
The Social Norm of Hematopoietic Stem Cells and Dysregulation in Leukemia


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Hematopoiesis Meets Demands
2.1. Tailoring of the Setup of HSC Compartment
2.2. Subsets of Lineage-Biased/Affiliated HSCs
2.3. HSC Behavior and Feedback Control from the Level Mature Cells
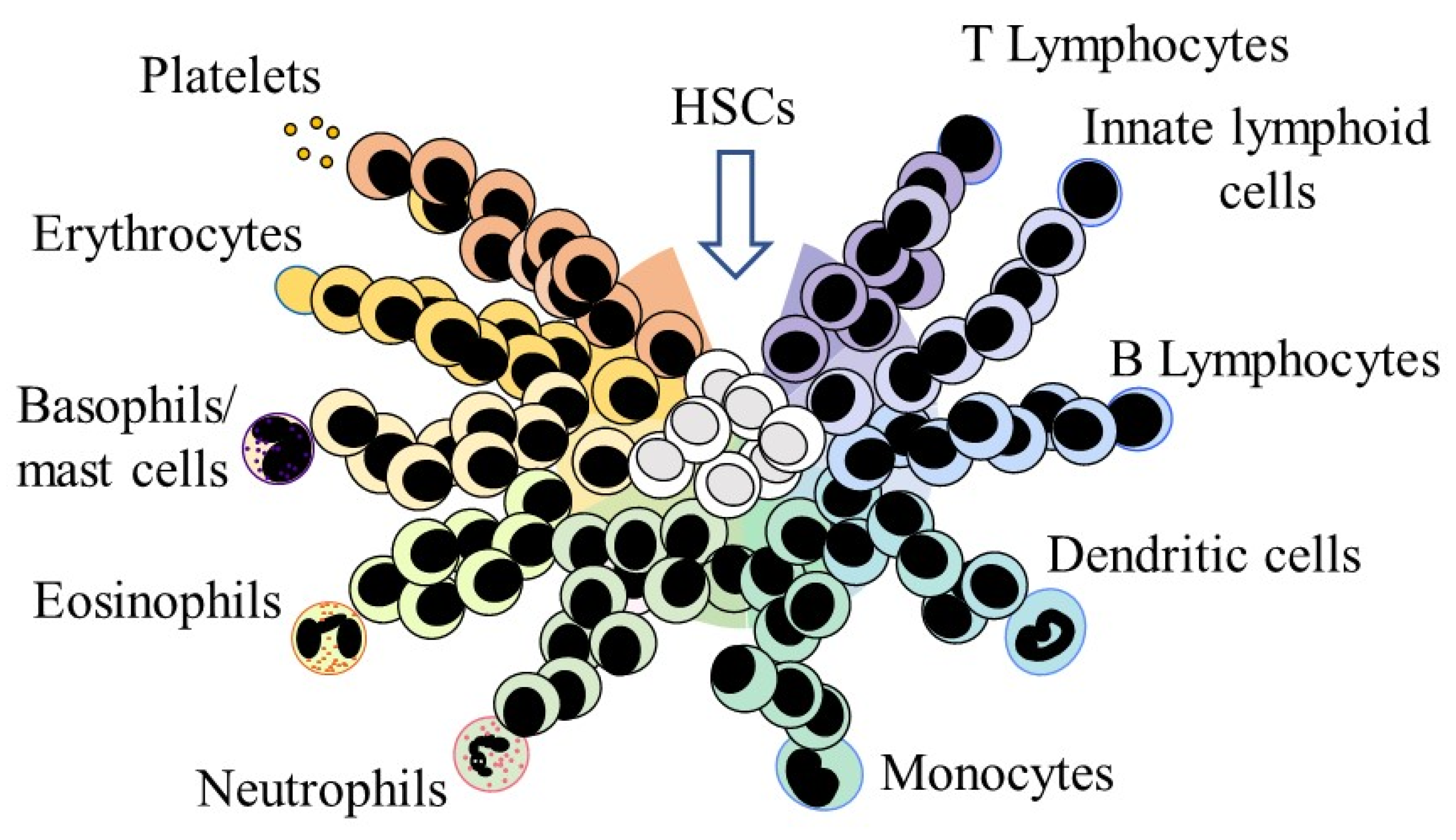
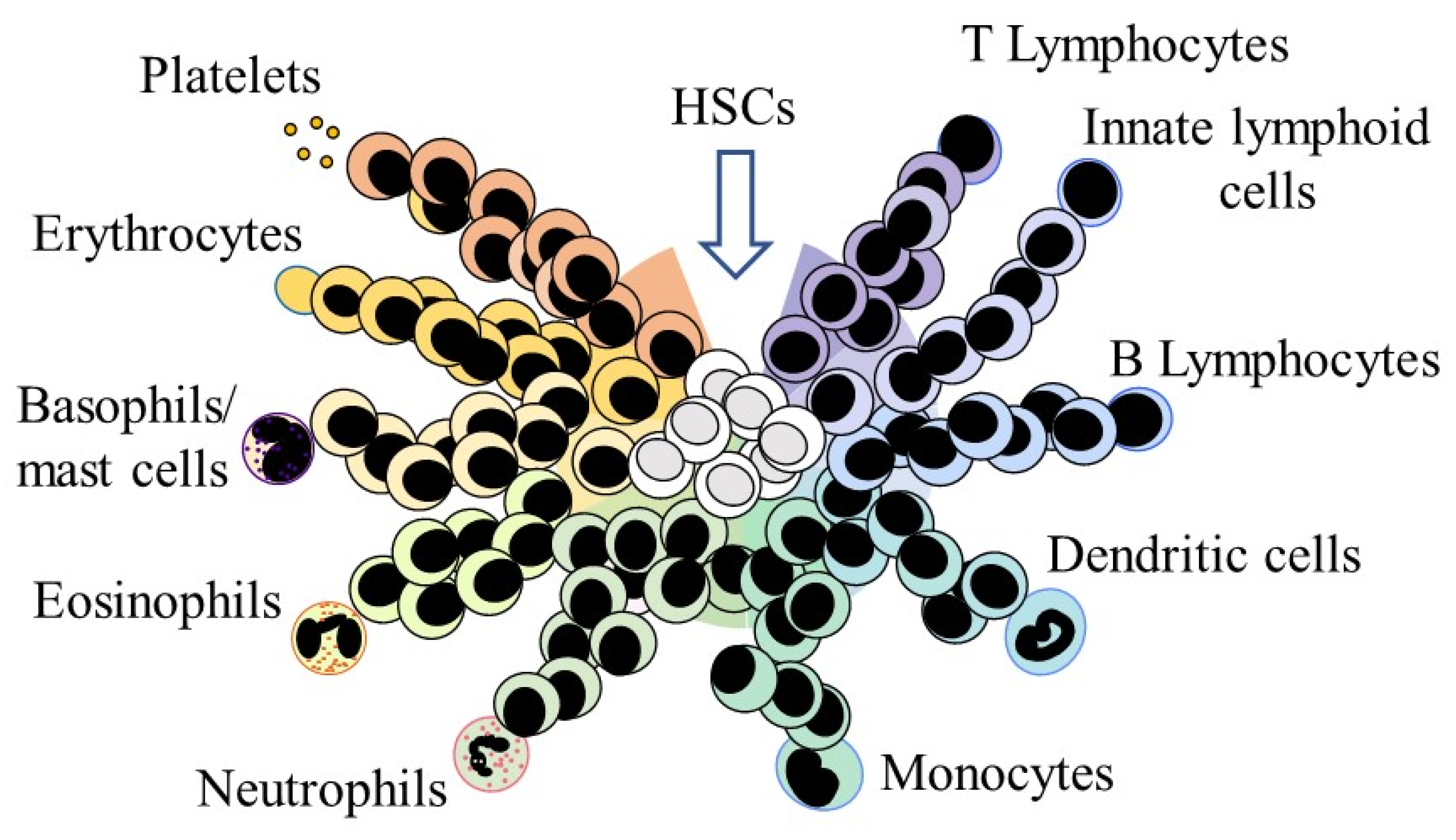
3. HSC Affiliation to a Cell Lineage
HSCs and Their Offspring Remain Versatile
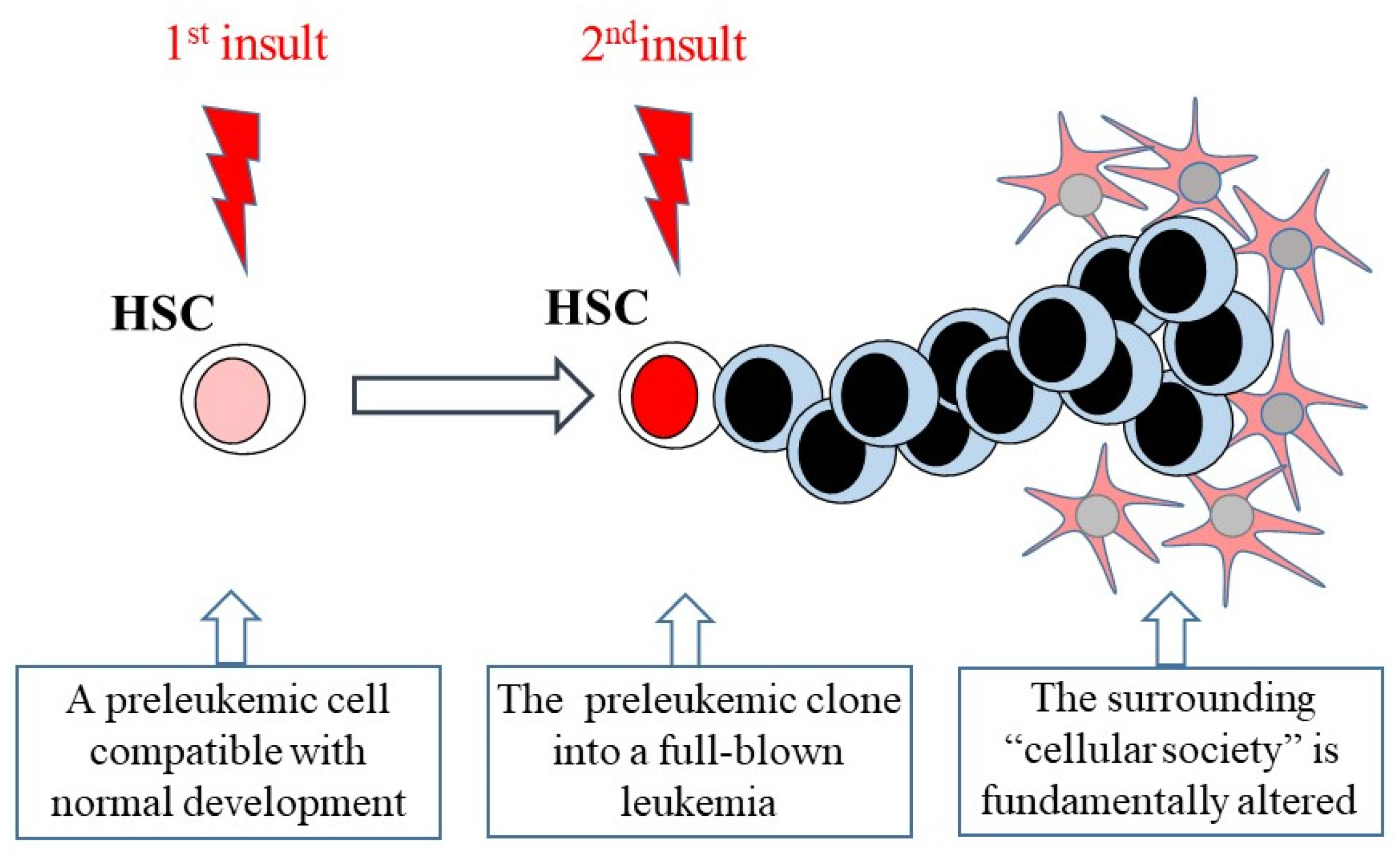
4. The Genesis of Leukemia
4.1. Some Leukemias Have a Specific Mutational Signature
4.2. The Cell of Origin for Some Leukemias
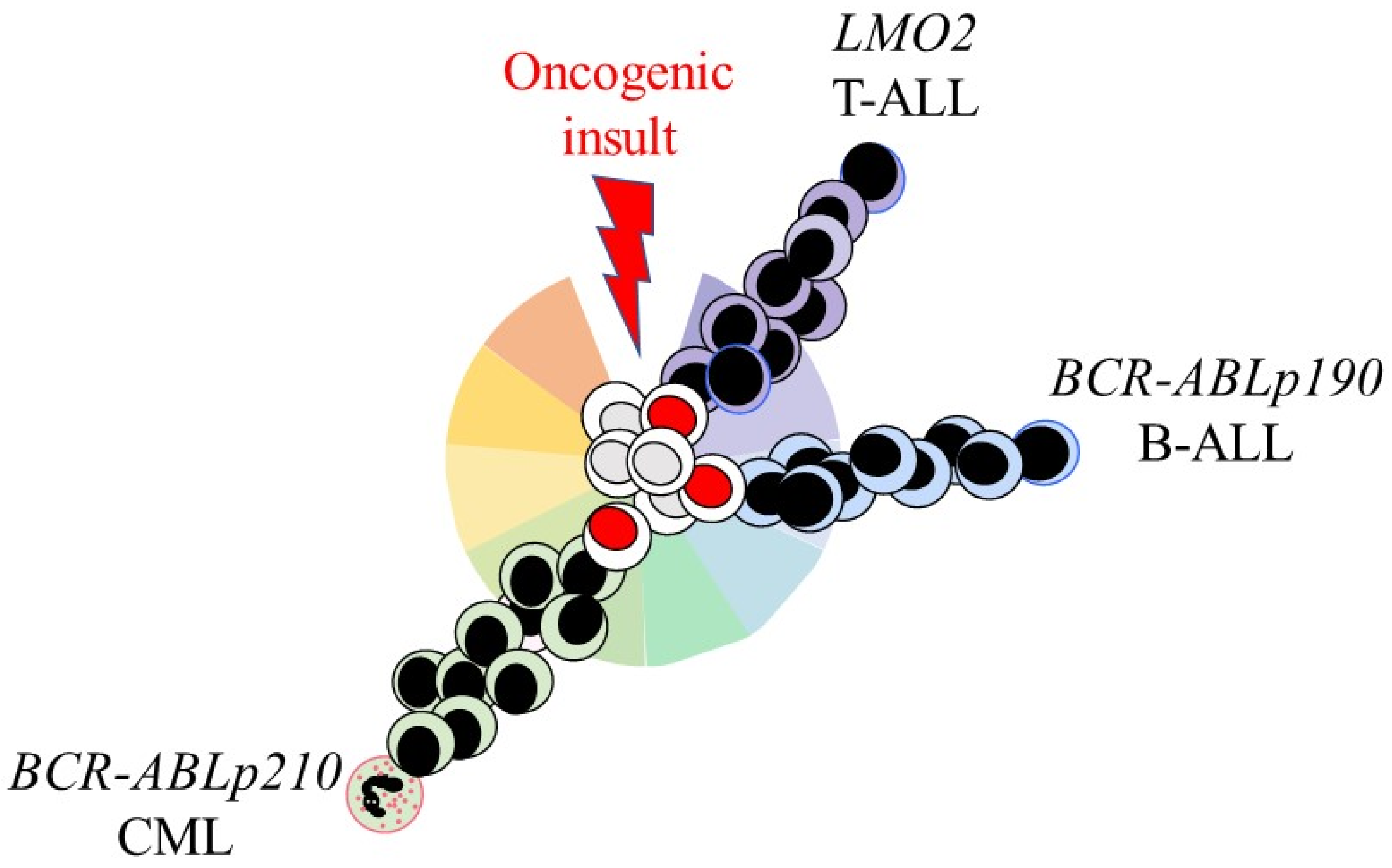
5. Some Oncogenes Restrict LSCs and Their Offspring to a Single Cell Lineage
6. The Antisocial Interplay between LSCs and Bone Marrow Niches
6.1. LSCs Disruption to Bone Marrow Niches
6.2. LSC Invasion of Distant Tissues
7. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organised as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 7, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Dick, J.E. Stem cell concepts renew cancer research. Blood 2008, 112, 4793–4807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Gutierrez, L.; Delgado, M.D.; Lean, J. MYC oncogene contributions to release of cell cycle brakes. Genes 2019, 10, 244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prior, I.A.; Levis, P.D.; Mattos, C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radha, G.; Raghavan, S.C. BCL2: A promising cancer therapeutic target. Biochim. Biophys. Acta 2017, 1868, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Danes, A.; Blanpain, C. Deciphering the cells of origin of squamous cell carcinomas. Nat. Rev. Cancer 2018, 18, 546–561. [Google Scholar] [CrossRef]
- Loeb, K.R.; Loeb, L.A. Significance of multiple mutations in cancer. Carcinogenesis 2000, 21, 379–385. [Google Scholar] [CrossRef]
- Manz, M.G.; Boettcher, S. Emergency granulopoiesis. Nature 2014, 14, 302–314. [Google Scholar] [CrossRef]
- Roberts, A.W. G-CSF: A key regulator of neutrophil production, but that’s not all! Growth Factors 2005, 23, 33–41. [Google Scholar] [CrossRef]
- Abercrombie, M.; Heaysman, J.E. Observations on the social behaviour of cells in tissue culture. II. Monolayering of fibroblasts. Exp. Cell Res. 1954, 6, 293–306. [Google Scholar] [CrossRef]
