
On the Base Composition of Transposable Elements


{kind=link}
Abstract
1. Introduction
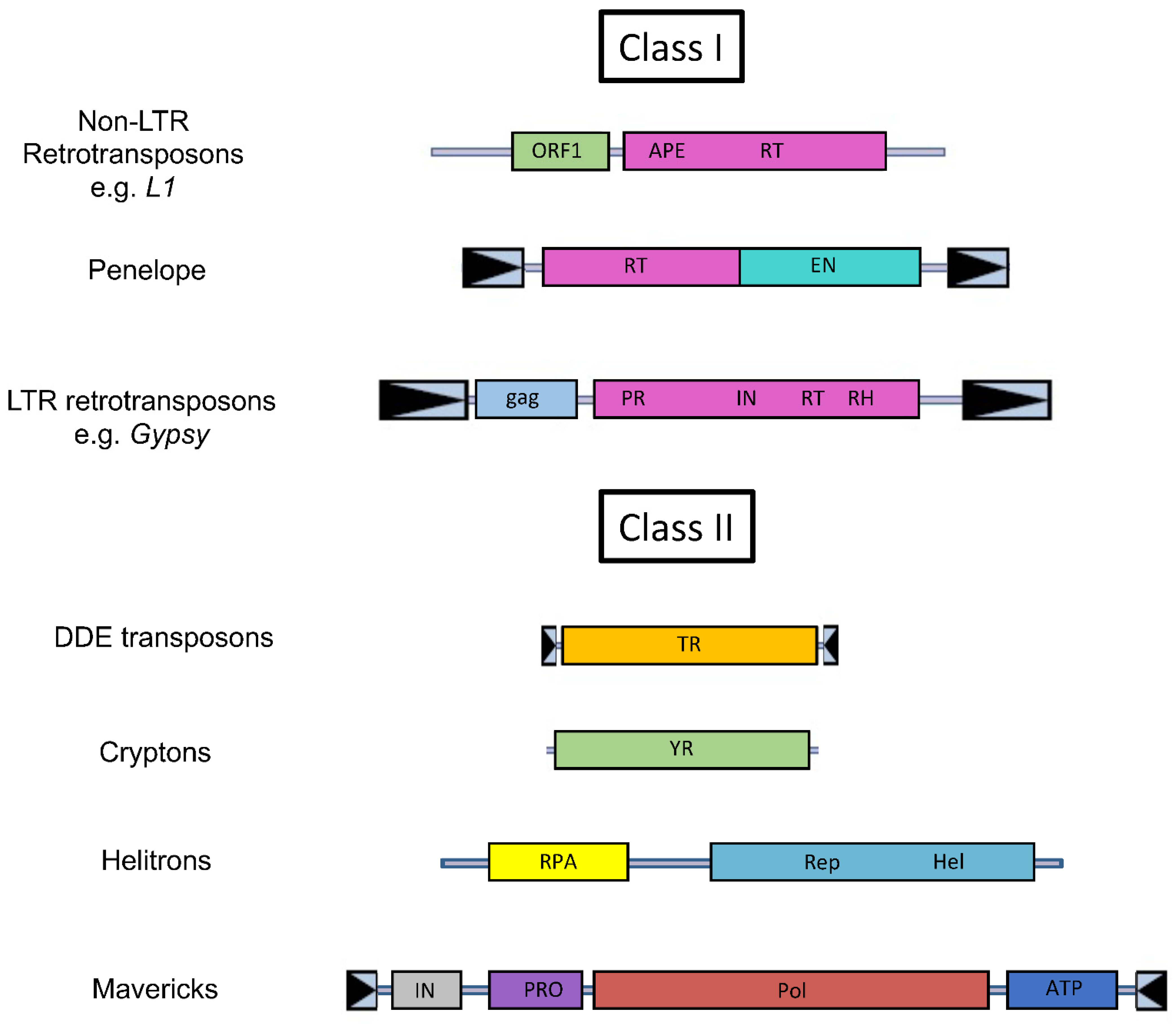
2. A Primer on Transposable Elements
3. Variation in the Base Composition of Transposable Elements
4. Consequences of the Unusual Base Composition of Transposable Elements
5. Why Do Transposable Elements Have Such Unusual Base Composition?
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Costantini, M.; Cammarano, R.; Bernardi, G. The evolution of isochore patterns in vertebrate genomes. BMC Genom. 2009, 10, 146. [Google Scholar] [CrossRef]
- Bernardi, G.; Bernardi, G. Compositional patterns in the nuclear genome of cold-blooded vertebrates. J. Mol. Evol. 1990, 31, 265–281. [Google Scholar] [CrossRef]
- Zhou, Z.; Dang, Y.; Zhou, M.; Li, L.; Yu, C.-H.; Fu, J.; Chen, S.; Liu, Y. Codon usage is an important determinant of gene expression levels largely through its effects on transcription. Proc. Natl. Acad. Sci. USA 2016, 113, E6117–E6125. [Google Scholar] [CrossRef]
- Han, J.S.; Szak, S.T.; Boeke, J.D. Transcriptional disruption by the L1 retrotransposon and implications for mammalian transcriptomes. Nature 2004, 429, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Akashi, H. Synonymous codon usage in Drosophila melanogaster: Natural selection and translational accuracy. Genetics 1994, 136, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Duret, L. Evolution of synonymous codon usage in metazoans. Curr. Opin. Genet. Dev. 2002, 12, 640–649. [Google Scholar] [CrossRef]
- Duret, L.; Mouchiroud, D. Expression pattern and, surprisingly, gene length shape codon usage in Caenorhabditis, Drosophila, and Arabidopsis. Proc. Natl. Acad. Sci. USA 1999, 96, 4482–4487. [Google Scholar] [CrossRef]
- Stoletzki, N.; Eyre-Walker, A. Synonymous codon usage in Escherichia coli: Selection for translational accuracy. Mol. Biol. Evol. 2007, 24, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Almojil, D.; Bourgeois, Y.; Falis, M.; Hariyani, I.; Wilcox, J.; Boissinot, S. The structural, functional and evolutionary impact of transposable elements in eukaryotes. Genes 2021, 12, 918. [Google Scholar] [CrossRef]
- Capy, P. Taming, Domestication and Exaptation: Trajectories of Transposable Elements in Genomes. Cells 2021, 10, 3590. [Google Scholar] [CrossRef]
- Bourque, G.; Burns, K.H.; Gehring, M.; Gorbunova, V.; Seluanov, A.; Hammell, M.; Imbeault, M.; Izsvák, Z.; Levin, H.L.; Macfarlan, T.S. Ten things you should know about transposable elements. Genome Biol. 2018, 19, 199. [Google Scholar] [CrossRef] [PubMed]
- Sinzelle, L.; Izsvak, Z.; Ivics, Z. Molecular domestication of transposable elements: From detrimental parasites to useful host genes. Cell. Mol. Life Sci. 2009, 66, 1073–1093. [Google Scholar] [CrossRef] [PubMed]
- Boissinot, S.; Sookdeo, A. The evolution of LINE-1 in vertebrates. Genome Biol. Evol. 2016, 8, 3485–3507. [Google Scholar] [CrossRef]
