
A Review of Toxicity Mechanism Studies of Electronic Cigarettes on Respiratory System



Abstract
1. Introduction
1.1. Brief Introduction to E-Cigarettes
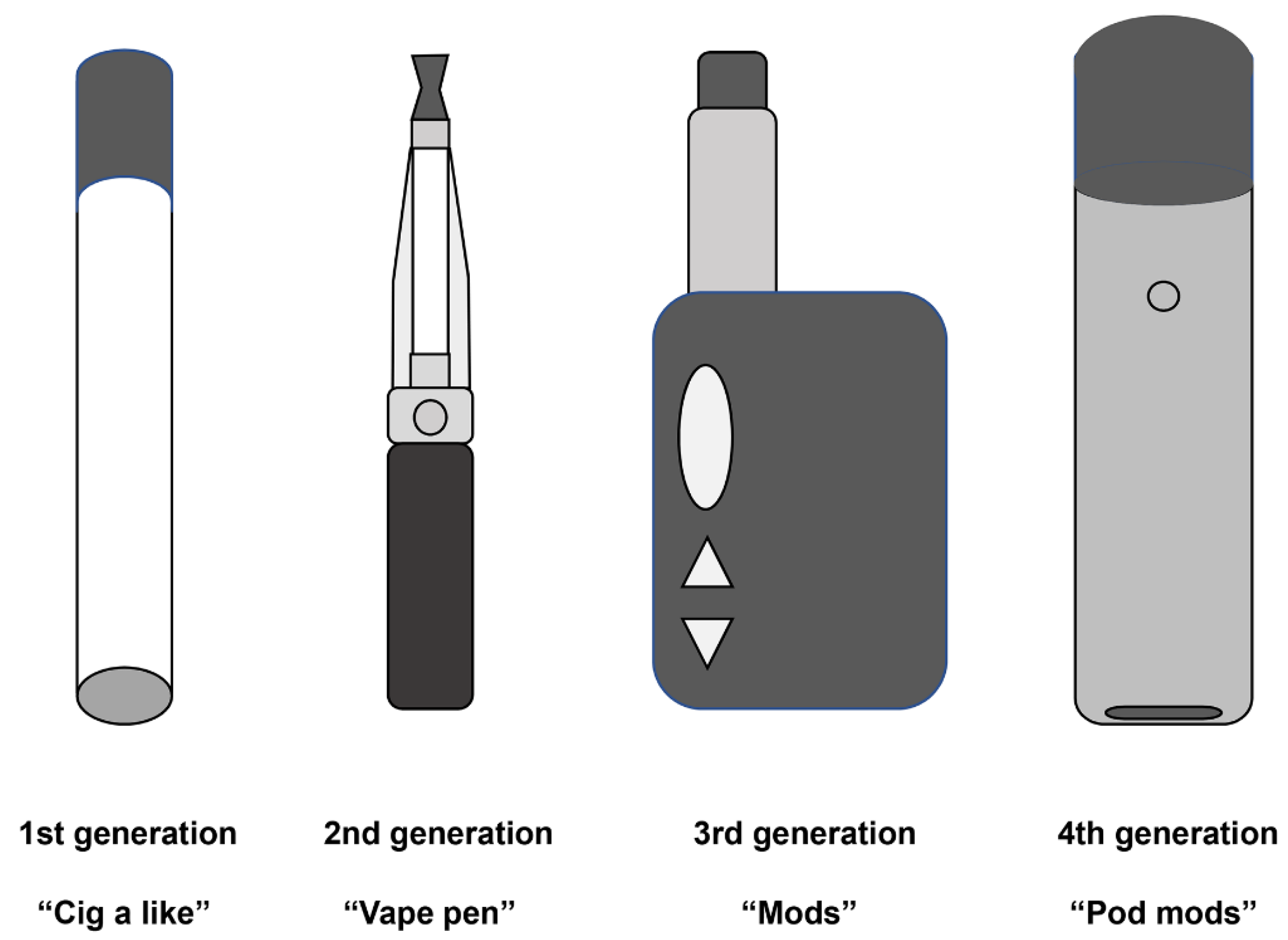
1.2. Evolution of E-Cigarettes
1.3. Population Analysis of E-Cigarettes
1.4. Composition Comparison of E-Cigarette and Conventional Cigarette Smoke
1.5. Toxicity Comparison of E-Cigarettes and Conventional Cigarettes
2. Effects of Conventional Cigarettes on the Respiratory System
2.1. Conventional Cigarettes and Respiratory Diseases
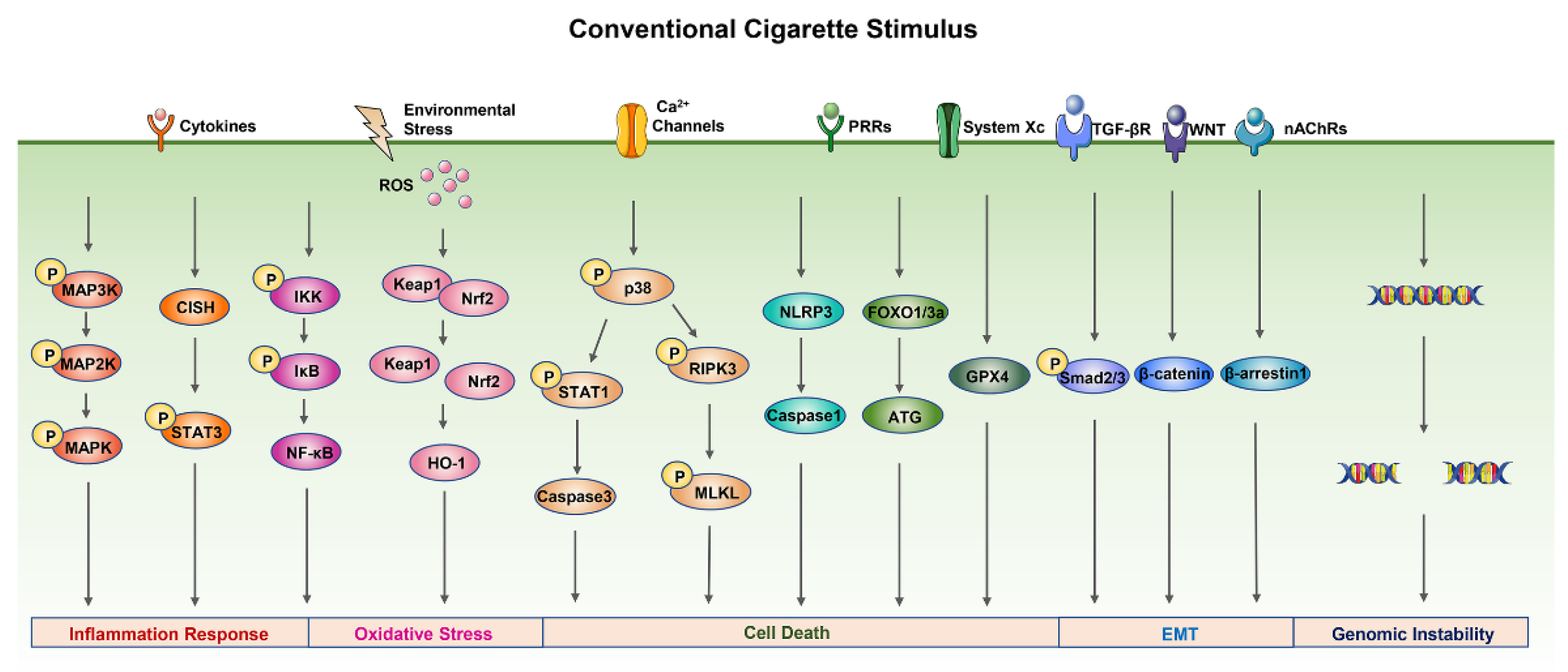
2.2. Conventional Cigarettes Related Toxicity Mechanisms and Signal Pathways
2.2.1. Inflammation Response
2.2.2. Oxidative Stress
2.2.3. Cell Death
2.2.4. Epithelial–Mesenchymal Transition
2.2.5. Genomic Instability
3. Effects of E-Cigarettes on the Respiratory System
3.1. E-Cigarettes and Respiratory Diseases
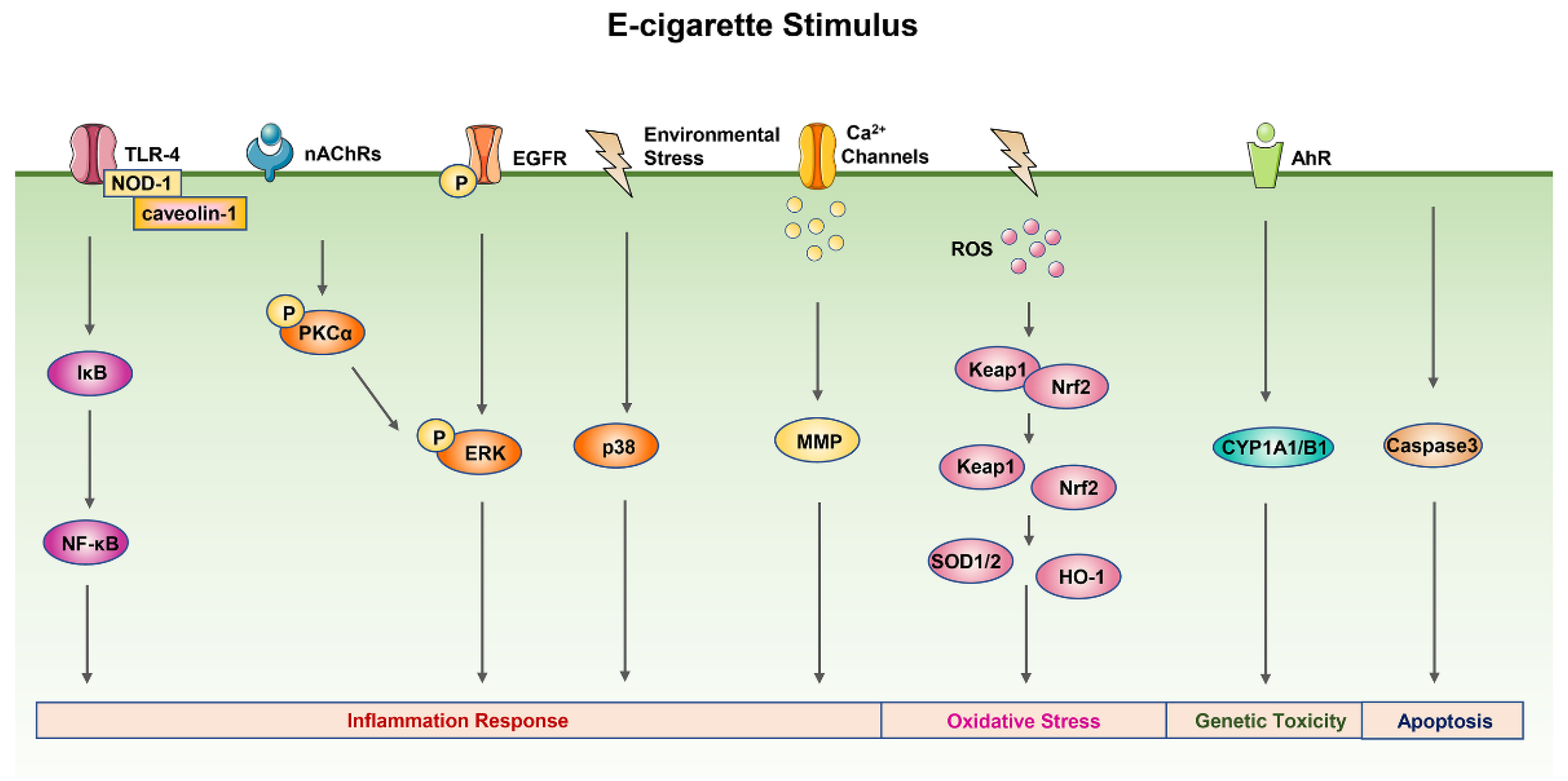
3.2. E-Cigarette-Related Toxicity Mechanisms
3.2.1. Inflammation Response
3.2.2. Oxidative Stress
3.2.3. DNA Damage
3.2.4. Other Mechanisms
3.3. E-Cigarette-Related Signal Pathways
3.3.1. MAPK Signal Pathway
3.3.2. NK-κB Signal Pathway
3.3.3. Nrf2 Signal Pathway
3.3.4. PKCα/ERK Signal Pathway
3.3.5. Ca2+ Signal Pathway
3.3.6. AhR Signal Pathway
3.3.7. EGFR Signal Pathway
4. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yamin, C.K.; Bitton, A.; Bates, D.W. E-Cigarettes: A Rapidly Growing Internet Phenomenon. Ann. Intern. Med. 2010, 153, 607–609. [Google Scholar] [CrossRef] [PubMed]
- Sapru, S.; Vardhan, M.; Li, Q.; Guo, Y.; Li, X.; Saxena, D. E-cigarettes use in the United States: Reasons for use, perceptions, and effects on health. BMC Public Health 2020, 20, 1518. [Google Scholar] [CrossRef] [PubMed]
- Protano, C.; Avino, P.; Manigrasso, M.; Vivaldi, V.; Perna, F.; Valeriani, F.; Vitali, M. Environmental Electronic Vape Exposure from Four Different Generations of Electronic Cigarettes: Airborne Particulate Matter Levels. Int. J. Environ. Res. Public Health 2018, 15, 2172. [Google Scholar] [CrossRef] [PubMed]
- Barrington-Trimis, J.L.; Leventhal, A.M. Adolescents’ Use of “Pod Mod” E-Cigarettes—Urgent Concerns. N. Engl. J. Med. 2018, 379, 1099–1102. [Google Scholar] [CrossRef] [PubMed]
- Gentzke, A.S.; Wang, T.W.; Jamal, A.; Park-Lee, E.; Ren, C.; Cullen, K.A.; Neff, L. Tobacco Product Use among Middle and High School Students—United States, 2020. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 1881–1888. [Google Scholar] [CrossRef] [PubMed]
- Bauld, L.; MacKintosh, A.M.; Eastwood, B.; Ford, A.; Moore, G.; Dockrell, M.; Arnott, D.; Cheeseman, H.; McNeill, A. Young People’s Use of E-Cigarettes across the United Kingdom: Findings from Five Surveys 2015–2017. Int. J. Environ. Res. Public Health 2017, 14, 973. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, I.B.; Smith, T.; Arrazola, R.A.; Palipudi, K.M.; Garcia, D.Q.I.; Prasad, V.M.; Commar, A.; Schotte, K.; Garwood, P.D.; Armour, B.S. Current Tobacco Smoking, Quit Attempts, and Knowledge About Smoking Risks Among Persons Aged ≥15 Years—Global Adult Tobacco Survey, 28 Countries, 2008–2016. MMWR Morb. Mortal. Wkly. Rep. 2018, 67, 1072–1076. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, M.; Wu, J.; Xu, X.; Yin, P.; Huang, Z.; Zhang, X.; Zhou, Y.; Zhang, X.; Li, C.; et al. E-cigarette use among adults in China: Findings from repeated cross-sectional surveys in 2015–2016 and 2018–2019. Lancet Public Health 2020, 5, e639–e649. [Google Scholar] [CrossRef]
- Romijnders, K.; van Osch, L.; de Vries, H.; Talhout, R. Perceptions and Reasons Regarding E-Cigarette Use among Users and Non-Users: A Narrative Literature Review. Int. J. Environ. Res. Public Health 2018, 15, 1190. [Google Scholar] [CrossRef]
- Dusautoir, R.; Zarcone, G.; Verriele, M.; Garçon, G.; Fronval, I.; Beauval, N.; Allorge, D.; Riffault, V.; Locoge, N.; Lo-Guidice, J.; et al. Comparison of the chemical composition of aerosols from heated tobacco products, electronic cigarettes and tobacco cigarettes and their toxic impacts on the human bronchial epithelial BEAS-2B cells. J. Hazard. Mater. 2021, 401, 123417. [Google Scholar] [CrossRef]
- Konstantinou, E.; Fotopoulou, F.; Drosos, A.; Dimakopoulou, N.; Zagoriti, Z.; Niarchos, A.; Makrynioti, D.; Kouretas, D.; Farsalinos, K.; Lagoumintzis, G.; et al. Tobacco-specific nitrosamines: A literature review. Food Chem. Toxicol. 2018, 118, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Kosmider, L.; Kimber, C.F.; Kurek, J.; Corcoran, O.; Dawkins, L.E. Compensatory Puffing with Lower Nicotine Concentration E-liquids Increases Carbonyl Exposure in E-cigarette Aerosols. Nicotine Tob. Res. 2018, 20, 998–1003. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Aravindakshan, A.; Hilpert, M.; Olmedo, P.; Rule, A.M.; Navas-Acien, A.; Aherrera, A. Metal/Metalloid Levels in Electronic Cigarette Liquids, Aerosols, and Human Biosamples: A Systematic Review. Environ. Health Perspect. 2020, 128, 36001. [Google Scholar] [CrossRef] [PubMed]