- Vedel, S.; Tay, S.; Johnson, D.M.; Bruus, H.; Quake, S.R. Migration of cells in a social context. Proc. Natl. Acad. Sci. USA 2013, 110, 129–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietras, E.M.; Reynaud, D.; Kang, Y.-A.; Carlin, D.; Calero-Nieto, F.J.; Leavitt, A.D.; Stuart, J.M.; Gottgens, B.; Passegue, E. Functional distinct subsets of lineage-biased multipotent progenitors control blood production in normal and regenerative conditions. Cell Stem Cell 2015, 17, 35–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Ramus, A.; Chapman, B.; Johnnidis, J.B.; Le, L.; Ho, Y.J.; Klein, A.; Hoffman, O.; Camargo, F.D. Clonal dynamics of native haematopoiesis. Nature 2014, 514, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Busch, K.; Klapproth, K.; Barile, M.; Flossdorf, M.; Holland-Letz, T.; Schlenner, S.M.; Reth, M.; Hofer, T.; Rodewald, H.R. Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature 2015, 518, 543–546. [Google Scholar] [CrossRef]
- Ema, H.; Morita, Y.; Suda, T. Heterogeneity and hierarchy of hematopoietic stem cells. Exp. Hematol. 2014, 42, 74–82. [Google Scholar] [CrossRef]
- Yamamoto, R.; Morita, Y.; Ooehara, J.; Hamanaka, S.; Onodera, M.; Rudolph, K.L.; Ema, H.; Nakauchi, H. Lineage-restricted progenitors generated directly from haematopoietic stem cells. Cell 2013, 154, 1112–1126. [Google Scholar] [CrossRef] [Green Version]
- Sanjuan-Pla, A.; Macauley, I.C.; Jensen, C.T.; Woll, P.S.; Luis, T.C.; Mead, A.; Moore, S.; Carella, O.; Matsuoka, S.; Jones, T.B.; et al. Platelet-biased stem cells reside at the apex of the haematopoietic stem-cell hierarchy. Nature 2013, 502, 232–236. [Google Scholar] [CrossRef]
- Mooney, C.J.; Cunningham, A.; Tsapogas, P.; Toellner, K.-M.; Brown, G. Selective expression of Flt3 within the mouse hematopoietic stem cell compartment. Int. J. Mol. Sci. 2017, 18, 1037. [Google Scholar] [CrossRef] [Green Version]
- Notta, F.; Zandi, S.; Takayama, N.; Dobson, S.; Gan, O.I.; Wilson, G.; Kaufmann, K.B.; McLeod, J.; Laurenti, E.; Dunant, C.F.; et al. Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science 2016, 351, 6269. [Google Scholar] [CrossRef] [Green Version]
- Mossedegh-Keller, N.; Sarrazins, S.; Kandella, P.K.; Espinosa, L.; Stanley, E.R.; Nutt, S.L.; Moore, L.; Siewke, M.H. M-CSF instructs myeloid lineage fate in single haematopoietic stem cells. Nature 2013, 497, 239–243. [Google Scholar] [CrossRef]
- Grover, R.; Mancini, I.C.; Moore, S.; Mead, A.J.; Atkinson, D.; Rasmussen, K.D.; O’Carrol, D.O.; Jacobsen, S.E.W.; Nerlov, C. Erythropoietin guides multipotent progenitors towards an erythroid fate. J. Exp. Med. 2014, 211, 181–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.P.; Grinenko, T.; Ramasz, B.; Franke, K.; Leshe, M.; Dahl, A.; Gassman, M.; Chavakis, T.; Henry, L.; Wielockx, B. Haematopoietic stem cells but not multipotent progenitors drive erythropoiesis during chronic erythroid stress in epo transgenic mice. Stem Cell Rep. 2018, 10, 1908–1919. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, D.; Burgess, A.W. Clonal analysis of progenitor cell commitment of granulocyte or macrophage production. J. Cell Physiol 1982, 111, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Rieger, M.A.; Hoppe, P.S.; Smejkal, B.M.; Eitelhuber, A.C.; Schroeder, T. Haematopoietic cytokines can instruct lineage choice. Science 2009, 325, 217–218. [Google Scholar] [CrossRef]
- Palis, J. Primitive and definitive erythropoiesis in mammals. Front. Physiol. 2014, 5, 3. [Google Scholar] [CrossRef] [Green Version]
- Cheshier, S.H.; Prahaska, S.S.; Weissman, I.L. The effect of bleeding on hematopoietic stem cell cycling and self-renewal. Stem Cells Dev. 2007, 16, 707–718. [Google Scholar] [CrossRef]
- Paulson, R.F.; Ruan, B.; Hao, S.; Chen, Y. Stress erythropoiesis is a key inflammatory response. Cells 2020, 9, 634. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.; Ohneda, K.; Hosaya-Ohmura, S.; Tsukamoto, S.; Ohneda, O.; Philipsen, S.; Yamamoto, M. Real-time monitoring of stress erythropoiesis in vivo using Gata1 and β-globin LCR Luciferase transgenic mice. Blood 2006, 108, 725–733. [Google Scholar] [CrossRef]
- Ceredig, R.; Rolink, A.G.; Brown, G. Models of haematopoiesis: Seeing the wood for the trees. Nat. Rev. Immunol. 2009, 9, 293–300. [Google Scholar] [CrossRef]
- Nestorowa, S.; Hamey, F.K.; Pijuan Sala, B.; Diamanti, F.; Shepherd, M.; Laurenti, E.; Wilson, N.K.; Kent, D.G.; Gottgens, B. A single-cell resolution map of mouse hematopoietic stem and progenitor cell differentiation. Blood 2016, 128, e20–e31. [Google Scholar] [CrossRef] [Green Version]
- Psaila, B.; Mead, A.J. Single cell approaches reveal novel cellular pathways for megakaryocyte and erythroid differentiation. Blood 2019, 133, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Velten, L.; Haas, S.F.; Raffel, S.; Blaszkiecz, S.; Islam, S.; Hennig, B.P.; Hirche, C.; Lutz, C.; Buss, E.C.; Nowak, D.; et al. Human haematopoietic stem cell lineage commitment is a continuous process. Nat. Cell Biol. 2017, 19, 271–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, J.J.; Bhandola, A. The earliest thymic progenitors for T cells possess myeloid lineage potential. Nature 2008, 452, 764–767. [Google Scholar] [CrossRef] [PubMed]