- Lerat, E.; Capy, P.; Biemont, C. Codon usage by transposable elements and their host genes in five species. J. Mol. Evol. 2002, 54, 625–637. [Google Scholar] [CrossRef]
- Ruggiero, R.P.; Boissinot, S. Variation in base composition underlies functional and evolutionary divergence in non-LTR retrotransposons. Mob. DNA 2020, 11, 14–18. [Google Scholar] [CrossRef]
- Shields, D.C.; Sharp, P.M. Evidence that mutation patterns vary among Drosophila transposable elements. J. Mol. Biol. 1989, 207, 843–846. [Google Scholar] [CrossRef]
- Jia, J.; Xue, Q. Codon usage biases of transposable elements and host nuclear genes in Arabidopsis thaliana and Oryza sativa. Genom. Proteom. Bioinform. 2009, 7, 175–184. [Google Scholar] [CrossRef]
- Andrieu, O.; Fiston, A.-S.; Anxolabéhère, D.; Quesneville, H. Detection of transposable elements by their compositional bias. BMC Bioinform. 2004, 5, 94. [Google Scholar] [CrossRef]
- Tollis, M.; Boissinot, S. The evolutionary dynamics of transposable elements in eukaryote genomes. Repetitive DNA 2012, 7, 68–91. [Google Scholar]
- Wells, J.N.; Feschotte, C. A Field Guide to Eukaryotic Transposable Elements. Annu. Rev. Genet. 2020, 54, 539–561. [Google Scholar] [CrossRef]
- Kojima, K.K. Structural and sequence diversity of eukaryotic transposable elements. Genes Genet. Syst. 2018, 94, 233–252. [Google Scholar] [CrossRef] [PubMed]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Eickbush, T.H.; Malik, H.S. Origins and evolution of retrotransposons. In Mobile DNA II; American Society of Microbiology: Washington, DC, USA, 2002; pp. 1111–1144. [Google Scholar]
- Dewannieux, M.; Esnault, C.; Heidmann, T. LINE-mediated retrotransposition of marked Alu sequences. Nat. Genet. 2003, 35, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Dewannieux, M.; Heidmann, T. LINEs, SINEs and processed pseudogenes: Parasitic strategies for genome modeling. Cytogenet. Genome Res. 2005, 110, 35–48. [Google Scholar] [CrossRef]
- Ohshima, K.; Okada, N. SINEs and LINEs: Symbionts of eukaryotic genomes with a common tail. Cytogenet. Genome Res. 2005, 110, 475–490. [Google Scholar] [CrossRef]
- Feschotte, C.; Pritham, E.J. DNA transposons and the evolution of eukaryotic genomes. Annu. Rev. Genet. 2007, 41, 331–368. [Google Scholar] [CrossRef]
- Kapitonov, V.V.; Jurka, J. Rolling-circle transposons in eukaryotes. Proc. Natl. Acad. Sci. USA 2001, 98, 8714–8719. [Google Scholar] [CrossRef]
- Kapitonov, V.V.; Jurka, J. Helitrons on a roll: Eukaryotic rolling-circle transposons. Trends Genet. 2007, 23, 521–529. [Google Scholar] [CrossRef]
- Kapitonov, V.V.; Jurka, J. Self-synthesizing DNA transposons in eukaryotes. Proc. Natl. Acad. Sci. USA 2006, 103, 4540–4545. [Google Scholar] [CrossRef]
- Pritham, E.J.; Putliwala, T.; Feschotte, C. Mavericks, a novel class of giant transposable elements widespread in eukaryotes and related to DNA viruses. Gene 2007, 390, 3–17. [Google Scholar] [CrossRef]