- Helen, G.S.; Liakoni, E.; Nardone, N.; Addo, N.; Jacob, P.R.; Benowitz, N.L. Comparison of Systemic Exposure to Toxic and/or Carcinogenic Volatile Organic Compounds (VOC) during Vaping, Smoking, and Abstention. Cancer Prev. Res. 2020, 13, 153–162. [Google Scholar] [CrossRef]
- Flora, J.W.; Meruva, N.; Huang, C.B.; Wilkinson, C.T.; Ballentine, R.; Smith, D.C.; Werley, M.S.; McKinney, W.J. Characterization of potential impurities and degradation products in electronic cigarette formulations and aerosols. Regul. Toxicol. Pharmacol. 2016, 74, 1–11. [Google Scholar] [CrossRef]
- Olmedo, P.; Goessler, W.; Tanda, S.; Grau-Perez, M.; Jarmul, S.; Aherrera, A.; Chen, R.; Hilpert, M.; Cohen, J.E.; Navas-Acien, A.; et al. Metal Concentrations in e-Cigarette Liquid and Aerosol Samples: The Contribution of Metallic Coils. Environ. Health Persp. 2018, 126, 27010. [Google Scholar] [CrossRef]
- Münzel, T.; Hahad, O.; Kuntic, M.; Keaney, J.F.; Deanfield, J.E.; Daiber, A. Effects of tobacco cigarettes, e-cigarettes, and waterpipe smoking on endothelial function and clinical outcomes. Eur. Heart J. 2020, 41, 4057–4070. [Google Scholar] [CrossRef]
- Gray, N.; Halstead, M.; Valentin-Blasini, L.; Watson, C.; Pappas, R.S. Toxic Metals in Liquid and Aerosol from Pod-Type Electronic Cigarettes. J. Anal. Toxicol. 2022, 46, 69–75. [Google Scholar] [CrossRef]
- Soleimani, F.; Dobaradaran, S.; De-la-Torre, G.E.; Schmidt, T.C.; Saeedi, R. Content of toxic components of cigarette, cigarette smoke vs. cigarette butts: A comprehensive systematic review. Sci. Total Environ. 2022, 813, 152667. [Google Scholar] [CrossRef]
- Wang, G.; Liu, W.; Song, W. Toxicity assessment of electronic cigarettes. Inhal. Toxicol. 2019, 31, 259–273. [Google Scholar] [CrossRef]
- Merecz-Sadowska, A.; Sitarek, P.; Zielinska-Blizniewska, H.; Malinowska, K.; Zajdel, K.; Zakonnik, L.; Zajdel, R. A Summary of In Vitro and In Vivo Studies Evaluating the Impact of E-Cigarette Exposure on Living Organisms and the Environment. Int. J. Mol. Sci. 2020, 21, 652. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, Y.; Chen, J.; Yang, X.; Jiang, X.; Liu, P.; Li, M. Comparison of biological and transcriptomic effects of conventional cigarette and electronic cigarette smoke exposure at toxicological dose in BEAS-2B cells. Ecotoxicol. Environ. Safe. 2021, 222, 112472. [Google Scholar] [CrossRef] [PubMed]
- US Centers for Disease and Control Prevention; US National Center for Chronic Disease Prevention and Health Promotion; US Office on Smoking and Health. How Tobacco Smoke Causes Disease: The Biology and Behavioral Basis for Smoking-Attributable Disease: A Report of the Surgeon General; US Centers for Disease Control and Prevention: Atlanta, GA, USA, 2010.
- Tsai, M.; Byun, M.K.; Shin, J.; Crotty, A.L. Effects of e-cigarettes and vaping devices on cardiac and pulmonary physiology. J. Physiol. 2020, 598, 5039–5062. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Hu, S.; Li, C.; Ma, H.; Wang, Q.; Meng, G.; Guo, T.; Zhang, J. Cigarette Smoke Induced Lung Barrier Dysfunction, EMT, and Tissue Remodeling: A Possible Link between COPD and Lung Cancer. Biomed Res. Int. 2019, 2019, 2025636. [Google Scholar] [CrossRef] [PubMed]
- GBD 2015 Mortality and Causes of Death Collaborators. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980-2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1459–1544. [Google Scholar] [CrossRef]
- Rabe, K.F.; Watz, H. Chronic obstructive pulmonary disease. Lancet 2017, 389, 1931–1940. [Google Scholar] [CrossRef]
- Dransfield, M.T.; Kunisaki, K.M.; Strand, M.J.; Anzueto, A.; Bhatt, S.P.; Bowler, R.P.; Criner, G.J.; Curtis, J.L.; Hanania, N.A.; Nath, H.; et al. Acute Exacerbations and Lung Function Loss in Smokers with and without Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2017, 195, 324–330. [Google Scholar] [CrossRef]
- Gibbs, K.; Collaco, J.M.; McGrath-Morrow, S.A. Impact of Tobacco Smoke and Nicotine Exposure on Lung Development. Chest 2016, 149, 552–561. [Google Scholar] [CrossRef]
- Alavinezhad, A.; Boskabady, M.H. The prevalence of asthma and related symptoms in Middle East countries. Clin. Respir. J. 2018, 12, 865–877. [Google Scholar] [CrossRef]
- Santos, V.; Moreira, M.; Rosa, A.; Sobragi, S.M.; Silva, C.; Dalcin, P. Association of quality of life and disease control with cigarette smoking in patients with severe asthma. Braz. J. Med. Biol. Res. 2022, 55, e11149. [Google Scholar] [CrossRef]
- Ross, K.R.; Gupta, R.; DeBoer, M.D.; Zein, J.; Phillips, B.R.; Mauger, D.T.; Li, C.; Myers, R.E.; Phipatanakul, W.; Fitzpatrick, A.M.; et al. Severe asthma during childhood and adolescence: A longitudinal study. J. Allergy Clin. Immunol. 2020, 145, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Grabenhenrich, L.B.; Gough, H.; Reich, A.; Eckers, N.; Zepp, F.; Nitsche, O.; Forster, J.; Schuster, A.; Schramm, D.; Bauer, C.P.; et al. Early-life determinants of asthma from birth to age 20 years: A German birth cohort study. J. Allergy Clin. Immunol. 2014, 133, 979–988. [Google Scholar] [CrossRef]