- Balciunaite, G.; Ceredig, R.; Rolink, A.G. The earliest subpopulation of mouse thymocytes contains potent T, significant macrophage and natural killer but no B lymphocyte potential. Blood 2005, 105, 1930–1936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Land, H.; Parada, L.F.; Weinberg, R.A. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature 1983, 304, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.F.; Maia, A.T.; Wiemels, J.; Ford, A.M. Leukemia in twins: Lessons in natural history. Blood 2003, 102, 2321–2333. [Google Scholar] [CrossRef] [PubMed]
- Schafer, D.; Olsen, M.; Lahnemann, D.; Stanulla, M.; Slany, R.; Schiegelow, A.; Fischer, U. Five percent of healthy newborns have an ETVG-RUNX1 fusion as revealed by DNA-based GIPFEL screening. Blood 2018, 131, 821–826. [Google Scholar] [CrossRef] [Green Version]
- Groffen, J.; Stephenson, J.R.; Heisterkamp, N.; de Klein, A.; Bartram, C.R.; Grosveld, G. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosomal 22. Cell 1984, 36, 93–94. [Google Scholar] [CrossRef]
- Shtivelman, E.; Lifshitz, B.; Gale, R.P.; Canaani, E. Fused transcript of abl and bcr genes in chronic myelogenous leukemia. Nature 1985, 315, 550–554. [Google Scholar] [CrossRef]
- Cazzaniga, G.; van Delti, F.W.; Lo Nigro, L.; Ford, A.M.; Score, J.; Iacobucci, I.; Mirabile, E.; Taj, M.; Colman, S.M.; Biondi, A.; et al. Developmental origins and impact of BCR-ABL1 fusion and IKZF1 delections in monozygotic twins with Ph+ acute lymphoblastic leukemia. Blood 2011, 118, 5559–5564. [Google Scholar] [CrossRef]
- Kosik, P.; Skorvaga, M.; Durdik, M.; Jakl, I.; Nikitina, E.; Markova, E.; Kozics, K.; Horvathova, E.; Belyaev, I. Low numbers of pre-leukemic fusion genes are frequently present in umbilical cord blood without affecting DNA damage response. Oncotarget 2017, 8, 35824–35834. [Google Scholar] [CrossRef] [Green Version]
- Boss, S.; Deininger, M.; Gora-Tybor, J.; Goldmann, J.M.; Melo, J.V. The presence of atypical BCR-ABL fusion genes in leukocytes of normal individuals: Biological significance and implications for the assessment of minimal residual disease. Blood 1998, 92, 3363–3367. [Google Scholar]
- Sanjuan-Pla, A.; Bueno, C.; Preito, C.; Acha, P.; Stam, R.W.; Marschalek, R.; Mendez, P. Revisiting the biology of infant t(4;11)/MLL-AF4+ B-cell acute lymphoblastic leukemia. Blood 2015, 126, 2676–2685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pui, C.H.; Nichols, K.E.; Yang, J.J. Somatic and germline genomics in paediatric acute lymphoblastic leukaemia. Nat. Rev. Clin. Oncol. 2019, 16, 238–240. [Google Scholar] [CrossRef] [PubMed]