- Hartl, D.; Lozovskaya, E.; Lawrence, J. Nonautonomous transposable elements in prokaryotes and eukaryotes. Genetica 1992, 86, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Novick, P.A.; Smith, J.D.; Floumanhaft, M.; Ray, D.A.; Boissinot, S. The evolution and diversity of DNA transposons in the genome of the lizard Anolis carolinensis. Genome Biol. Evol. 2011, 3, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Nagel, D.H.; Feschotte, C.; Hancock, C.N.; Wessler, S.R. Tuned for transposition: Molecular determinants underlying the hyperactivity of a Stowaway MITE. Science 2009, 325, 1391–1394. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.F.; Tóth, G.; Riggs, A.D.; Jurka, J. Ancestral, mammalian-wide subfamilies of LINE-1 repetitive sequences. J. Mol. Biol. 1995, 246, 401–417. [Google Scholar] [CrossRef] [PubMed]
- Furano, A.V. The biological properties and evolutionary dynamics of mammalian LINE-1 retrotransposons. Prog. Nucleic Acid Res. Mole. Biol. 2000, 64, 255–294. [Google Scholar]
- Furano, A.V.; Duvernell, D.D.; Boissinot, S. L1 (LINE-1) retrotransposon diversity differs dramatically between mammals and fish. Trends Genet. 2004, 20, 9–14. [Google Scholar] [CrossRef]
- Khan, H.; Smit, A.; Boissinot, S. Molecular evolution and tempo of amplification of human LINE-1 retrotransposons since the origin of primates. Genome Res. 2006, 16, 78–87. [Google Scholar] [CrossRef]
- Sookdeo, A.; Hepp, C.M.; Boissinot, S. Contrasted patterns of evolution of the LINE-1 retrotransposon in perissodactyls: The history of a LINE-1 extinction. Mob. DNA 2018, 9, 12. [Google Scholar] [CrossRef]
- Sookdeo, A.; Hepp, C.M.; McClure, M.A.; Boissinot, S. Revisiting the evolution of mouse LINE-1 in the genomic era. Mob. DNA 2013, 4, 3. [Google Scholar] [CrossRef]
- Boissinot, S.; Furano, A.V. Adaptive evolution in LINE-1 retrotransposons. Mol. Biol. Evol. 2001, 18, 2186–2194. [Google Scholar] [CrossRef][Green Version]
- Jacobs, F.M.; Greenberg, D.; Nguyen, N.; Haeussler, M.; Ewing, A.D.; Katzman, S.; Paten, B.; Salama, S.R.; Haussler, D. An evolutionary arms race between KRAB zinc-finger genes ZNF91/93 and SVA/L1 retrotransposons. Nature 2014, 516, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Blumenstiel, J.P. Birth, school, work, death, and resurrection: The life stages and dynamics of transposable element proliferation. Genes 2019, 10, 336. [Google Scholar] [CrossRef] [PubMed]
- Schaack, S.; Gilbert, C.; Feschotte, C. Promiscuous DNA: Horizontal transfer of transposable elements and why it matters for eukaryotic evolution. Trends Ecol. Evol. 2010, 25, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, C.; Feschotte, C. Horizontal acquisition of transposable elements and viral sequences: Patterns and consequences. Curr. Opin. Genet. Dev. 2018, 49, 15–24. [Google Scholar] [CrossRef]
- Bourgeois, Y.; Boissinot, S. On the population dynamics of junk: A review on the population genomics of transposable elements. Genes 2019, 10, 419. [Google Scholar] [CrossRef]
