × 105 cells were aliquoted into duplicate sterile Eppendorf tubes. Cells were lysed and ATP was extracted by the addition of 100 µL 1% (w/v) TCA. The TCA was neutralized by the addition of 900 µL of 28 mM tricine buffer pH 8.25 and 5 µL of supernatant was transferred to a fresh microcentrifuge tube and 45 µL of reconstituted luciferin reaction buffer was added and immediately measured. The intensity of the light emitted which is proportional to the amount of ATP was measured in a Modulus Fluorometer (Turner Biosystems, Sunnyvale, CA, USA) using the luminometer module. A separate calibration curve relating the luminescence signal with known amounts of ATP was used to estimate the total amount of ATP in each sample.
4.15. Reactive Oxygen Species (ROS) Assay
ROS measurements were performed using DCFDA (2′,7′-dichlorofluorescein diacetate, Sigma-Aldrich, St. Louis, MI, USA), a fluorogenic, cell-permeant reagent that is able to detect hydroxyl, peroxyl and other reactive oxygen species within cells. Once DFCDA diffuses into the cells, it is deacetylated by cellular esterases and then oxidized by the intracellular ROS into the green fluorescent DCF. D. discoideum AX2 and transformed strains grown axenically were harvested by centrifugation at 1000× g for 1 min, resuspended in LoFlo HL5 at a density of 1 × 106 /mL and 100 μL of the cell suspension was plated into 4 wells of a 96 well black, clear flat bottom plate (Corning Inc., Corning, NY, USA). After one hour of incubation at RT, 100 μL of DCFDA was added to two wells to a final concentration of 20 μM, and two wells were used with no reaction mix for background subtraction. The fluorescence was measured using a Clariostar plate reader at 485 nm excitation, 535 nm emission. The background values were subtracted from the stained values and then averaged and normalized to values from parental AX2 strain.
4.16. Legionella Proliferation Assay
The
Legionella proliferation assay was performed as described previously [
22]. Axenically grown cells were harvested by centrifugation at 1000×
g/3 min, washed twice with Sorensen 1×C buffer (17 mM KH
2PO
4/Na
2PO
4, 50 mM CaCl
2, pH 6.0) and the pellet was resuspended in MB medium (0.7% yeast extract, 1.4% proteose peptone, 0.062% Na
2HPO
4·2H
2O, 0.049% KH
2PO
4, pH 6.9) at a density of 5 × 10
5 cells/mL. For each strain, 1 × 10
5 cells were transferred into 6 wells of a 96-well tissue culture plate (Corning Inc., Corning, NY, USA) and allowed to adhere for 30 min. The
Legionella pneumophila Corby strain was grown on Buffered Charcoal Yeast Extract Agar (BCYE agar) supplemented with chloramphenicol 5 μg/mL at 37 °C with 5% CO
2 for 3 days. The bacteria were harvested and resuspended in distilled water, diluted in MB to a density of 1 × 10
6 bacteria/mL and used to infect
D. discodeium in each well at an MOI of 1:1. This was achieved by reading the OD of the bacterial suspension, assuming that an OD
600 of 1 equates to 10
9 bacteria/mL. To ensure initial adherence the plate was centrifuged at 700×
g/10 min. At each time point (0, 2, 24, 48, 72 and 96 h) 50 µg/mL of gentamycin was added to each of the wells and incubated for 30 more min to achieve the lysis of any uningested bacterial cells. The
D. discoideum amoebae were resuspended, transferred to 1.5 mL microfuge tubes and pelleted by centrifugation at 13,000×
g for 10 min. The cells were washed twice in ice-cold Sorensen’s 1 ×C buffer and resuspended in 300 µL Sorensen’s 1×C buffer, supplemented with 0.02% saponin and lysed by vigorous vortexing for 10 sec. A dilution series of the lysate from 10
−1 to 10
−4 was prepared and spread-plated on BCYE plates without chloramphenicol. The plates were incubated at 37 °C with 5% CO
2 for 3–4 days prior to counting the number of colonies to calculate the number of viable
L. pneumophila for each time point.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}