- Rabbits, T.H. LMO T-cell translocation oncogenes typify genes activated by chromosomal translocations that alter transcription and development processes. Genes Dev. 1998, 12, 2651–2657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murga Penas, E.M.; Hinz, K.; Roser, K.; Copie-Bergman, C.; Wlodorska, I.; Marynen, P.; Hagemeijar, A.; Gaulard, P.; Loning, T.; Hassfeld, D.K.; et al. Translocations t(14;18)(q21;q21) and t(14;18)(q32;q21) are the main chromosomal abnormalities involving MLT/MALT1 in MALT lymphomas. Leukemia 2003, 17, 2225–2229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sankar, S.; Lessnick, S.L. Promiscuous partnerships in Ewing’s sarcoma. Cancer Genet. 2011, 204, 351–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, J.; Rocques, P.J.; Crew, A.J.; Gill, S.; Shipley, J.; Chan, A.M.; Gusterson, B.A.; Cooper, C.S. Identification of the novel genes, SYT and SSX, involved in the t(X;18)(p11.2;q11.2) translocation found in human synovial sarcoma. Nat. Genet. 1994, 7, 502–508. [Google Scholar] [CrossRef]
- Fialkow, P.J.; Denman, A.M.; Jacobson, G.J.; Lowenthal, M.N. Chronic myelocytic leukaemia: Origin of some lymphocytes from leukaemic stem cell. J. Clin. Investig. 1978, 62, 815–823. [Google Scholar] [CrossRef] [Green Version]
- Malouf, C.; Ottersbach, K. The fetal liver lymphoid-primed multipotent progenitor provides the prerequisites for the initiation of t(4;11) MLL-AT4 infant leukemia. Haematologica 2018, 103, e573. [Google Scholar] [CrossRef] [Green Version]
- Adolfsson, J.; Mansson, R.; Buza-Vidas, N.; Hultquist, A.; Liuba, K.; Jensen, C.T.; Bryder, D.; Yang, L.; Borge, O.-J.; Thoren, L.A.M.; et al. Identification of Flt3+ lympho-myeloid stem cells lacking erythro-megakaryocytic potential: A revised road map for adult blood lineage commitment. Cell 2005, 121, 295–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greaves, M.F. Analysis of the clinical and biological significance of lymphoid phenotypes in acute leukemia. Cancer Res. 1981, 41, 4752–4766. [Google Scholar] [PubMed]
- Quijano, C.A.; Moore II, D.; Arthur, D.; Feusner, J.; Winter, S.S.; Pallavicini, M.C. Cytogenetically aberrant cells are present in the CD34+ CD33− 38− 19− marrow compartment in children with acute lymphoblastic leukemia. Leukemia 1997, 11, 1508–1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, C.V.; Blair, A. A primitive cell origin for B-cell precursor ALL. Stem Cell Rev. 2005, 1, 189–196. [Google Scholar] [CrossRef]
- Hirt, A.; Schmid, A.-M.; Amman, R.A.; Leibundgut, K. In pediatric lymphoblastic leukemia of B cell origin, a small population of primitive blast cells is noncycling, suggesting them to be leukemia stem candidates. Pediatric Res. 2011, 69, 194–199. [Google Scholar] [CrossRef] [Green Version]