- Goodier, J.L. Restricting retrotransposons: A review. Mob. DNA 2016, 7, 16. [Google Scholar] [CrossRef]
- Czech, B.; Hannon, G.J. One Loop to Rule Them All: The Ping-Pong Cycle and piRNA-Guided Silencing. Trends Biochem. Sci. 2016, 41, 324–337. [Google Scholar] [CrossRef]
- Deniz, O.; Frost, J.M.; Branco, M.R. Regulation of transposable elements by DNA modifications. Nat. Rev. Genet. 2019, 20, 417–431. [Google Scholar] [CrossRef]
- Yoder, J.A.; Walsh, C.P.; Bestor, T.H. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 1997, 13, 335–340. [Google Scholar] [CrossRef]
- Bourgeois, Y.; Ruggiero, R.; Hariyani, I.; Boissinot, S. Disentangling the determinants of transposable elements dynamics in vertebrate genomes using empirical evidences and simulations. PLoS Genet. 2020, 16, e1009082. [Google Scholar] [CrossRef]
- Ruggiero, R.P.; Bourgeois, Y.; Boissinot, S. LINE insertion polymorphisms are abundant but at low frequencies across populations of Anolis carolinensis. Front. Genet. 2017, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Biémont, C.; Lemeunier, F.; Guerreiro, M.G.; Brookfield, J.; Gautier, C.; Aulard, S.; Pasyukova, E. Population dynamics of the copia, mdg1, mdg3, gypsy, and P transposable elements in a natural population of Drosophila melanogaster. Genet. Res. 1994, 63, 197–212. [Google Scholar] [CrossRef] [PubMed]
- Boissinot, S.; Entezam, A.; Furano, A.V. Selection against deleterious LINE-1-containing loci in the human lineage. Mol. Biol. Evol. 2001, 18, 926–935. [Google Scholar] [CrossRef]
- Charlesworth, B.; Langley, C.H. The population genetics of Drosophila transposable elements. Annu. Rev. Genet. 1989, 23, 251–287. [Google Scholar] [CrossRef] [PubMed]
- Xue, A.T.; Ruggiero, R.P.; Hickerson, M.J.; Boissinot, S. Differential effect of selection against LINE retrotransposons among vertebrates inferred from whole-genome data and demographic modeling. Genome Biol. Evol. 2018, 10, 1265–1281. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, J.; Macpherson, J.M.; Messer, P.W.; Petrov, D.A. Inferring the strength of selection in Drosophila under complex demographic models. Mol. Biol. Evol. 2009, 26, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Lockton, S.; Ross-Ibarra, J.; Gaut, B.S. Demography and weak selection drive patterns of transposable element diversity in natural populations of Arabidopsis lyrata. Proc. Natl. Acad. Sci. USA 2008, 105, 13965–13970. [Google Scholar] [CrossRef] [PubMed]
- Garcia Guerreiro, M.P.; Chavez-Sandoval, B.E.; Balanya, J.; Serra, L.; Fontdevila, A. Distribution of the transposable elements bilbo and gypsy in original and colonizing populations of Drosophila subobscura. BMC Evol. Biol. 2008, 8, 234. [Google Scholar] [CrossRef]
- Lynch, M.; Conery, J.S. The origins of genome complexity. Science 2003, 302, 1401–1404. [Google Scholar] [CrossRef]
- Blass, E.; Bell, M.; Boissinot, S. Accumulation and rapid decay of non-LTR retrotransposons in the genome of the three-spine stickleback. Genome Biol. Evol. 2012, 4, 687–702. [Google Scholar] [CrossRef]
