
Characteristics of microRNAs and Target Genes in Maize Root under Drought Stress

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
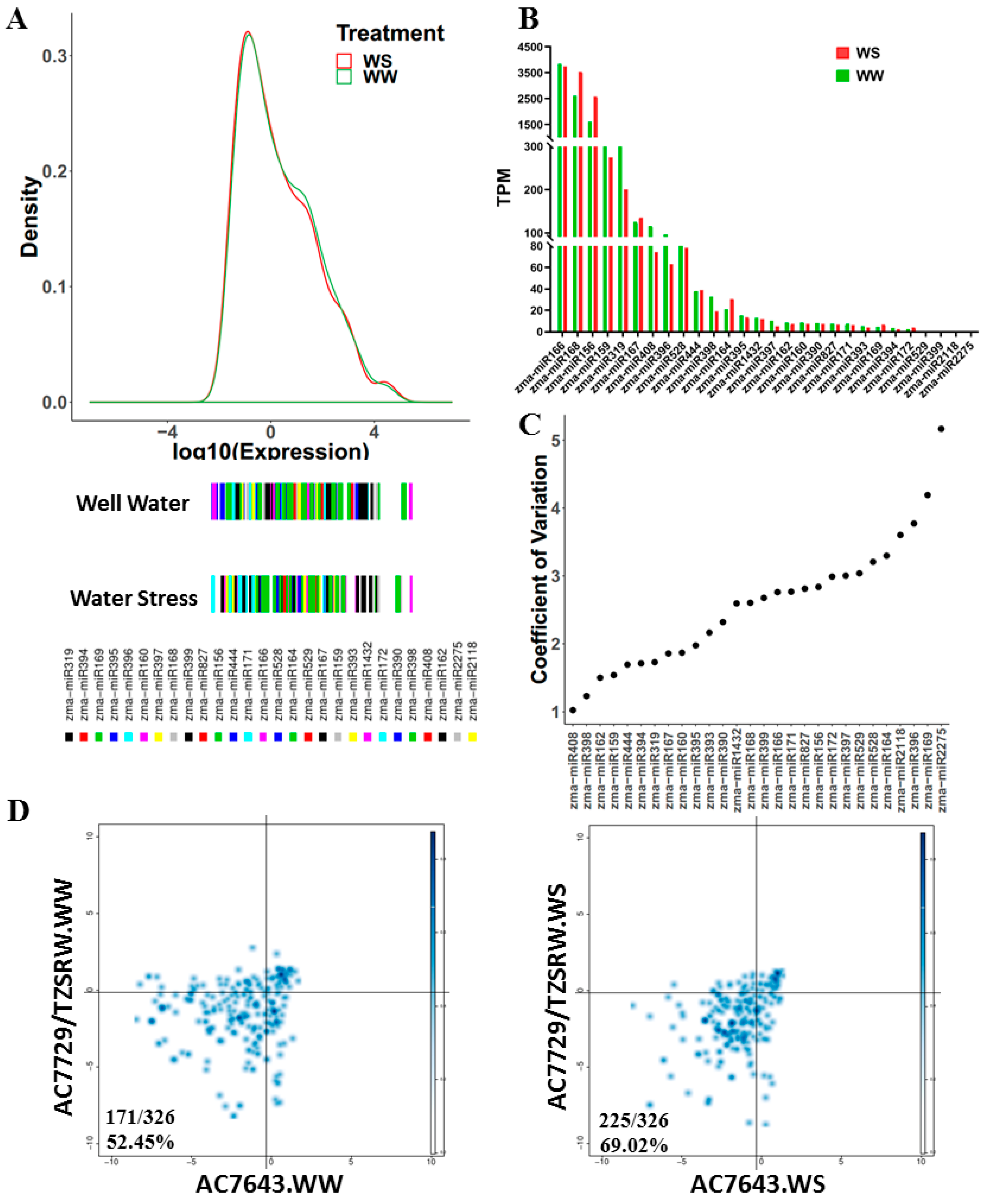
2.1. Identification of miRNAs in Maize Root
2.2. Expression Pattern of Identified miRNAs in Maize Root
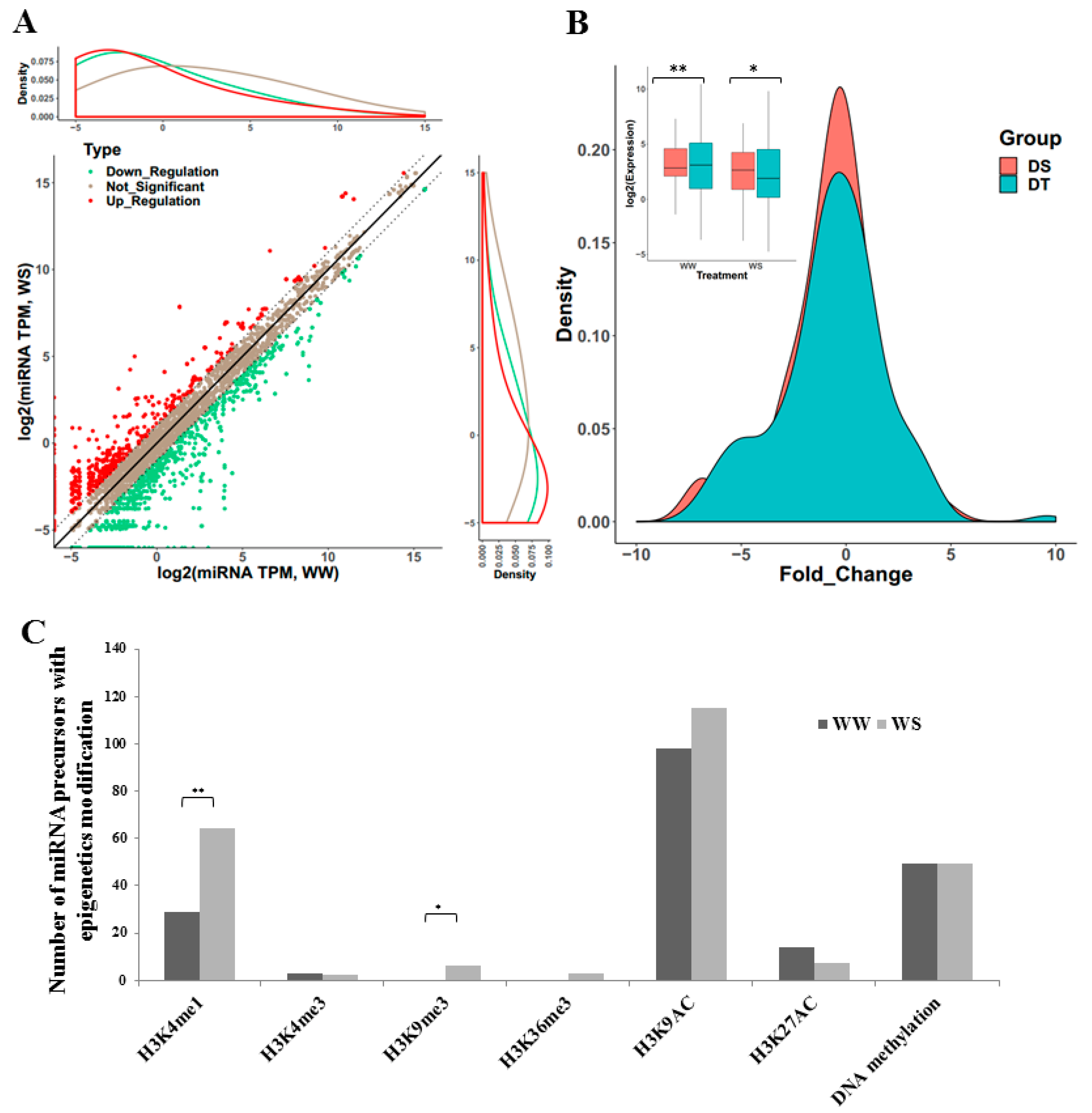
2.3. Differential Expression of miRNAs under Drought Condition
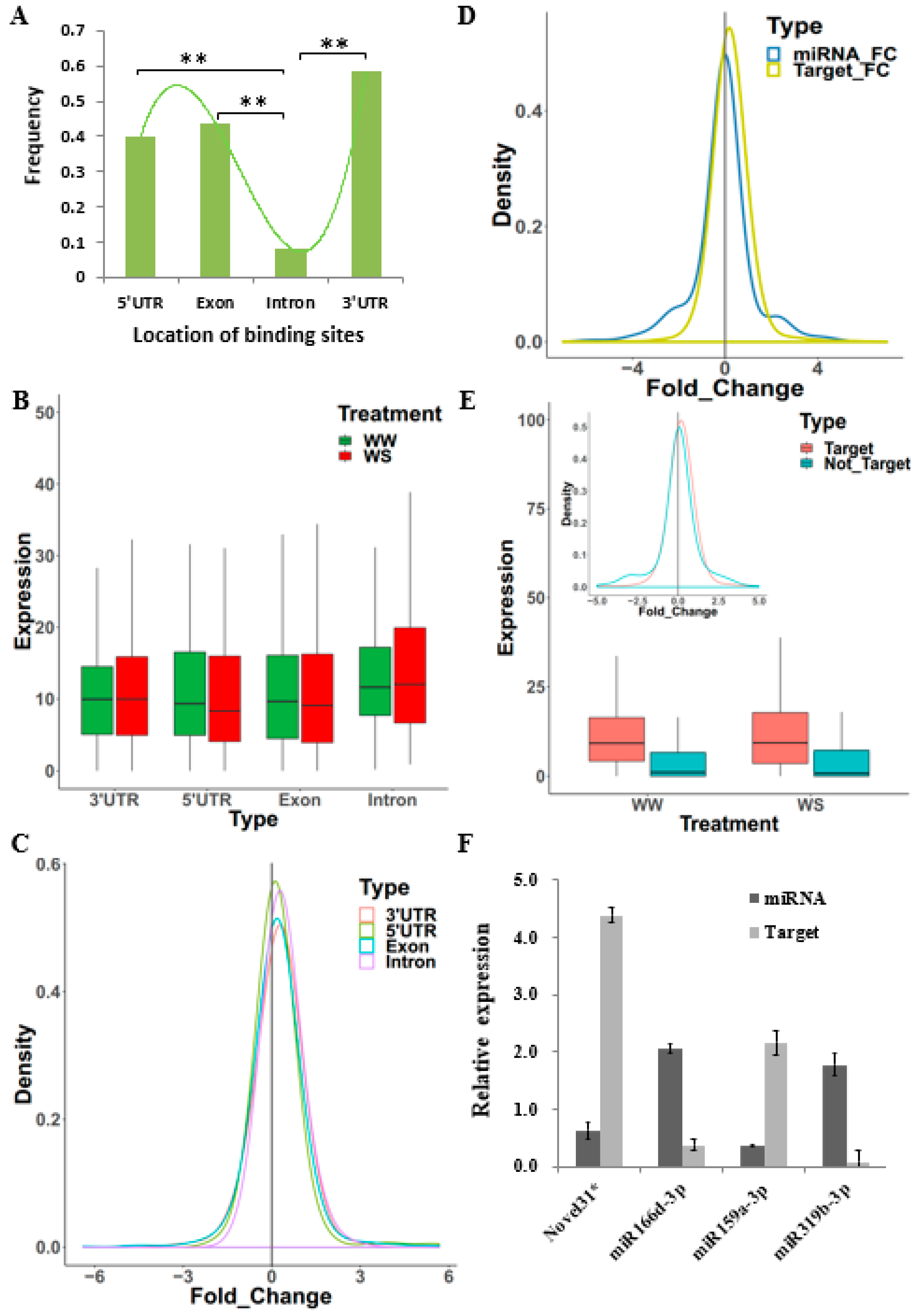
2.4. The Characteristics of Target Genes Regulated by miRNAs
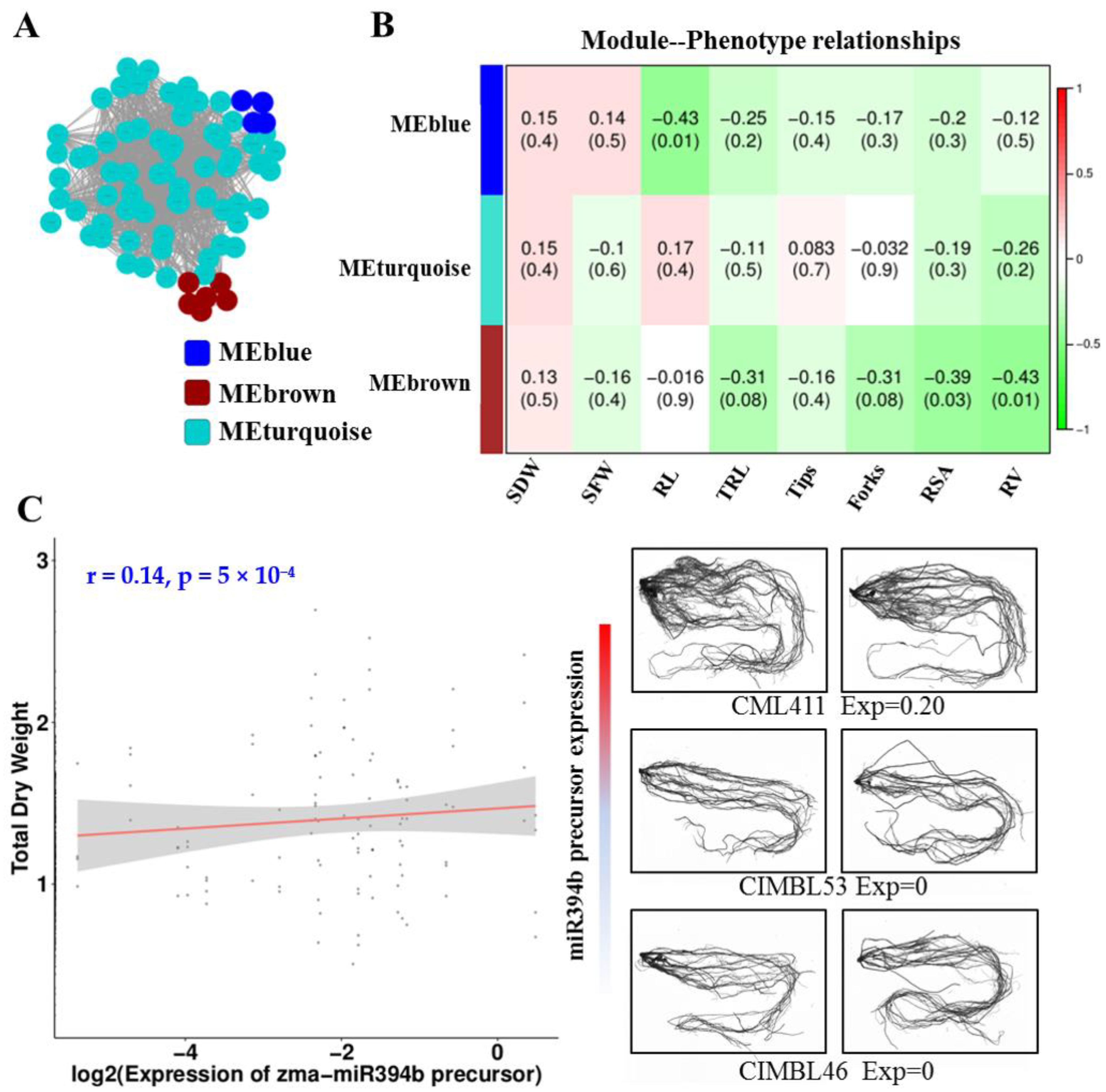
2.5. Co-Expression Network Analysis of miRNAs and Targets
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Plant Material and Treatment
5.2. Library Construction and Sequencing
5.3. miRNA Identification and Data Processing
5.4. RNA Analysis
5.5. WGCNA Network Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Zhou, H.; Zhang, Q.; Zhang, J.; Ni, F.; Liu, C.; Qi, Y. DNA methylation mediated by a microRNA pathway. Mol. Cell 2010, 38, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Wang, Y.; Xue, D.; Wang, J.; Yan, M.; Liu, G.; Dong, G.; Zeng, D.; Lu, Z.; Zhu, X.; et al. Regulation of OsSPL14 by OsmiR156 defines ideal plant architecture in rice. Nat. Genet. 2010, 42, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Guo, G.; Guo, W.; Guo, G.; Tong, D.; Ni, Z.; Sun, Q.; Yao, Y. miRNA164-directed cleavage of ZmNAC1 confers lateral root development in maize (Zea mays L.). BMC Plant Biol. 2012, 12, 220. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Li, D.; Mao, D.; Liu, X.U.E.; Ji, C.; Li, X.; Zhao, X.; Cheng, Z.; Chen, C.; Zhu, L. Overexpression of microRNA319 impacts leaf morphogenesis and leads to enhanced cold tolerance in rice (Oryza sativa L.). Plant Cell Environ. 2013, 36, 2207–2218. [Google Scholar] [CrossRef]