- Wang, B.; Chen, H.; Chan, Y.L.; Wang, G.; Oliver, B.G. Why Do Intrauterine Exposure to Air Pollution and Cigarette Smoke Increase the Risk of Asthma? Front. Cell Dev. Biol. 2020, 8, 38. [Google Scholar] [CrossRef] [PubMed]
- Mortaz, E.; Adcock, I.M.; Ito, K.; Kraneveld, A.D.; Nijkamp, F.P.; Folkerts, G. Cigarette smoke induces CXCL8 production by human neutrophils via activation of TLR9 receptor. Eur. Respir. J. 2010, 36, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Sun, Y.; Wu, B.; Xu, D.; Yang, J.; Gu, L.; Du, C. Overexpression of FOXA2 attenuates cigarette smoke-induced cellular senescence and lung inflammation through inhibition of the p38 and Erk1/2 MAPK pathways. Int. Immunopharmacol. 2021, 94, 107427. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.F.; Ma, N.; He, Z.Y.; Zhong, X.N.; Zhang, J.Q.; Bai, J.; Deng, J.M.; Tang, X.J.; Luo, Z.L.; Huang, M.; et al. Erythromycin inhibits cigarette smoke-induced inflammation through regulating the PPARγ/NF-κB signaling pathway in macrophages. Int. Immunopharmacol. 2021, 96, 107775. [Google Scholar] [CrossRef]
- Peng, H.Y.; Hsiao, J.R.; Chou, S.T.; Hsu, Y.M.; Wu, G.H.; Shieh, Y.S.; Shiah, S.G. MiR-944/CISH mediated inflammation via STAT3 is involved in oral cancer malignance by cigarette smoking. Neoplasia 2020, 22, 554–565. [Google Scholar] [CrossRef]
- Xu, L.; Li, X.; Wang, H.; Xie, F.; Liu, H.; Xie, J. Cigarette smoke triggers inflammation mediated by autophagy in BEAS-2B cells. Ecotoxicol. Environ. Saf. 2019, 184, 109617. [Google Scholar] [CrossRef]
- Schuliga, M. NF-kappaB Signaling in Chronic Inflammatory Airway Disease. Biomolecules 2015, 5, 1266–1283. [Google Scholar] [CrossRef]
- Jarnicki, A.; Putoczki, T.; Ernst, M. Stat3: Linking inflammation to epithelial cancer—More than a “gut” feeling? Cell Div. 2010, 5, 14. [Google Scholar] [CrossRef]
- Barnes, P.J. Oxidative stress-based therapeutics in COPD. Redox Biol. 2020, 33, 101544. [Google Scholar] [CrossRef] [PubMed]
- Dang, X.; He, B.; Ning, Q.; Liu, Y.; Guo, J.; Niu, G.; Chen, M. Alantolactone suppresses inflammation, apoptosis and oxidative stress in cigarette smoke-induced human bronchial epithelial cells through activation of Nrf2/HO-1 and inhibition of the NF-κB pathways. Respir. Res. 2020, 21, 95. [Google Scholar] [CrossRef] [PubMed]
- Sauler, M.; Bazan, I.S.; Lee, P.J. Cell Death in the Lung: The Apoptosis-Necroptosis Axis. Annu. Rev. Physiol. 2019, 81, 375–402. [Google Scholar] [CrossRef]
- Feng, H.; Li, M.; Altawil, A.; Yin, Y.; Zheng, R.; Kang, J. Cigarette smoke extracts induce apoptosis in Raw264.7 cells via endoplasmic reticulum stress and the intracellular Ca(2+)/P38/STAT1 pathway. Toxicol. Vitr. 2021, 77, 105249. [Google Scholar] [CrossRef] [PubMed]
- Luan, G.; Zhu, Z.; Wu, K.; Yin, S. Theaflavin-3,3′-digallate attenuates cigarette smoke extract-induced pulmonary emphysema in mice by suppressing necroptosis. Exp. Ther. Med. 2022, 23, 11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.Y.; Jiang, Y.X.; Yang, Y.C.; Liu, J.Y.; Huo, C.; Ji, X.L.; Qu, Y.Q. Cigarette smoke extract induces pyroptosis in human bronchial epithelial cells through the ROS/NLRP3/caspase-1 pathway. Life Sci. 2021, 269, 119090. [Google Scholar] [CrossRef]
- Yoshida, M.; Minagawa, S.; Araya, J.; Sakamoto, T.; Hara, H.; Tsubouchi, K.; Hosaka, Y.; Ichikawa, A.; Saito, N.; Kadota, T.; et al. Involvement of cigarette smoke-induced epithelial cell ferroptosis in COPD pathogenesis. Nat. Commun. 2019, 10, 3145. [Google Scholar] [CrossRef]
- Bagam, P.; Kaur, G.; Singh, D.P.; Batra, S. In vitro study of the role of FOXO transcription factors in regulating cigarette smoke extract-induced autophagy. Cell Biol. Toxicol. 2020, 37, 531–553. [Google Scholar] [CrossRef]
- Vij, N.; Chandramani-Shivalingappa, P.; Van Westphal, C.; Hole, R.; Bodas, M. Cigarette smoke-induced autophagy impairment accelerates lung aging, COPD-emphysema exacerbations and pathogenesis. Am. J. Physiol. Cell Physiol. 2018, 314, C73–C87. [Google Scholar] [CrossRef]
- Vu, T.; Jin, L.; Datta, P.K. Effect of Cigarette Smoking on Epithelial to Mesenchymal Transition (EMT) in Lung Cancer. J. Clin. Med. 2016, 5, 44. [Google Scholar] [CrossRef]
- Zuo, H.; Trombetta-Lima, M.; Heijink, I.H.; van der Veen, C.; Hesse, L.; Faber, K.N.; Poppinga, W.J.; Maarsingh, H.; Nikolaev, V.O.; Schmidt, A.M. A-Kinase Anchoring Proteins Diminish TGF-β(1)/Cigarette Smoke-Induced Epithelial-To-Mesenchymal Transition. Cells 2020, 9, 356. [Google Scholar] [CrossRef] [PubMed]
- Carlier, F.M.; Dupasquier, S.; Ambroise, J.; Detry, B.; Lecocq, M.; Biétry-Claudet, C.; Boukala, Y.; Gala, J.L.; Bouzin, C.; Verleden, S.E.; et al. Canonical WNT pathway is activated in the airway epithelium in chronic obstructive pulmonary disease. EBioMedicine 2020, 61, 103034. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Ju, Y.S.; Haase, K.; Van Loo, P.; Martincorena, I.; Nik-Zainal, S.; Totoki, Y.; Fujimoto, A.; Nakagawa, H.; Shibata, T.; et al. Mutational signatures associated with tobacco smoking in human cancer. Science 2016, 354, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Tian, B.; Geng, X.; Zhou, H.; Xu, Z.; Lai, T.; Wu, Y.; Bao, Z.; Chen, Z.; Li, W.; et al. IL-17-Mediated Inflammation Promotes Cigarette Smoke-Induced Genomic Instability. Cells 2021, 10, 1173. [Google Scholar] [CrossRef]
