
Current Methods for Recombination Detection in Bacteria



Abstract
1. Introduction
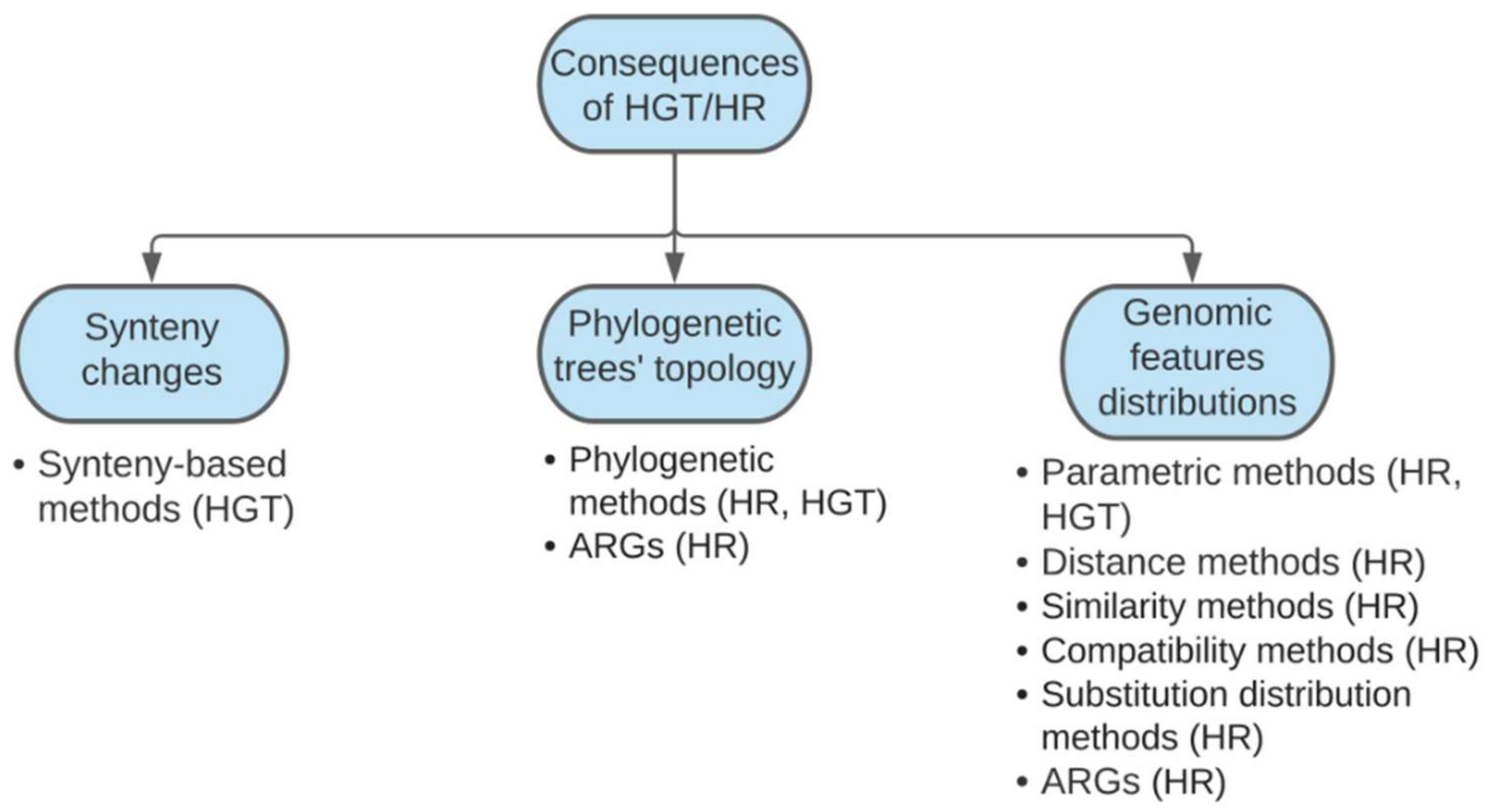
2. A Brief Overview of Conventional Methods for Detecting Homologous Recombination and Horizontal Gene Transfer
- Similarity methods are designed to reveal gene conversion by tracking anomalous identity in variable parts of the genome [37];
- Distance methods find local dissimilarities between sequences using a sliding window technique [38];
- Substitution distribution approaches group together sequences with similar patterns of integral substitution properties through comparison with the calculated model distribution [41];
3. Current Bioinformatics Tools for Recombination Analysis
3.1. Synteny-Based Methods
3.2. Phylogenetic Methods
3.2.1. Phylogenetic Methods for HR Detection
3.2.2. Implicit Phylogenetic Methods to Reveal HGT
3.3. Methods Based on Genetic Features
3.3.1. Compatibility Methods to Reveal HR
3.3.2. Substitution Distribution-Based HR Detection Approaches
3.3.3. Parametric Methods for HR Identification
3.3.4. Ancestral Recombination Graphs
3.3.5. Parametric Methods for Finding HGT Events
4. Assessing the Effectiveness of Recombination Detection Software
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| HR | Homologous recombination |
| NHR | Non-homologous recombination |
| HGT | Horizontal gene transfer |
| LGT | Lateral gene transfer |
| SSAPs | Single-strand annealing proteins |
| SSA | Single-strand annealing |
| ARGs | Ancestral recombination graphs |
| MLST | Multilocus sequence typing |
| CGP | Coarse-graining approach for phylogenetic reconstruction |
| CRM | Constant relative mutability |
| SI | Synteny index |
| PRRSV | Porcine reproductive and respiratory syndrome virus |
| GPSCs | Global pneumococcal sequence clusters |
| SNPs | Single nucleotide polymorphisms |
| HMM | Hidden Markov model |
| TDA | Topological data analysis |
| MCMC | Markov chain Monte Carlo |
| tARG | Topological ARG |
| OGFs | Orthologous gene families |
References
- Posada, D.; Crandall, K.A.; Holmes, E.C. Recombination in evolutionary genomics. Annu. Rev. Genet. 2002, 36, 75–97. [Google Scholar] [CrossRef]
- Ravenhall, M.; Škunca, N.; Lassalle, F.; Dessimoz, C. Inferring horizontal gene transfer. PLoS Comput. Biol. 2015, 11, e1004095. [Google Scholar] [CrossRef] [PubMed]
- Lemey, P.; Posada, D. Introduction to recombination detection. In The Phylogenetic Handbook; Vandamme, A.-M., Salemi, M., Lemey, P., Eds.; Cambridge University Press: Cambridge, UK, 2012; pp. 493–518. ISBN 9780511819049. [Google Scholar]
- Vos, M. Why do bacteria engage in homologous recombination? Trends Microbiol. 2009, 17, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Didelot, X.; Maiden, M.C.J. Impact of recombination on bacterial evolution. Trends Microbiol. 2010, 18, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Rong, X.; Huang, Y. Widespread interspecies homologous recombination reveals reticulate evolution within the genus Streptomyces. Mol. Phylogenet. Evol. 2016, 102, 246–254. [Google Scholar] [CrossRef]