- Chuck, G.; Cigan, A.M.; Saeteurn, K.; Hake, S. The heterochronic maize mutant Corngrass1 results from overexpression of a tandem microRNA. Nat. Genet. 2007, 39, 544–549. [Google Scholar] [CrossRef]
- Akdogan, G.; Tufekci, E.D.; Uranbey, S.; Unver, T. miRNA-based drought regulation in wheat. Funct. Integr. Genom. 2016, 16, 221–233. [Google Scholar] [CrossRef]
- Fang, Y.; Xie, K.; Xiong, L. Conserved miR164-targeted NAC genes negatively regulate drought resistance in rice. J. Exp. Bot. 2014, 65, 2119–2135. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zou, Z.; Gong, P.; Zhang, J.; Ziaf, K.; Li, H.; Xiao, F.; Ye, Z. Over-expression of microRNA169 confers enhanced drought tolerance to tomato. Biotechnol. Lett. 2011, 33, 403–409. [Google Scholar] [CrossRef]
- Wei, L.; Zhang, D.; Xiang, F.; Zhang, Z. Differentially expressed miRNAs potentially involved in the regulation of defense mechanism to drought stress in maize seedlings. Int. J. Plant Sci. 2009, 170, 979–989. [Google Scholar] [CrossRef]
- Millar, A.A.; Waterhouse, P.M. Plant and animal microRNAs: Similarities and differences. Funct. Integr. Genom. 2005, 5, 129–135. [Google Scholar] [CrossRef]
- Pasquinelli, A.E. MicroRNAs and their targets: Recognition, regulation and an emerging reciprocal relationship. Nat. Rev. Genet. 2012, 13, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.Y.; Lee, W.C.; Chou, H.C.; Chen, A.P.; Chou, S.J.; Chen, H.M. Global Analysis of Truncated RNA Ends Reveals New Insights into Ribosome Stalling in Plants. Plant Cell 2016, 28, 2398–2416. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Willmann, M.R.; Anderson, S.J.; Gregory, B.D. Genome-Wide Mapping of Uncapped and Cleaved Transcripts Reveals a Role for the Nuclear mRNA Cap-Binding Complex in Cotranslational RNA Decay in Arabidopsis. Plant Cell 2016, 28, 2385–2397. [Google Scholar] [CrossRef] [PubMed]
- Lanet, E.; Delannoy, E.; Sormani, R.; Floris, M.; Brodersen, P.; Crete, P.; Voinnet, O.; Robaglia, C. Biochemical evidence for translational repression by Arabidopsis microRNAs. Plant Cell 2009, 21, 1762–1768. [Google Scholar] [CrossRef] [Green Version]
- Iwakawa, H.-o.; Tomari, Y. Molecular Insights into microRNA-Mediated Translational Repression in Plants. Mol. Cell 2013, 52, 591–601. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Bai, X.; Yang, L.; Lv, D.; Li, Y.; Cai, H.; Ji, W.; Guo, D.; Zhu, Y. Over-expression of osa-MIR396c decreases salt and alkali stress tolerance. Planta 2010, 231, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, F.; Cao, H.; Peng, H.; Ni, Z.; Sun, Q.; Yao, Y. TamiR159 directed wheat TaGAMYB cleavage and its involvement in anther development and heat response. PLoS ONE 2012, 7, e48445. [Google Scholar] [CrossRef] [Green Version]
- Uga, Y.; Sugimoto, K.; Ogawa, S.; Rane, J.; Ishitani, M.; Hara, N.; Kitomi, Y.; Inukai, Y.; Ono, K.; Kanno, N.; et al. Control of root system architecture by DEEPER ROOTING 1 increases rice yield under drought conditions. Nat. Genet. 2013, 45, 1097. [Google Scholar] [CrossRef]
- Oh, J.E.; Kwon, Y.; Kim, J.H.; Noh, H.; Hong, S.W.; Lee, H. A dual role for MYB60 in stomatal regulation and root growth of Arabidopsis thaliana under drought stress. Plant Mol. Biol. 2011, 77, 91–103. [Google Scholar] [CrossRef]