- Jiao, Y.; Liu, C.; Cui, F.M.; Xu, J.Y.; Tong, J.; Qi, X.F.; Wang, L.L.; Zhu, W. Long intergenic non-coding RNA induced by X-ray irradiation regulates DNA damage response signaling in the human bronchial epithelial BEAS-2B cell line. Oncol. Lett. 2015, 9, 169–176. [Google Scholar] [CrossRef][Green Version]
- Gao, S.; Lin, H.; Yu, W.; Zhang, F.; Wang, R.; Yu, H.; Qian, B. LncRNA LCPAT1 is involved in DNA damage induced by CSE. Biochem. Biophys. Res. Commun. 2019, 508, 512–515. [Google Scholar] [CrossRef]
- Palamidas, A.; Tsikrika, S.; Katsaounou, P.A.; Vakali, S.; Gennimata, S.A.; Kaltsakas, G.; Gratziou, C.; Koulouris, N. Acute effects of short term use of ecigarettes on Airways Physiology and Respiratory Symptoms in Smokers with and without Airways Obstructive Diseases and in Healthy non smokers. Tob. Prev. Cessat 2017, 3, 5. [Google Scholar] [CrossRef]
- McConnell, R.; Barrington-Trimis, J.L.; Wang, K.; Urman, R.; Hong, H.; Unger, J.; Samet, J.; Leventhal, A.; Berhane, K. Electronic Cigarette Use and Respiratory Symptoms in Adolescents. Am. J. Respir. Crit. Care Med. 2017, 195, 1043–1049. [Google Scholar] [CrossRef]
- Bowler, R.P.; Hansel, N.N.; Jacobson, S.; Graham, B.R.; Make, B.J.; Han, M.K.; O’Neal, W.K.; Oelsner, E.C.; Casaburi, R.; Barjaktarevic, I.; et al. Electronic Cigarette Use in US Adults at Risk for or with COPD: Analysis from Two Observational Cohorts. J. Gen. Intern. Med. 2017, 32, 1315–1322. [Google Scholar] [CrossRef]
- Garcia-Arcos, I.; Geraghty, P.; Baumlin, N.; Campos, M.; Dabo, A.J.; Jundi, B.; Cummins, N.; Eden, E.; Grosche, A.; Salathe, M.; et al. Chronic electronic cigarette exposure in mice induces features of COPD in a nicotine-dependent manner. Thorax 2016, 71, 1119–1129. [Google Scholar] [CrossRef]
- Reinikovaite, V.; Rodriguez, I.E.; Karoor, V.; Rau, A.; Trinh, B.B.; Deleyiannis, F.W.; Taraseviciene-Stewart, L. The effects of electronic cigarette vapour on the lung: Direct comparison to tobacco smoke. Eur. Respir. J. 2018, 51, 1701661. [Google Scholar] [CrossRef] [PubMed]
- Olfert, I.M.; DeVallance, E.; Hoskinson, H.; Branyan, K.W.; Clayton, S.; Pitzer, C.R.; Sullivan, D.P.; Breit, M.J.; Wu, Z.; Klinkhachorn, P.; et al. Chronic exposure to electronic cigarettes results in impaired cardiovascular function in mice. J. Appl. Physiol. 2018, 124, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Reid, K.M.; Forrest, J.R.; Porter, L. Tobacco Product Use among Youths with and Without Lifetime Asthma—Florida, 2016. MMWR Morb. Mortal. Wkly. Rep. 2018, 67, 599–601. [Google Scholar] [CrossRef] [PubMed]
- Bircan, E.; Bezirhan, U.; Porter, A.; Fagan, P.; Orloff, M.S. Electronic cigarette use and its association with asthma, chronic obstructive pulmonary disease (COPD) and asthma-COPD overlap syndrome among never cigarette smokers. Tob. Induc. Dis. 2021, 19, 75. [Google Scholar] [CrossRef]
- Lappas, A.S.; Tzortzi, A.S.; Konstantinidi, E.M.; Teloniatis, S.I.; Tzavara, C.K.; Gennimata, S.A.; Koulouris, N.G.; Behrakis, P.K. Short-term respiratory effects of e-cigarettes in healthy individuals and smokers with asthma. Respirology 2018, 23, 291–297. [Google Scholar] [CrossRef]
- Polosa, R.; Morjaria, J.B.; Caponnetto, P.; Caruso, M.; Campagna, D.; Amaradio, M.D.; Ciampi, G.; Russo, C.; Fisichella, A. Persisting long term benefits of smoking abstinence and reduction in asthmatic smokers who have switched to electronic cigarettes. Discov. Med. 2016, 21, 99–108. [Google Scholar]
- Iskandar, A.R.; Gonzalez-Suarez, I.; Majeed, S.; Marescotti, D.; Sewer, A.; Xiang, Y.; Leroy, P.; Guedj, E.; Mathis, C.; Schaller, J.P.; et al. A framework for in vitro systems toxicology assessment of e-liquids. Toxicol. Mech. Methods 2016, 26, 389–413. [Google Scholar] [CrossRef]
- Anthérieu, S.; Garat, A.; Beauval, N.; Soyez, M.; Allorge, D.; Garçon, G.; Lo-Guidice, J. Comparison of cellular and transcriptomic effects between electronic cigarette vapor and cigarette smoke in human bronchial epithelial cells. Toxicol. Vitr. 2017, 45, 417–425. [Google Scholar] [CrossRef]
- Lerner, C.A.; Sundar, I.K.; Yao, H.; Gerloff, J.; Ossip, D.J.; McIntosh, S.; Robinson, R.; Rahman, I. Vapors Produced by Electronic Cigarettes and E-Juices with Flavorings Induce Toxicity, Oxidative Stress, and Inflammatory Response in Lung Epithelial Cells and in Mouse Lung. PLoS ONE 2015, 10, e116732. [Google Scholar] [CrossRef]
- Scott, A.; Lugg, S.T.; Aldridge, K.; Lewis, K.E.; Bowden, A.; Mahida, R.Y.; Grudzinska, F.S.; Dosanjh, D.; Parekh, D.; Foronjy, R.; et al. Pro-inflammatory effects of e-cigarette vapour condensate on human alveolar macrophages. Thorax 2018, 73, 1161–1169. [Google Scholar] [CrossRef]