- Ochman, H.; Lawrence, J.G.; Groisman, E.A. Lateral gene transfer and the nature of bacterial innovation. Nature 2000, 405, 299–304. [Google Scholar] [CrossRef]
- Lassalle, F.; Didelot, X. Bacterial microevolution and the pangenome. In The Pangenome: Diversity, Dynamics and Evolution of Genomes; Springer: Cham, Switzerland, 2020; pp. 129–149. [Google Scholar] [CrossRef]
- Avni, E.; Montoya, D.; Lopez, D.; Modlin, R.; Pellegrini, M.; Snir, S. A phylogenomic study quantifies competing mechanisms for pseudogenization in prokaryotes. The Mycobacterium leprae case. PLoS ONE 2018, 13, e0204322. [Google Scholar] [CrossRef]
- Mostowy, R.; Croucher, N.J.; Andam, C.P.; Corander, J.; Hanage, W.P.; Marttinen, P. Efficient Inference of Recent and Ancestral Recombination within Bacterial Populations. Mol. Biol. Evol. 2017, 34, 1167–1182. [Google Scholar] [CrossRef]
- Steczkiewicz, K.; Prestel, E.; Bidnenko, E.; Szczepankowska, A.K. Expanding Diversity of Firmicutes Single-Strand Annealing Proteins: A Putative Role of Bacteriophage-Host Arms Race. Front. Microbiol. 2021, 12, 644622. [Google Scholar] [CrossRef]
- Subramaniam, S.; Erler, A.; Fu, J.; Kranz, A.; Tang, J.; Gopalswamy, M.; Ramakrishnan, S.; Keller, A.; Grundmeier, G.; Müller, D.; et al. DNA annealing by Redβ is insufficient for homologous recombination and the additional requirements involve intra- and intermolecular interactions. Sci. Rep. 2016, 6, 34525. [Google Scholar] [CrossRef]
- Iranzo, J.; Wolf, Y.I.; Koonin, E.V.; Sela, I. Gene gain and loss push prokaryotes beyond the homologous recombination barrier and accelerate genome sequence divergence. Nat. Commun. 2019, 10, 5376. [Google Scholar] [CrossRef]
- Ely, B. Recombination and gene loss occur simultaneously during bacterial horizontal gene transfer. PLoS ONE 2020, 15, 4–6. [Google Scholar] [CrossRef]
- Levin, B.R.; Cornejo, O.E. The population and evolutionary dynamics of homologous gene recombination in bacteria. PLoS Genet. 2009, 5, e1000601. [Google Scholar] [CrossRef]
- Gürtler, V.; Mayall, B.C. Genomic approaches to typing, taxonomy and evolution of bacterial isolates. Int. J. Syst. Evol. Microbiol. 2001, 51, 3–16. [Google Scholar] [CrossRef]
- Aujoulat, F.; Romano-Bertrand, S.; Masnou, A.; Marchandin, H.; Jumas-Bilak, E. Niches, population structure and genome reduction in Ochrobactrum intermedium: Clues to technology-driven emergence of pathogens. PLoS ONE 2014, 9, e0171448. [Google Scholar] [CrossRef]