- Jeong, J.S.; Kim, Y.S.; Baek, K.H.; Jung, H.; Ha, S.H.; Do Choi, Y.; Kim, M.; Reuzeau, C.; Kim, J.K. Root-specific expression of OsNAC10 improves drought tolerance and grain yield in rice under field drought conditions. Plant Physiol. 2010, 153, 185–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axtell, M.J. Classification and comparison of small RNAs from plants. Annu. Rev. Plant Biol. 2013, 64, 137–159. [Google Scholar] [CrossRef] [Green Version]
- Zhao, P.; Ding, D.; Zhang, F.; Zhao, X.; Xue, Y.; Li, W.; Fu, Z.; Li, H.; Tang, J. Investigating the molecular genetic basis of heterosis for internode expansion in maize by microRNA transcriptomic deep sequencing. Funct. Integr. Genom. 2015, 15, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Liu, D.; Wu, J.; Zhao, X.; Hao, M.; Geng, S.; Yan, J.; Jiang, X.; Zhang, L.; Wu, J.; et al. mRNA and Small RNA Transcriptomes Reveal Insights into Dynamic Homoeolog Regulation of Allopolyploid Heterosis in Nascent Hexaploid Wheat. Plant Cell 2014, 26, 1878–1900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, R.; Li, L.; Li, J. Spatial and temporal expression modes of MicroRNAs in an elite rice hybrid and its parental lines. Planta 2013, 238, 259–269. [Google Scholar] [CrossRef]
- Cui, L.G.; Shan, J.X.; Shi, M.; Gao, J.P.; Lin, H.X. The miR156-SPL9-DFR pathway coordinates the relationship between development and abiotic stress tolerance in plants. Plant J. 2014, 80, 1108–1117. [Google Scholar] [CrossRef] [PubMed]
- Arshad, M.; Feyissa, B.A.; Amyot, L.; Aung, B.; Hannoufa, A. MicroRNA156 improves drought stress tolerance in alfalfa (Medicago sativa) by silencing SPL13. Plant Sci. Int. J. Exp. Plant Biol. 2017, 258, 122–136. [Google Scholar] [CrossRef]