- O’Farrell, H.E.; Brown, R.; Brown, Z.; Milijevic, B.; Ristovski, Z.D.; Bowman, R.V.; Fong, K.M.; Vaughan, A.; Yang, I.A. E-cigarettes induce toxicity comparable to tobacco cigarettes in airway epithelium from patients with COPD. Toxicol. Vitr. 2021, 75, 105204. [Google Scholar] [CrossRef] [PubMed]
- Larcombe, A.N.; Janka, M.A.; Mullins, B.J.; Berry, L.J.; Bredin, A.; Franklin, P.J. The effects of electronic cigarette aerosol exposure on inflammation and lung function in mice. Am. J. Physiol.-Lung C 2017, 313, L67–L79. [Google Scholar] [CrossRef] [PubMed]
- Glynos, C.; Bibli, S.; Katsaounou, P.; Pavlidou, A.; Magkou, C.; Karavana, V.; Topouzis, S.; Kalomenidis, I.; Zakynthinos, S.; Papapetropoulos, A. Comparison of the effects of e-cigarette vapor with cigarette smoke on lung function and inflammation in mice. Am. J. Physiol.-Lung C 2018, 315, L662–L672. [Google Scholar] [CrossRef] [PubMed]
- Crotty Alexander, L.E.; Drummond, C.A.; Hepokoski, M.; Mathew, D.; Moshensky, A.; Willeford, A.; Das, S.; Singh, P.; Yong, Z.; Lee, J.H.; et al. Chronic inhalation of e-cigarette vapor containing nicotine disrupts airway barrier function and induces systemic inflammation and multiorgan fibrosis in mice. Am. J. Physiol. -Regul. Integr. Comp. Physiol. 2018, 314, R834–R847. [Google Scholar] [CrossRef]
- Lerner, C.A.; Rutagarama, P.; Ahmad, T.; Sundar, I.K.; Elder, A.; Rahman, I. Electronic cigarette aerosols and copper nanoparticles induce mitochondrial stress and promote DNA fragmentation in lung fibroblasts. Biochem. Biophys. Res. Commun. 2016, 477, 620–625. [Google Scholar] [CrossRef]
- Sun, Y.W.; Chen, K.M.; Atkins, H.; Aliaga, C.; Gordon, T.; Guttenplan, J.B.; El-Bayoumy, K. Effects of E-Cigarette Aerosols with Varying Levels of Nicotine on Biomarkers of Oxidative Stress and Inflammation in Mice. Chem. Res. Toxicol. 2021, 34, 1161–1168. [Google Scholar] [CrossRef]
- Yogeswaran, S.; Muthumalage, T.; Rahman, I. Comparative Reactive Oxygen Species (ROS) Content among Various Flavored Disposable Vape Bars, including Cool (Iced) Flavored Bars. Toxics 2021, 9, 235. [Google Scholar] [CrossRef]
- Lamb, T.; Muthumalage, T.; Rahman, I. Pod-based menthol and tobacco flavored e-cigarettes cause mitochondrial dysfunction in lung epithelial cells. Toxicol. Lett. 2020, 333, 303–311. [Google Scholar] [CrossRef]
- Wavreil, F.D.M.; Heggland, S.J. Cinnamon-flavored electronic cigarette liquids and aerosols induce oxidative stress in human osteoblast-like MG-63 cells. Toxicol. Rep. 2020, 7, 23–29. [Google Scholar] [CrossRef]
- Muthumalage, T.; Prinz, M.; Ansah, K.O.; Gerloff, J.; Sundar, I.K.; Rahman, I. Inflammatory and Oxidative Responses Induced by Exposure to Commonly Used e-Cigarette Flavoring Chemicals and Flavored e-Liquids without Nicotine. Front. Physiol. 2017, 8, 1130. [Google Scholar] [CrossRef]
- Ganapathy, V.; Manyanga, J.; Brame, L.; McGuire, D.; Sadhasivam, B.; Floyd, E.; Rubenstein, D.A.; Ramachandran, I.; Wagener, T.; Queimado, L. Electronic cigarette aerosols suppress cellular antioxidant defenses and induce significant oxidative DNA damage. PLoS ONE 2017, 12, e177780. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.; Majeste, A.; Hanus, J.; Wang, S. E-Cigarette Aerosol Exposure Induces Reactive Oxygen Species, DNA Damage, and Cell Death in Vascular Endothelial Cells. Toxicol. Sci. 2016, 154, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Park, S.H.; Weng, M.W.; Wang, H.T.; Huang, W.C.; Lepor, H.; Wu, X.R.; Chen, L.C.; Tang, M.S. E-cigarette smoke damages DNA and reduces repair activity in mouse lung, heart, and bladder as well as in human lung and bladder cells. Proc. Natl. Acad. Sci. USA 2018, 115, E1560–E1569. [Google Scholar] [CrossRef] [PubMed]
- Canistro, D.; Vivarelli, F.; Cirillo, S.; Babot Marquillas, C.; Buschini, A.; Lazzaretti, M.; Marchi, L.; Cardenia, V.; Rodriguez-Estrada, M.T.; Lodovici, M.; et al. E-cigarettes induce toxicological effects that can raise the cancer risk. Sci. Rep. 2017, 7, 2028. [Google Scholar] [CrossRef] [PubMed]
- Alanazi, H.; Park, H.J.; Chakir, J.; Semlali, A.; Rouabhia, M. Comparative study of the effects of cigarette smoke and electronic cigarettes on human gingival fibroblast proliferation, migration and apoptosis. Food Chem. Toxicol. 2018, 118, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Rouabhia, M.; Park, H.J.; Semlali, A.; Zakrzewski, A.; Chmielewski, W.; Chakir, J. E-Cigarette Vapor Induces an Apoptotic Response in Human Gingival Epithelial Cells through the Caspase-3 Pathway. J. Cell. Physiol. 2017, 232, 1539–1547. [Google Scholar] [CrossRef]
- Zahedi, A.; Phandthong, R.; Chaili, A.; Remark, G.; Talbot, P. Epithelial-to-mesenchymal transition of A549 lung cancer cells exposed to electronic cigarettes. Lung Cancer 2018, 122, 224–233. [Google Scholar] [CrossRef]
- Park, H.R.; Vallarino, J.; O’Sullivan, M.; Wirth, C.; Panganiban, R.A.; Webb, G.; Shumyatcher, M.; Himes, B.E.; Park, J.A.; Christiani, D.C.; et al. Electronic cigarette smoke reduces ribosomal protein gene expression to impair protein synthesis in primary human airway epithelial cells. Sci. Rep. 2021, 11, 17517. [Google Scholar] [CrossRef]