- Hao, L.; Holden, M.T.G.; Wang, X.; Andrew, L.; Wellnitz, S.; Hu, F.; Whaley, M.; Sammons, S.; Knipe, K.; Frace, M.; et al. Distinct evolutionary patterns of Neisseria meningitidis serogroup B disease outbreaks at two universities in the USA. Microb. Genom. 2018, 4, 1–10. [Google Scholar] [CrossRef]
- Nudel, K.; Zhao, X.; Basu, S.; Dong, X.; Hoffmann, M.; Feldgarden, M.; Allard, M.; Klompas, M.; Bry, L. Genomics of Corynebacterium striatum, an emerging multidrug-resistant pathogen of immunocompromised patients. Clin. Microbiol. Infect. 2018, 24, 1016.e7–1016.e13. [Google Scholar] [CrossRef]
- Liu, L.; Cui, Y.; Zheng, B.; Jiang, S.; Yu, W.; Shen, P.; Ji, J.; Li, L.; Qin, N.; Xiao, Y. Analysis of tigecycline resistance development in clinical Acinetobacter baumannii isolates through a combined genomic and transcriptomic approach. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef]
- Štaudová, B.; Strouhal, M.; Zobaníková, M.; Čejková, D.; Fulton, L.L.; Chen, L.; Giacani, L.; Centurion-Lara, A.; Bruisten, S.M.; Sodergren, E.; et al. Whole Genome Sequence of the Treponema pallidum subsp. endemicum Strain Bosnia A: The Genome Is Related to Yaws Treponemes but Contains Few Loci Similar to Syphilis Treponemes. PLoS Negl. Trop. Dis. 2014, 8. [Google Scholar] [CrossRef]
- Guo, Q.; Mustapha, M.M.; Chen, M.; Qu, D.; Zhang, X.; Chen, M.; Doi, Y.; Wang, M.; Harrison, L.H. Evolution of sequence type 4821 clonal complex meningococcal strains in China from prequinolone to quinolone era, 1972–2013. Emerg. Infect. Dis. 2018, 24, 683–690. [Google Scholar] [CrossRef]
- Potnis, N.; Kandel, P.P.; Merfa, M.V.; Retchless, A.C.; Parker, J.K.; Stenger, D.C.; Almeida, R.P.P.; Bergsma-Vlami, M.; Westenberg, M.; Cobine, P.A.; et al. Patterns of inter- and intrasubspecific homologous recombination inform eco-evolutionary dynamics of Xylella fastidiosa. ISME J. 2019, 13, 2319–2333. [Google Scholar] [CrossRef]
- Rounge, T.B.; Rohrlack, T.; Kristensen, T.; Jakobsen, K.S. Recombination and selectional forces in cyanopeptolin NRPS operons from highly similar, but geographically remote Planktothrix strains. BMC Microbiol. 2008, 8, 1–10. [Google Scholar] [CrossRef]
- Bosch, R.; García-Valdés, E.; Moore, E.R.B. Complete nucleotide sequence and evolutionary significance of a chromosomally encoded naphthalene-degradation lower pathway from Pseudomonas stutzeri AN10. Gene 2000, 245, 65–74. [Google Scholar] [CrossRef]
- Martin, D.P.; Lemey, P.; Posada, D. Analysing recombination in nucleotide sequences. Mol. Ecol. Resour. 2011, 11, 943–955. [Google Scholar] [CrossRef]
- Archibald, J.M.; Roger, A.J. Gene duplication and gene conversion shape the evolution of archaeal chaperonins. J. Mol. Biol. 2002, 316, 1041–1050. [Google Scholar] [CrossRef]
- Schierup, M.H.; Hein, J. Consequences of recombination on traditional phylogenetic analysis. Genetics 2000, 156, 879–891. [Google Scholar] [CrossRef]
- Gribaldo, S.; Philippe, H. Ancient phylogenetic relationships. Theor. Popul. Biol. 2002, 61, 391–408. [Google Scholar] [CrossRef]