- Gutierrez, L.; Bussell, J.D.; Pacurar, D.I.; Schwambach, J.; Pacurar, M.; Bellini, C. Phenotypic plasticity of adventitious rooting in Arabidopsis is controlled by complex regulation of AUXIN RESPONSE FACTOR transcripts and microRNA abundance. Plant Cell 2009, 21, 3119–3132. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Roy, S.; Singh, S.; Das, S.S.; Gautam, V.; Yadav, S.; Kumar, A.; Singh, A.; Samantha, S.; Sarkar, A.K. Phytohormonal crosstalk modulates the expression of miR166/165s, target Class III HD-ZIPs, and KANADI genes during root growth in Arabidopsis thaliana. Sci. Rep. 2017, 7, 3408. [Google Scholar] [CrossRef] [Green Version]
- Berger, F.; Zhang, K.; Sridhar, V.V.; Zhu, J.; Kapoor, A.; Zhu, J.-K. Distinctive Core Histone Post-Translational Modification Patterns in Arabidopsis thaliana. PLoS ONE 2007, 2, e1210. [Google Scholar] [CrossRef]
- Chinnusamy, V.; Zhu, J.-K. Epigenetic regulation of stress responses in plants. Curr. Opin. Plant Biol. 2009, 12, 133–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nathan, D. Histone sumoylation is a negative regulator in Saccharomyces cerevisiae and shows dynamic interplay with positive-acting histone modifications. Genes Dev. 2006, 20, 966–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, T.; Liu, Z.; Dai, X.; Xiang, F. Primary root growth in Arabidopsis thaliana is inhibited by the miR159 mediated repression of MYB33, MYB65 and MYB101. Plant Sci. Int. J. Exp. Plant Biol. 2017, 262, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Song, J.B.; Gao, S.; Sun, D.; Li, H.; Shu, X.X.; Yang, Z.M. miR394 