- Petrov, D.A.; Sangster, T.A.; Johnston, J.S.; Hartl, D.L.; Shaw, K.L. Evidence for DNA loss as a determinant of genome size. Science 2000, 287, 1060–1062. [Google Scholar] [CrossRef] [PubMed]
- Kapusta, A.; Suh, A.; Feschotte, C. Dynamics of genome size evolution in birds and mammals. Proc. Natl. Acad. Sci. USA 2017, 114, E1460–E1469. [Google Scholar] [CrossRef] [PubMed]
- Hershberg, R.; Petrov, D.A. Selection on codon bias. Annu. Rev. Genet. 2008, 42, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Ingvarsson, P.K. Gene expression and protein length influence codon usage and rates of sequence evolution in Populus tremula. Mol. Biol. Evol. 2007, 24, 836–844. [Google Scholar] [CrossRef]
- Shields, D.C.; Sharp, P.M.; Higgins, D.G.; Wright, F. “Silent” sites in Drosophila genes are not neutral: Evidence of selection among synonymous codons. Mol. Biol. Evol. 1988, 5, 704–716. [Google Scholar]
- Gaffaroglu, M.; Majtanova, Z.; Symonova, R.; Pelikanova, S.; Unal, S.; Lajbner, Z.; Rab, P. Present and Future Salmonid Cytogenetics. Genes 2020, 11, 1462. [Google Scholar] [CrossRef]
- Symonova, R.; Suh, A. Nucleotide composition of transposable elements likely contributes to AT/GC compositional homogeneity of teleost fish genomes. Mob. DNA 2019, 10, 49. [Google Scholar] [CrossRef]
- Besansky, N. Codon usage patterns in chromosomal and retrotransposon genes of the mosquito Anopheles gambiae. Insect Mol. Biol. 1993, 1, 171–178. [Google Scholar] [CrossRef]
- Southworth, J.; Grace, C.A.; Marron, A.O.; Fatima, N.; Carr, M. A genomic survey of transposable elements in the choanoflagellate Salpingoeca rosetta reveals selection on codon usage. Mob. DNA 2019, 10, 44. [Google Scholar] [CrossRef]
- Jiang, R.H.; Govers, F. Nonneutral GC3 and retroelement codon mimicry in Phytophthora. J. Mol. Evol. 2006, 63, 458–472. [Google Scholar] [CrossRef]
- Aerts, S.; Thijs, G.; Dabrowski, M.; Moreau, Y.; De Moor, B. Comprehensive analysis of the base composition around the transcription start site in Metazoa. BMC Genom. 2004, 5, 34. [Google Scholar] [CrossRef] [PubMed]
- Basame, S.; Li, P.W.-l.; Howard, G.; Branciforte, D.; Keller, D.; Martin, S.L. Spatial assembly and RNA binding stoichiometry of a LINE-1 protein essential for retrotransposition. J. Mol. Biol. 2006, 357, 351–357. [Google Scholar] [CrossRef]
- Doucet, A.J.; Hulme, A.E.; Sahinovic, E.; Kulpa, D.A.; Moldovan, J.B.; Kopera, H.C.; Athanikar, J.N.; Hasnaoui, M.; Bucheton, A.; Moran, J.V. Characterization of LINE-1 ribonucleoprotein particles. PLoS Genet. 2010, 6, e1001150. [Google Scholar] [CrossRef] [PubMed]