- Arenas, M.; Posada, D. The effect of recombination on the reconstruction of ancestral sequences. Genetics 2010, 184, 1133–1139. [Google Scholar] [CrossRef]
- Shriner, D.; Nickle, D.C.; Jensen, M.A.; Mullins, J.I. Potential impact of recombination on sitewise approaches for detecting positive natural selection. Genet. Res. 2003, 81, 115–121. [Google Scholar] [CrossRef]
- Hedge, J.; Wilson, D.J. Bacterial phylogenetic reconstruction from whole genomes is robust to recombination but demographic inference is not. mBio 2014, 5, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Stott, C.M.; Bobay, L.M. Impact of homologous recombination on core genome phylogenies. BMC Genom. 2020, 21, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pang, T.Y. A coarse-graining, ultrametric approach to resolve the phylogeny of prokaryotic strains with frequent homologous recombination. BMC Evol. Biol. 2020, 20, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, T.G.; Welch, D.; Drummond, A.J.; Biggs, P.J.; George, T.; French, N.P. Inferring ancestral recombination graphs from bacterial genomic data. Genetics 2017, 205, 857–870. [Google Scholar] [CrossRef]
- Hanage, W.P. Not so simple after all: Bacteria, their population genetics, and recombination. Cold Spring Harb. Perspect. Biol. 2016, 8, 1–18. [Google Scholar] [CrossRef]
- Ohta, T.; Basten, C.J. Gene conversion generates hypervariability at the variable regions of kallikreins and their inhibitors. Mol. Phylogenet. Evol. 1992, 1, 87–90. [Google Scholar] [CrossRef]
- Weiller, G.F. Phylogenetic profiles: A graphical method for detecting genetic recombinations in homologous sequences. Mol. Biol. Evol. 1998, 15, 326–335. [Google Scholar] [CrossRef]
- Bruen, T.C.; Philippe, H.; Bryant, D. A simple and robust statistical test for detecting the presence of recombination. Genetics 2006, 172, 2665–2681. [Google Scholar] [CrossRef]
- Lai, Y.P.; Ioerger, T.R. A statistical method to identify recombination in bacterial genomes based on SNP incompatibility. BMC Bioinform. 2018, 19, 450. [Google Scholar] [CrossRef]
- Gibbs, M.J.; Armstrong, J.S.; Gibbs, A.J. Sister-scanning: A Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 2000, 16, 573–582. [Google Scholar] [CrossRef]
- Taylor, J.C.; Martin, H.C.; Lise, S.; Broxholme, J.; Cazier, J.-B.; Rimmer, A.; Kanapin, A.; Lunter, G.; Fiddy, S.; Allan, C.; et al. Factors influencing success of clinical genome sequencing across a broad spectrum of disorders. Nat. Genet. 2015, 47, 717–726. [Google Scholar] [CrossRef]
- Maynard Smith, J.; Smith, N.H. Detecting recombination from gene trees. Mol. Biol. Evol. 1998, 15, 590–599. [Google Scholar] [CrossRef]
- Croucher, N.J.; Page, A.J.; Connor, T.R.; Delaney, A.J.; Keane, J.A.; Bentley, S.D.; Parkhill, J.; Harris, S.R. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 2015, 43, e15. [Google Scholar] [CrossRef]