and LCR are involved in Arabidopsis salt and drought stress responses in an abscisic acid-dependent manner. BMC Plant Biol. 2013, 13, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.; Li, D.; Li, Z.; Hu, Q.; Yang, C.; Zhu, L.; Luo, H. Constitutive Expression of a miR319 Gene Alters Plant Development and Enhances Salt and Drought Tolerance in Transgenic Creeping Bentgrass. Plant Physiol. 2013, 161, 1375–1391. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Chung, P.J.; Liu, J.; Jang, I.C.; Kean, M.J.; Xu, J.; Chua, N.H. Genome-wide identification of long noncoding natural antisense transcripts and their responses to light in Arabidopsis. Genome Res. 2014, 24, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.-C.; Lin, F.-M.; Chang, W.-C.; Lin, K.-Y.; Huang, H.-D.; Lin, N.-S. miRExpress: Analyzing high-throughput sequencing data for profiling microRNA expression. BMC Bioinform. 2009, 10, 328. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.J.; Ma, Y.K.; Chen, T.; Wang, M.; Wang, X.J. PsRobot: A web-based plant small RNA meta-analysis toolbox. Nucleic Acids Res. 2012, 40, W22–W28. [Google Scholar] [CrossRef]
- Axtell, M.J. ShortStack: Comprehensive annotation and quantification of small RNA genes. RNA 2013, 19, 740–751. [Google Scholar] [CrossRef] [Green Version]
- Moxon, S.; Schwach, F.; Dalmay, T.; MacLean, D.; Studholme, D.J.; Moulton, V. A toolkit for analysing large-scale plant small RNA datasets. Bioinformatics 2008, 24, 2252–2253. [Google Scholar] [CrossRef] [Green Version]