- Bernt, K.M.; Armstrong, S.A. Leukemia stem cells and acute lymphoblastic leukemia. Semin. Hematol. 2009, 46, 33–38. [Google Scholar] [CrossRef] [Green Version]
- Alizadeh, A.A.; Majeti, R. Surprise! HSCs are aberrant in chronic lymphocytic leukemia. Cancer Cell 2011, 20, 135–136. [Google Scholar] [CrossRef] [Green Version]
- Kikushige, Y.; Iskikawa, F.; Miyamoto, T.; Shima, T.; Urata, S.; Yashimoto, T.; Mari, Y.; Iiono, T.; Yamauchi, T.; Eto, T.; et al. Self-renewing hematopoietic stem cell is the primary target in pathogenesis of human chronic lymphocytic leukemia. Cancer Cell 2011, 246–259. [Google Scholar] [CrossRef] [Green Version]
- Perez-Caro, M.; Cobaleda, C.; Gonzalez-Herrero, I.; Vicente-Duenas, C.; Bermejo-Rodriguez, C.; Sanchez-Beato, M.; Orfao, A.; Pintado, B.; Flores, T.; Sanchez-Martin, M.; et al. Cancer induction by restriction of oncogene expression to the stem cell compartment. EMBO J. 2009, 28, 8–20. [Google Scholar] [CrossRef] [Green Version]
- Vicente-Duenas, C.; Gonzalez-Herrero, I.; Sehgal, L.; Garcia-Ramirez, I.; Rodriguez-Hernandez, G.; Pintado, B.; Blanco, O.; Criado, F.J.G.; Cenador, M.B.G.; Green, M.R.; et al. Dnmt1 links BCR-ABLp210 to epigenetic tumor stem cell priming in myeloid leukemia. Leukemia 2019, 33, 249–278. [Google Scholar] [CrossRef]
- Martin-Lorenzo, A.; Auer, F.; Chan, L.N.; Garcia-Ramirez, I.; Gonzalez-Herrero, I.; Rodriguez-Hernandez, G.; Bartenhagen, C.; Dugas, M.; Gombert, M.; Ginzel, S.; et al. Loss of Pax5 Exploits Sca1-BCR-ABLp190 Susceptibility to Confer the Metabolic Shift Essential for pB-ALL. Cancer Res. 2018, 78, 2669–2679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Ramirez, L.; Bhatia, S.; Rodriguez-Hermandez, G.; Gonzalez-Herrero, L.; Walter, C.; Gonzales de Tena-Davilo, S.; Parvin, S.; Haas, O.; Woessmann, W.; Stanulla, M.; et al. Lmo2 expression defines tumor cell identity during T-cell leukemogenesis. EMBO J. 2018, 37, e98783. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Hernandez, G.; Hauer, J.; Martin-Lorenzo, A.; Schafer, D.; Bartenhagen, C.; Garcia-Ramirez, I.; Auer, F.; Gonzalez-Herrero, T.; Ruiz-Roca, L.; Gombert, M.; et al. Infection exposure promotes ETV6-RUNX1 precursor B cell leukaemia via impaired H3K4 demethylases. Cancer Res. 2017, 77, 4265–4277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Hernandez, G.; Casado-Garcia, A.; Isidro-Hernandez, M.; Picard, D.; Raboso-Gallego, T.; Aleman-Arteaga, S.; Orfao, A.; Blanco, O.; Riesco, S.; Prieto-Matos, P.; et al. The second oncogenic hit determines the cell fate of ETV6-RUNX1 positive leukemia. Front. Cell Dev. Biol. 2021, 9, 704591. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Niu, C.; Ye, L.; Huang, H.; He, X.; Tong, W.-G.; Ross, J.; Haug, J.; Johnson, T.; Feng, J.Q.; et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature 2003, 425, 836–841. [Google Scholar] [CrossRef] [Green Version]