- Daubin, V.; Lerat, E.; Perrière, G. The source of laterally transferred genes in bacterial genomes. Genome Biol. 2003, 4, R57. [Google Scholar] [CrossRef]
- Vernikos, G.S.; Parkhill, J. Interpolated variable order motifs for identification of horizontally acquired DNA: Revisiting the Salmonella pathogenicity islands. Bioinformatics 2006, 22, 2196–2203. [Google Scholar] [CrossRef]
- Worning, P.; Jensen, L.J.; Nelson, K.E.; Brunak, S.; Ussery, D.W. Structural analysis of DNA sequence: Evidence for lateral gene transfer in Thermotoga maritima. Nucleic Acids Res. 2000, 28, 706–709. [Google Scholar] [CrossRef]
- Vernikos, G.S.; Parkhill, J. Resolving the structural features of genomic islands: A machine learning approach. Genome Res. 2008, 18, 331–342. [Google Scholar] [CrossRef]
- Adato, O.; Ninyo, N.; Gophna, U.; Snir, S. Detecting horizontal gene transfer between closely related taxa. PLoS Comput. Biol. 2015, 11, e1004408. [Google Scholar] [CrossRef]
- Sevillya, G.; Adato, O.; Snir, S. Detecting horizontal gene transfer: A probabilistic approach. BMC Genom. 2020, 21, 106. [Google Scholar] [CrossRef]
- Shifman, A.; Ninyo, N.; Gophna, U.; Snir, S. Phylo SI: A new genome-wide approach for prokaryotic phylogeny. Nucleic Acids Res. 2014, 42, 2391–2404. [Google Scholar] [CrossRef]
- Hepp, B.; Da Cunha, V.; Lorieux, F.; Oberto, J. BAGET 2.0: An updated web tool for the effortless retrieval of prokaryotic gene context and sequence. Bioinformatics 2021, 37, 2750–2752. [Google Scholar] [CrossRef]
- Farrer, R.A. Synima: A Synteny imaging tool for annotated genome assemblies. BMC Bioinform. 2017, 18, 507. [Google Scholar] [CrossRef]
- Stahlecker, J.; Mingyar, E.; Ziemert, N.; Mungan, M.D. SYN-View: A Phylogeny-Based Synteny Exploration Tool for the Identification of Gene Clusters Linked to Antibiotic Resistance. Molecules 2021, 26, 144. [Google Scholar] [CrossRef]
- Minkin, I.; Patel, A.; Kolmogorov, M.; Vyahhi, N.; Pham, S. Sibelia: A Scalable and Comprehensive Synteny Block Generation Tool for Closely Related Microbial Genomes. In Proceedings of the Algorithms in Bioinformatics; Darling, A., Stoye, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 215–229. [Google Scholar]
- Oberto, J. SyntTax: A web server linking synteny to prokaryotic taxonomy. BMC Bioinform. 2013, 14, 4. [Google Scholar] [CrossRef]
- Enav, H.; Ley, R.E. SynTracker: A synteny based tool for tracking microbial strains. bioRxiv 2021. [Google Scholar] [CrossRef]
- Zhou, Z.; Charlesworth, J.; Achtman, M. Accurate reconstruction of bacterial pan- and core genomes with PEPPAN. Genome Res. 2020, 30, 1667–1679. [Google Scholar] [CrossRef]