- Axtell, M.J.; Meyers, B.C. Revisiting Criteria for Plant MicroRNA Annotation in the Era of Big Data. Plant Cell 2018, 30, 272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server. Nucleic Acids Res. 2011, 39 (Suppl. S2), W155–W159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, I.; Robert, G. R: A language and environment for statistical computing. R Found. Stat. Comput. 2008, 5, 299–314. [Google Scholar]
- Chen, C.; Ridzon, D.A.; Broomer, A.J.; Zhou, Z.; Lee, D.H.; Nguyen, J.T.; Barbisin, M.; Xu, N.L.; Mahuvakar, V.R.; Andersen, M.R.; et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005, 33, e179. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, Q.; Lv, H.; Li, Q.; Zhang, X.; Li, L.; Xu, J.; Wu, F.; Wang, Q.; Feng, X.; Lu, Y. Characteristics of microRNAs and Target Genes in Maize Root under Drought Stress. Int. J. Mol. Sci. 2022, 23, 4968. https://doi.org/10.3390/ijms23094968
Tang Q, Lv H, Li Q, Zhang X, Li L, Xu J, Wu F, Wang Q, Feng X, Lu Y. Characteristics of microRNAs and Target Genes in Maize Root under Drought Stress. International Journal of Molecular Sciences. 2022; 23(9):4968. https://doi.org/10.3390/ijms23094968
Chicago/Turabian StyleTang, Qi, Haozhe Lv, Qimeng Li, Xiaoyue Zhang, Le Li, Jie Xu, Fengkai Wu, Qingjun Wang, Xuanjun Feng, and Yanli Lu. 2022. "Characteristics of microRNAs and Target Genes in Maize Root under Drought Stress" International Journal of Molecular Sciences 23, no. 9: 4968. https://doi.org/10.3390/ijms23094968
APA StyleTang, Q., Lv, H., Li, Q., Zhang, X., Li, L., Xu, J., Wu, F., Wang, Q., Feng, X., & Lu, Y. (2022). Characteristics of microRNAs and Target Genes in Maize Root under Drought Stress. International Journal of Molecular Sciences, 23(9), 4968. https://doi.org/10.3390/ijms23094968