- Lowenberg, B.; Delwei, R.; Touw, I. Hematopoietic growth factors and in vitro growth of acute myeloblastic leukaemia. Crit. Rev. Oncol. Heamtol. 1990, 10, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Nishi, K.; Katayama, N.; Miwa, H.; Shikami, M.; Masuya, M.; Shiki, H. The survival of human leukaemic B-cell precursors is supported by stromal cells and cytokines associated with the expression of bcl-2. Br. J. Haematol. 1995, 105, 701–710. [Google Scholar] [CrossRef]
- Duarte, D.; Hawkins, E.D.; Lo Celso, C. The interplay of leukaemia cells and the bone marrow microenvironment. Blood 2018, 131, 1507–1511. [Google Scholar] [CrossRef]
- Walkley, C.R.; Olsen, G.H.; Dworkin, S.; Fabb, S.A.; Swann, J.; McArthur, G.A.; Westmoreland, S.V.; Chambon, P.; Scadden, D.J.; Purton, L.E. A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor gamma deficiency. Cell 2007, 126, 1092–1110. [Google Scholar]
- Sanchez-Aguilera, A.; Mendez-Ferrer, S. The hematopoietic stem-cell niche in health and leukemia. Cell Mol. Life Sci. 2017, 74, 579–590. [Google Scholar] [CrossRef] [Green Version]
- Greaves, M. Darwinian medicine: A case for cancer. Nat. Rev. Cancer 2007, 7, 213–221. [Google Scholar] [CrossRef] [PubMed]
- DeGregori, J. Connecting cancer to its cause requires incorporation of effects on tissue microenvironments. Cancer Res. 2017, 77, 6065–6068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyd, A.L.; Campbell, C.J.V.; Hopkins, C.J.; Fiebig-Comyn, A.; Russel, J.; Ulemek, T.; Foley, R.; Leber, B.; Xenocostas, A.; Collins, T.J.; et al. Niche displacement of human leukemic stem cells uniquely allows their competitive replacement with healthy HSPCs. J. Exp. Med. 2014, 211, 1925–1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, E.D.; Duarte, D.; Akinduro, O.; Kharshed, R.A.; Passaro, D.; Nowicka, M.; Straszkowski, L.; Scott, M.K.; Rothery, S.; Ruivo, N.; et al. T-cell acute leukaemia exhibits dynamic interactions with bone marrow microenvironments. Nature 2016, 532, 518–522. [Google Scholar] [CrossRef] [Green Version]
- Schepers, K.; Pietras, E.M.; Reynaud, D.; Flach, J.; Binnewies, M.; Garg, T.; Wagers, A.J.; Hsiao, E.C.; Passegue, E. Myeloblastic neoplasia remodels the endosteal bone marrow nice into a self-reinforcing leukemic niche. Cell Stem Cell 2013, 13, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Hanoun, M.; Zhang, D.; Mizoguchi, T.; Pinho, S.; Pierce, H.; Kunisaki, Y.; Lacombe, J.; Armstrong, S.A.; Duhrsen, U.; Frenette, P.S. Acute myelogenous leukaemia-induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell niche. Cell Stem Cell 2014, 15, 365–375. [Google Scholar] [CrossRef] [Green Version]