- Perepelitsa-Belancio, V.; Deininger, P. RNA truncation by premature polyadenylation attenuates human mobile element activity. Nat. Genet. 2003, 35, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Han, J.S.; Boeke, J.D. A highly active synthetic mammalian retrotransposon. Nature 2004, 429, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Han, J.S.; Boeke, J.D. LINE-1 retrotransposons: Modulators of quantity and quality of mammalian gene expression? Bioessays 2005, 27, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Roy-Engel, A.; El-Sawy, M.; Farooq, L.; Odom, G.; Perepelitsa-Belancio, V.; Bruch, H.; Oyeniran, O.; Deininger, P. Human retroelements may introduce intragenic polyadenylation signals. Cytogenet. Genome Res. 2005, 110, 365–371. [Google Scholar] [CrossRef]
- Medstrand, P.; van de Lagemaat, L.N.; Mager, D.L. Retroelement distributions in the human genome: Variations associated with age and proximity to genes. Genome Res. 2002, 12, 1483–1495. [Google Scholar] [CrossRef]
- Cutter, A.D.; Good, J.M.; Pappas, C.T.; Saunders, M.A.; Starrett, D.M.; Wheeler, T.J. Transposable element orientation bias in the Drosophila melanogaster genome. J. Mol. Evol. 2005, 61, 733–741. [Google Scholar] [CrossRef]
- van de Lagemaat, L.N.; Landry, J.R.; Mager, D.L.; Medstrand, P. Transposable elements in mammals promote regulatory variation and diversification of genes with specialized functions. Trends Genet. 2003, 19, 530–536. [Google Scholar] [CrossRef]
- Slotkin, R.K.; Martienssen, R. Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 2007, 8, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, Y.; Saze, H.; Kinoshita, T.; Miura, A.; Soppe, W.J.; Koornneef, M.; Kakutani, T. Control of FWA gene silencing in Arabidopsis thaliana by SINE-related direct repeats. Plant J. 2007, 49, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Parinov, S.; Sundaresan, V. Functional genomics in Arabidopsis: Large-scale insertional mutagenesis complements the genome sequencing project. Curr. Opin. Biotechnol. 2000, 11, 157–161. [Google Scholar] [CrossRef]
- Hollister, J.D.; Gaut, B.S. Epigenetic silencing of transposable elements: A trade-off between reduced transposition and deleterious effects on neighboring gene expression. Genome Res. 2009, 19, 1419–1428. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C.G.; Karpen, G.H. Pervasive epigenetic effects of Drosophila euchromatic transposable elements impact their evolution. eLife 2017, 6, e25762. [Google Scholar] [CrossRef]
- Vitte, C.; Panaud, O. LTR retrotransposons and flowering plant genome size: Emergence of the increase/decrease model. Cytogenet. Genome Res. 2005, 110, 91–107. [Google Scholar] [CrossRef]
- Hawkins, J.S.; Kim, H.; Nason, J.D.; Wing, R.A.; Wendel, J.F. Differential lineage-specific amplification of transposable elements is responsible for genome size variation in Gossypium. Genome Res. 2006, 16, 1252–1261. [Google Scholar] [CrossRef]
- Piegu, B.; Guyot, R.; Picault, N.; Roulin, A.; Sanyal, A.; Kim, H.; Collura, K.; Brar, D.S.; Jackson, S.; Wing, R.A.; et al. Doubling genome size without polyploidization: Dynamics of retrotransposition-driven genomic expansions in Oryza australiensis, a wild relative of rice. Genome Res. 2006, 16, 1262–1269. [Google Scholar] [CrossRef]