- Solleti, S.K.; Bhattacharya, S.; Ahmad, A.; Wang, Q.; Mereness, J.; Rangasamy, T.; Mariani, T.J. MicroRNA expression profiling defines the impact of electronic cigarettes on human airway epithelial cells. Sci. Rep. 2017, 7, 1081. [Google Scholar] [CrossRef]
- Le, H.H.T.; Liu, C.W.; Denaro, P.R.; Jousma, J.; Shao, N.Y.; Rahman, I.; Lee, W.H. Genome-wide differential expression profiling of lncRNAs and mRNAs in human induced pluripotent stem cell-derived endothelial cells exposed to e-cigarette extract. Stem Cell Res. Ther. 2021, 12, 593. [Google Scholar] [CrossRef]
- Ferger, B.; Spratt, C.; Earl, C.D.; Teismann, P.; Oertel, W.H.; Kuschinsky, K. Effects of nicotine on hydroxyl free radical formation in vitro and on MPTP-induced neurotoxicity in vivo. Naunyn Schmiedebergs Arch. Pharm. 1998, 358, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Vijg, J. From DNA damage to mutations: All roads lead to aging. Ageing Res. Rev. 2021, 68, 101316. [Google Scholar] [CrossRef] [PubMed]
- Higham, A.; Rattray, N.J.W.; Dewhurst, J.A.; Trivedi, D.K.; Fowler, S.J.; Goodacre, R.; Singh, D. Electronic cigarette exposure triggers neutrophil inflammatory responses. Respir. Res. 2016, 17, 56. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.; Todd, I.; Tighe, P.J.; Fairclough, L.C. Electronic cigarette vapour moderately stimulates pro-inflammatory signalling pathways and interleukin-6 production by human monocyte-derived dendritic cells. Arch. Toxicol. 2020, 94, 2097–2112. [Google Scholar] [CrossRef]
- Singh, D.P.; Begum, R.; Kaur, G.; Bagam, P.; Kambiranda, D.; Singh, R.; Batra, S. E-cig vapor condensate alters proteome and lipid profiles of membrane rafts: Impact on inflammatory responses in A549 cells. Cell Biol. Toxicol. 2021, 37, 773–793. [Google Scholar] [CrossRef]
- Hubner, R.H.; Schwartz, J.D.; De Bishnu, P.; Ferris, B.; Omberg, L.; Mezey, J.G.; Hackett, N.R.; Crystal, R.G. Coordinate control of expression of Nrf2-modulated genes in the human small airway epithelium is highly responsive to cigarette smoking. Mol. Med. 2009, 15, 203–219. [Google Scholar] [CrossRef]
- Marshall, K.; Liu, Z.; Olfert, I.M.; Gao, W. Chronic electronic cigarette use elicits molecular changes related to pulmonary pathogenesis. Toxicol. Appl. Pharmacol. 2020, 406, 115224. [Google Scholar] [CrossRef]
- Kuntic, M.; Oelze, M.; Steven, S.; Kröller-Schön, S.; Stamm, P.; Kalinovic, S.; Frenis, K.; Vujacic-Mirski, K.; Bayo Jimenez, M.T.; Kvandova, M.; et al. Short-term e-cigarette vapour exposure causes vascular oxidative stress and dysfunction: Evidence for a close connection to brain damage and a key role of the phagocytic NADPH oxidase (NOX-2). Eur. Heart J. 2020, 41, 2472–2483. [Google Scholar] [CrossRef]
- Singh, R.K.; Kumar, S.; Gautam, P.K.; Tomar, M.S.; Verma, P.K.; Singh, S.P.; Kumar, S.; Acharya, A. Protein kinase C-alpha and the regulation of diverse cell responses. Biomol. Concepts 2017, 8, 143–153. [Google Scholar] [CrossRef]
- Bootman, M.D.; Bultynck, G. Fundamentals of Cellular Calcium Signaling: A Primer. Cold Spring Harb. Perspect. Biol. 2020, 12, a038802. [Google Scholar] [CrossRef]
- Ghosh, A.; Beyazcicek, O.; Davis, E.S.; Onyenwoke, R.U.; Tarran, R. Cellular effects of nicotine salt-containing e-liquids. J. Appl. Toxicol. 2021, 41, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Coakley, R.D.; Ghio, A.J.; Muhlebach, M.S.; Esther, C.R.; Alexis, N.E.; Tarran, R. Chronic E-Cigarette Use Increases Neutrophil Elastase and Matrix Metalloprotease Levels in the Lung. Am. J. Respir. Crit. Care 2019, 200, 1392–1401. [Google Scholar] [CrossRef] [PubMed]
- Ung, T.T.; Nguyen, T.T.; Li, S.; Han, J.; Jung, Y.D. Nicotine stimulates CYP1A1 expression in human hepatocellular carcinoma cells via AP-1, NF-κB, and AhR. Toxicol. Lett. 2021, 349, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.W.; Kosinska, W.; Guttenplan, J.B. E-cigarette Aerosol Condensate Enhances Metabolism of Benzo(a)pyrene to Genotoxic Products, and Induces CYP1A1 and CYP1B1, Likely by Activation of the Aryl Hydrocarbon Receptor. Int. J. Environ. Res. Public Health 2019, 16, 2468. [Google Scholar] [CrossRef]
- Kwon, H.J.; Oh, Y.T.; Park, S.; Kim, S.S.; Park, J.; Yin, J.; Hong, J.H.; Kim, C.I.; Ryu, H.; Park, J.B.; et al. Analysis of electric cigarette liquid effect on mouse brain tumor growth through EGFR and ERK activation. PLoS ONE 2021, 16, e256730. [Google Scholar] [CrossRef]
- Wang, T.W.; Neff, L.J.; Park-Lee, E.; Ren, C.; Cullen, K.A.; King, B.A. E-cigarette Use among Middle and High School Students—United States, 2020. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 1310–1312. [Google Scholar] [CrossRef]
- Chand, H.S.; Muthumalage, T.; Maziak, W.; Rahman, I. Pulmonary Toxicity and the Pathophysiology of Electronic Cigarette, or Vaping Product, Use Associated Lung Injury. Front. Pharmacol. 2020, 10, 1619. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Toxic Compound Type | Toxic Compound | Concentration Range Cigarette (/Puff) | Concentration Range E-Cigarette (/Puff) |