- Colmore, A.; Amorim, M.; Pontier, A.L.; Wang, S.; Jablonski, E.; Sipkins, D.A. Leukemic cells create bone marrow niches that disrupt the behaviour of normal hematopoietic progenitor cells. Science 2008, 322, 1861. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Gurudutta, G.U.; Satija, N.K.; Pati, S.; Afrin, F.; Gupta, P.; Verma, Y.K.; Singh, V.K.; Tripathi, R.P. Stem cell c-KIT and HOXB4 genes: Critical roles and mechanisms in self-renewal, proliferation, and differentiation. Stem Cells Dev. 2006, 15, 755–778. [Google Scholar] [CrossRef]
- Whiteley, A.E.; Price, T.T.; Cantelli, G.; Sipkins, D.A. Leukaemia: A model metastatic disease. Nat. Rev. Cancer 2021, 21, 461–475. [Google Scholar] [CrossRef]
- Novak, J.P.; Stewart, C.C. Stochastic versus deterministic haematopoiesis. Br. J. Haematol. 1991, 78, 149–154. [Google Scholar] [CrossRef]
- Wu, S.; Zhou, T.; Tian, T. A robust method for designing multistable systems. Syst. Biol. Appl. 2022, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Hamey, F.K.; Gottgens, B. Demystifying blood stem cells fates. Nat. Cell Biol. 2017, 19, 261–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurenti, L.; Gottgens, B. From haematopoietic stem cells to complex differentiation landscapes. Nature 2018, 553, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Vincente-Duenas, C.; Perez-Caro, M.; Abollo-Jimenez, F.; Cobaleda, C.; Sanchez-Garcia, I. Stem cell driven cancer: “hands-off” regulation of cancer development. Cell Cycle 2009, 8, 1314–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brown, G. The Social Norm of Hematopoietic Stem Cells and Dysregulation in Leukemia. Int. J. Mol. Sci. 2022, 23, 5063. https://doi.org/10.3390/ijms23095063
Brown G. The Social Norm of Hematopoietic Stem Cells and Dysregulation in Leukemia. International Journal of Molecular Sciences. 2022; 23(9):5063. https://doi.org/10.3390/ijms23095063
Chicago/Turabian StyleBrown, Geoffrey. 2022. "The Social Norm of Hematopoietic Stem Cells and Dysregulation in Leukemia" International Journal of Molecular Sciences 23, no. 9: 5063. https://doi.org/10.3390/ijms23095063
APA StyleBrown, G. (2022). The Social Norm of Hematopoietic Stem Cells and Dysregulation in Leukemia. International Journal of Molecular Sciences, 23(9), 5063. https://doi.org/10.3390/ijms23095063