- Grandaubert, J.; Lowe, R.G.; Soyer, J.L.; Schoch, C.L.; Van de Wouw, A.P.; Fudal, I.; Robbertse, B.; Lapalu, N.; Links, M.G.; Ollivier, B. Transposable element-assisted evolution and adaptation to host plant within the Leptosphaeria maculans-Leptosphaeria biglobosa species complex of fungal pathogens. BMC Genom. 2014, 15, 891. [Google Scholar] [CrossRef]
- Symonova, R.; Majtanova, Z.; Arias-Rodriguez, L.; Morkovsky, L.; Korinkova, T.; Cavin, L.; Pokorna, M.J.; Dolezalkova, M.; Flajshans, M.; Normandeau, E.; et al. Genome Compositional Organization in Gars Shows More Similarities to Mammals than to Other Ray-Finned Fish. J. Exp. Zool. B Mol. Dev. Evol. 2017, 328, 607–619. [Google Scholar] [CrossRef]
- Costantini, M.; Auletta, F.; Bernardi, G. Isochore patterns and gene distributions in fish genomes. Genomics 2007, 90, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Mugal, C.F.; Weber, C.C.; Ellegren, H. GC-biased gene conversion links the recombination landscape and demography to genomic base composition: GC-biased gene conversion drives genomic base composition across a wide range of species. Bioessays 2015, 37, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Boissinot, S. Selection against LINE-1 retrotransposons results principally from their ability to mediate ectopic recombination. Gene 2007, 390, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Matoulek, D.; Borůvková, V.; Ocalewicz, K.; Symonová, R. GC and repeats profiling along chromosomes—The future of fish compositional cytogenomics. Genes 2021, 12, 50. [Google Scholar] [CrossRef] [PubMed]
- Rödelsperger, C.; Sommer, R.J. Computational archaeology of the Pristionchus pacificus genome reveals evidence of horizontal gene transfers from insects. BMC Evol. Biol. 2011, 11, 239. [Google Scholar] [CrossRef]
- McHale, M.T.; Roberts, I.N.; Noble, S.M.; Beaumont, C.; Whitehead, M.P.; Seth, D.; Oliver, R.P. CfT-I: An LTR-retrotransposon in Cladosporium fulvum, a fungal pathogen of tomato. Mol. Gen. Genet. MGG 1992, 233, 337–347. [Google Scholar] [CrossRef]
- Wallau, G.L.; Ortiz, M.F.; Loreto, E.L.S. Horizontal transposon transfer in eukarya: Detection, bias, and perspectives. Genome Biol. Evol. 2012, 4, 801–811. [Google Scholar] [CrossRef]
- Powell, J.R.; Gleason, J.M. Codon usage and the origin of P elements. Mol. Biol. Evol. 1996, 13, 278–279. [Google Scholar] [CrossRef][Green Version]
- Springer, M.S.; Tusneem, N.A.; Davidson, E.H.; Britten, R.J. Phylogeny, rates of evolution, and patterns of codon usage among sea urchin retroviral-like elements, with implications for the recognition of horizontal transfer. Mol. Biol. Evol. 1995, 12, 219–230. [Google Scholar]
- Lerat, E.; Biémont, C.; Capy, P. Codon usage and the origin of P elements. Mol. Biol. Evol. 2000, 17, 467–468. [Google Scholar] [CrossRef][Green Version]
- Kordis, D.; Gubensek, F. Unusual horizontal transfer of a long interspersed nuclear element between distant vertebrate classes. Proc. Natl. Acad. Sci. USA 1998, 95, 10704–10709. [Google Scholar] [CrossRef] [PubMed]