|---|---|---|---|
| Carbonyls | Formaldehyde | <10 µg | <82 µg |
| Acetaldehyde | <140 µg | <53 µg | |
| Acrolein | <14 µg | <3.3 µg | |
| Propionaldehyde | <5.9 µg | <1.79 µg | |
| Crotonaldehyde | <2 µg | <0.04 µg | |
| N-nitrosamines | N’-nitrosonornicotine (NNN) | <370 ng | <0.029 ng |
| N’-nitrosoanabasine (NAB) | <15 ng | <0.01 ng | |
| 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) | <77 ng | <0.019 ng | |
| N’-nitrosoanatabine (NAT) | <16 ng | <0.085 ng | |
| Volatile organic compounds (VOCs) | Toluene | <6.9 µg | <1.53 µg |
| Benzene | <4.5 µg | <0.41 µg | |
| Inorganic compounds | Nickel | <60 ng | <6.4 ng |
| Cobalt | <0.02 ng | <0.58 ng | |
| Chromium | <7 ng | <9 ng | |
| Lead | <8.5 ng | <3.8 ng | |
| Cadmium | <35 ng | - | |
| Zinc | <1370 ng | <458 ng | |
| Cuprum | <130 ng | <20.9 ng | |
| Carbon monoxide (CO) | <2.3 mg | - | |
| Polycyclic aromatic hydrocarbons and heterocyclic aromatic hydrocarbons (PAHs) | Benz[a]anthracene | <7 ng | - |
| Benzo[b + k]fluoranthene | <3.4 ng | - | |
| Benzo[a]pyrene | <4 ng | - | |
| Dibenzo[a, h]anthracene | <0.4 ng | - | |
| Nicotine | <0.3 mg | <0.142 mg | |
| Particulate matter | Total particulate matter (TPM) | <1.7 mg | <5.8 mg |
| Toxicity Mechanism | Cells/Animals | E-Cigarette Model | Exposure Method | Toxicity Findings | Reference |
|---|---|---|---|---|---|
| Inflammation response Oxidative stress | NHBE cells Human 3D bronchial epithelial tissue | PG/VG: 70:30 Nicotine: 2% Flavors: flavorless | Incubation with media containing e-liquids for 24 h | Decreased cell viability, increase in G-CSF, CXCL1, and IL-8 and levels of GSH and ROS | [68] |
| Inflammation response | BEAS-2B cells | Lounge model designed with 2.8 Ω coil and 3.6 V power supply PG/VG: 65:35 Nicotine: 0 and 16 mg/mL Flavors: blond tobacco, chlorophyll mint, and unflavored | Air–liquid interface for 8 or 48 min (35 mL puff volume, 2 s draw, 60 s puff interval) | Low increase in IL-6 | [69] |
| Inflammation response Oxidative stress | H292 cells HFL1 cells C57BL/6J mice | Refillable ENDS with 2.2 Ω Nicotine: 0 and 16 mg/mL Flavors: classic tobacco, cinnamon roll, grape vape, American tobacco, etc. | Air–liquid interface for 5, 10, and 15 min (a puff of 3–4 s, 30 s puff interval) | Decreased cell viability, increase in IL-6 and IL-8, promotion of OX/ROS generation, and lung inflammation in mice | [70] |
| Inflammation response Oxidative stress Apoptosis | Human alveolar macrophages | Second-generation END with 650 mAh battery and 1.8 Ω coil PG/VG: 50:50 Nicotine: 0 and 36 mg/mL Flavors: flavorless | Incubation with media containing e-cigarette vapor condensate for 24 h | Decreased cell viability, increased apoptosis, increased ROS production and levels of IL-6, TNF-α, CXCL8, MCP-1, and MMP-9 | [71] |
| Inflammation response DNA damage | Bronchial epithelial cells 16HBE cells | JUUL® e-cigarette Nicotine: 5% Flavors: Virginia tobacco and menthol | Air–liquid interface for 30 min (55 mL puff volume, 4 s draw, 30 s puff interval) | Decreased cell viability, increase in IL-6, IL-8, and 8-OHdG | [72] |
| Inflammation response | BALB/c mice | Four different varieties of e-cigarette (Mt. Baker Vapor, Lynden, WA, USA) Nicotine: 0 and 12 mg/mL Flavors: American Tobacco | Whole-body exposure for 8 weeks | Increase in pulmonary inflammation and responsiveness to methacholine | [73] |
| Inflammation response | C57BL/6J mice | PG/VG: 1:1 Nicotine: 0 and 18 mg/mL Flavors: tobacco blend | Whole-body exposure for 3 days or 4 weeks | Increase in BALF cellularity, levels of IL-1β, IL-6, pulmonary inflammation, and responsiveness to methacholine | [74] |
| Inflammation response | C57BL/6 mice CD-1 mice | E-liquid was placed in a standard tank (1.8 Ω) with a rechargeable battery (3.4 V) PG/VG: 50:50 Nicotine: 24 mg/mL Flavors: flavorless | Nose-only inExpose system exposure for 3–6 months | Increase in circulating inflammatory cytokines | [75] |
| Oxidative stress DNA damage | HFL-1 cells | Lorillard Blu Classic Tobacco E-cigarette Nicotine: 16 mg/mL Flavors: classic tobacco | Air–liquid interface for 5, 10, 15, or 20 min (a puff of 3–4 s, 30 s puff interval) | Increase in mtROS, nuclear DNA fragmentation, and decrease in stability of an electron transport chain (ETC) complex IV subunit | [76] |
| Oxidative stress | BEAS-2B cells | Second-generation “Lounge” model with a 2.8 Ω nichrome coil and 4.6 W power supply and third-generation “ModBox” model with a 0.5 Ω Kanthal coil PG/VG: 65:35 Nicotine: 16 mg/mL Flavors: blond tobacco | Air–liquid interface for 40, 80, and 120 puffs (55 mL puff volume, 2 s draw, 30 s puff interval) | Increase of GSSG/GSH ratio at higher power settings | [10] |
| Oxidative stress DNA damage | B6C3F1 mice | PG/VG: 1:1 Nicotine: 0, 12, and 24 mg/mL Flavors: flavorless | Whole-body exposure for 8 weeks | Increase in 8-OHdG | [77] |
| Oxidative stress | Acellular ROS assay | PG/VG: 1:1 Nicotine: 0%–6.8% Flavors: tobacco, minty fruit, fruity, minty/cool (iced), desserts, and drinks/beverages | Incubation with media containing of e-cigarette vapor condensate for 15 min | Increase in ROS | [78] |
| Oxidative stress | BEAS-2B cells | JUUL® pod Nicotine: 5% Flavors: menthol and Virginia tobacco | Air–liquid interface for 30 min (55 mL puff volume, 3 s draw, 30 s puff interval) | Change of mitochondrial bioenergetics and decrease in mitochondrial respiration | [79] |
| Oxidative stress | MG-63 cells | Mister-E-Liquid and Vape Dudes PG/VG: 1:1 Nicotine: 0 mg/mL Flavors: flavorless and cinnamon | Incubation with media containing e-cigarette vapor condensate for 24 or 48 h | Decreased cell viability, increase in ROS | [80] |
| Oxidative stress Inflammation response | U937 cells Mono Mac 6 cells | Nicotine: 0 mg/mL Flavors: strawberry zing, café latte, pineapple coconut, cinnamon roll, etc. | Incubation with media containing of e-cigarette vapor condensate for 24 h | Decreased cell viability, increase in ROS and IL-8 | [81] |
| DNA damage | Human epithelial normal bronchial cells (Nuli1) Human premalignant dysplastic oral mucosal keratinocyte cells (POE9n) | Brands NJoy and eGo-T PG/VG: 50:50 Nicotine: 0, 12, and 18 mg/mL Flavors: traditional tobacco and desert sands | Incubation with media containing of e-cigarette vapor condensate for 2 weeks (1 h per day) | Increase in 8-oxo-dG and ROS, decrease in the expression of ERCC1 and OGG1 | [82] |
| DNA damage Oxidative stress Apoptosis | HUVEC cells | Brands Blu, Vuse, Green Smoke, and NJoy Nicotine: 2.4%, 4.5%, and 4.8% Flavors: tobacco | Incubation with media containing e-cigarette vapor condensate for 24 or 72 h | Decreased cell viability, increase in DNA damage, apoptosis, and ROS | [83] |
| DNA damage | BEAS-2B cells UROtsa cells FVBN mice | Brand NJoy PG/VG: 50:50 Nicotine: 10 mg/mL | Whole-body exposure for 12 weeks and cell exposure for 1 h | Increase in γ-OH-PdG and O6-MedG, decrease in the expression of repair proteins XPC and OGG1/2 | [84] |
| DNA damage | Sprague Dawley rats | Brand Essential cloud with a 2000 mAh battery and 2 Ω coil Nicotine: 18 mg/mL Flavors: red fruit | Whole-body exposure for 4 weeks | Increase in the free radical content, 8-OHdG, and DNA fragmentation | [85] |
| Apoptosis | Human primary gingival fibroblasts | Brand EMOW Nicotine: 12 mg/mL Flavors: smooth Canadian tobacco | Incubation with media containing e-cigarette vapor condensate for 24 h | Decrease in cell density and altered cell morphology, increase in cell apoptosis | [86] |
| Apoptosis | Human primary gingival epithelial cells | Brand EMOW Nicotine: 12 mg/mL Flavors: smooth Canadian tobacco | Air–liquid interface for 1, 2, or 3 days with 15 min per day (5 s draw, 30 s puff interval) | Increase in cell apoptosis and caspase-3 activity | [87] |
| Epithelial–mesenchymal transition | A549 cells | Nicotine: 48 mg/mL Flavors: menthol and tobacco | Incubation with media containing e-cigarette vapor condensate for 3–4 days | Acquisition of a fibroblast-like morphology, loss of cell-to-cell junctions, internalization of E-cadherin, increased motility, and upregulation of EMT markers | [88] |
| Transcriptomic changes | NHBE cells | No mention | Air–liquid interface for 6 or 24 h | Inducement of significant transcriptomic changes, increase in expression of ribosomal protein genes, change of ribosomal RNA transcription and protein synthesis | [89] |
| Transcriptomic changes | NHBE cells | E-cigarette liquid Nicotine: 0% or 2.4% | Incubation with media containing e-cigarette vapor condensate for 48 h | Change of microRNA expression profiling and increase in expression of multiple miRNAs | [90] |
| Transcriptomic changes | iPSC-EC cells | Vape Dudes E-cigarette PG/VG: 50:50 Nicotine: 24 mg/mL Flavors: menthol | Incubation with media containing e-cigarette vapor condensate for 24 h | Change of expression profiling of lncRNAs and mRNAs | [91] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Wang, Y.; Chen, J.; Liu, P.; Li, M. A Review of Toxicity Mechanism Studies of Electronic Cigarettes on Respiratory System. Int. J. Mol. Sci. 2022, 23, 5030. https://doi.org/10.3390/ijms23095030
Wang L, Wang Y, Chen J, Liu P, Li M. A Review of Toxicity Mechanism Studies of Electronic Cigarettes on Respiratory System. International Journal of Molecular Sciences. 2022; 23(9):5030. https://doi.org/10.3390/ijms23095030
Chicago/Turabian StyleWang, Lilan, Yao Wang, Jianwen Chen, Peiqing Liu, and Min Li. 2022. "A Review of Toxicity Mechanism Studies of Electronic Cigarettes on Respiratory System" International Journal of Molecular Sciences 23, no. 9: 5030. https://doi.org/10.3390/ijms23095030
APA StyleWang, L., Wang, Y., Chen, J., Liu, P., & Li, M. (2022). A Review of Toxicity Mechanism Studies of Electronic Cigarettes on Respiratory System. International Journal of Molecular Sciences, 23(9), 5030. https://doi.org/10.3390/ijms23095030