- Walsh, A.M.; Kortschak, R.D.; Gardner, M.G.; Bertozzi, T.; Adelson, D.L. Widespread horizontal transfer of retrotransposons. Proc. Natl. Acad. Sci. USA 2013, 110, 1012–1016. [Google Scholar] [CrossRef]
- Preston, B.D.; Poiesz, B.J.; Loeb, L.A. Fidelity of HIV-1 reverse transcriptase. Science 1988, 242, 1168–1171. [Google Scholar] [CrossRef] [PubMed]
- Carmi, S.; Church, G.M.; Levanon, E.Y. Large-scale DNA editing of retrotransposons accelerates mammalian genome evolution. Nat. Commun. 2011, 2, 519. [Google Scholar] [CrossRef] [PubMed]
- Lindič, N.; Budič, M.; Petan, T.; Knisbacher, B.A.; Levanon, E.Y.; Lovšin, N. Differential inhibition of LINE1 and LINE2 retrotransposition by vertebrate AID/APOBEC proteins. Retrovirology 2013, 10, 156. [Google Scholar] [CrossRef]
- Duret, L.; Marais, G.; Biémont, C. Transposons but not retrotransposons are located preferentially in regions of high recombination rate in Caenorhabditis elegans. Genetics 2000, 156, 1661–1669. [Google Scholar] [CrossRef]
- Kawakami, T.; Mugal, C.F.; Suh, A.; Nater, A.; Burri, R.; Smeds, L.; Ellegren, H. Whole-genome patterns of linkage disequilibrium across flycatcher populations clarify the causes and consequences of fine-scale recombination rate variation in birds. Mol. Ecol. 2017, 26, 4158–4172. [Google Scholar] [CrossRef]
- Myers, S.; Freeman, C.; Auton, A.; Donnelly, P.; McVean, G. A common sequence motif associated with recombination hot spots and genome instability in humans. Nat. Genet. 2008, 40, 1124. [Google Scholar] [CrossRef]
- Lerat, E.; Capy, P.; Biémont, C. The relative abundance of dinucleotides in transposable elements in five species. Mol. Biol. Evol. 2002, 19, 964–967. [Google Scholar] [CrossRef]
- Cosby, R.L.; Chang, N.-C.; Feschotte, C. Host–transposon interactions: Conflict, cooperation, and cooption. Genes Dev. 2019, 33, 1098–1116. [Google Scholar] [CrossRef]
- Venner, S.; Feschotte, C.; Biémont, C. Dynamics of transposable elements: Towards a community ecology of the genome. Trends Genet. 2009, 25, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Brookfield, J.F. The ecology of the genome—Mobile DNA elements and their hosts. Nat. Rev. Genet. 2005, 6, 128–136. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boissinot, S. On the Base Composition of Transposable Elements. Int. J. Mol. Sci. 2022, 23, 4755. https://doi.org/10.3390/ijms23094755
Boissinot S. On the Base Composition of Transposable Elements. International Journal of Molecular Sciences. 2022; 23(9):4755. https://doi.org/10.3390/ijms23094755
Chicago/Turabian StyleBoissinot, Stéphane. 2022. "On the Base Composition of Transposable Elements" International Journal of Molecular Sciences 23, no. 9: 4755. https://doi.org/10.3390/ijms23094755
APA StyleBoissinot, S. (2022). On the Base Composition of Transposable Elements. International Journal of Molecular Sciences, 23(9), 4755. https://doi.org/10.3390/ijms23094755