- Tonkin-Hill, G.; MacAlasdair, N.; Ruis, C.; Weimann, A.; Horesh, G.; Lees, J.A.; Gladstone, R.A.; Lo, S.; Beaudoin, C.; Floto, R.A.; et al. Producing polished prokaryotic pangenomes with the Panaroo pipeline. Genome Biol. 2020, 21, 180. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Delcher, A.L.; Salzberg, S.L.; Phillippy, A.M. Using MUMmer to identify similar regions in large sequence sets. Curr. Protoc. Bioinform. 2003, 1, 10.3. [Google Scholar] [CrossRef]
- Tan, M.; Long, H.; Liao, B.; Cao, Z.; Yuan, D.; Tian, G.; Zhuang, J.; Yang, J. QS-Net: Reconstructing phylogenetic networks based on quartet and sextet. Front. Genet. 2019, 10, 1–9. [Google Scholar] [CrossRef]
- Bloomquist, E.W.; Suchard, M.A. Unifying vertical and nonvertical evolution: A stochastic arg-based framework. Syst. Biol. 2010, 59, 27–41. [Google Scholar] [CrossRef] [PubMed]
- De Been, M.; Van Schaik, W.; Cheng, L.; Corander, J.; Willems, R.J. Recent recombination events in the core genome are associated with adaptive evolution in Enterococcus faecium. Genome Biol. Evol. 2013, 5, 1524–1535. [Google Scholar] [CrossRef] [PubMed]
- Milne, I.; Lindner, D.; Bayer, M.; Husmeier, D.; McGuire, G.; Marshall, D.F.; Wright, F. TOPALi v2: A rich graphical interface for evolutionary analyses of multiple alignments on HPC clusters and multi-core desktops. Bioinformatics 2009, 25, 126–127. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, 1–5. [Google Scholar] [CrossRef]
- Martin, D.P.; Varsani, A.; Roumagnac, P.; Botha, G.; Maslamoney, S.; Schwab, T.; Kelz, Z.; Kumar, V.; Murrell, B. RDP5: A computer program for analyzing recombination in, and removing signals of recombination from, nucleotide sequence datasets. Virus Evol. 2021, 7, veaa087. [Google Scholar] [CrossRef]
- Hudson, R.R.; Kaplan, N.L. Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics 1985, 111, 147–164. [Google Scholar] [CrossRef]
- Vandenbussche, F.; Mathijs, E.; Tignon, M.; Vandersmissen, T.; Cay, A.B. WGS- versus ORF5-Based Typing of PRRSV: A Belgian Case Study. Viruses 2021, 13, 2419. [Google Scholar] [CrossRef]
- Islam, A.; Ferdous, J.; Sayeed, M.A.; Islam, S.; Kaisar Rahman, M.; Abedin, J.; Saha, O.; Hassan, M.M.; Shirin, T. Spatial epidemiology and genetic diversity of SARS-CoV-2 and related coronaviruses in domestic and wild animals. PLoS ONE 2021, 16, e0260635. [Google Scholar] [CrossRef]
- Luka, M.M.; Kamau, E.; de Laurent, Z.R.; Morobe, J.M.; Alii, L.K.; Nokes, D.J.; Agoti, C.N. Whole genome sequencing of two human rhinovirus A types (A101 and A15) detected in Kenya, 2016–2018. Wellcome Open Res. 2021, 6, 178. [Google Scholar] [CrossRef]
- Tucciarone, C.M.; Franzo, G.; Legnardi, M.; Lazzaro, E.; Zoia, A.; Petini, M.; Furlanello, T.; Caldin, M.; Cecchinato, M.; Drigo, M. Genetic Insights into Feline Parvovirus: Evaluation of Viral Evolutionary Patterns and Association between Phylogeny and Clinical Variables. Viruses 2021, 13, 1033. [Google Scholar] [CrossRef]
- Didelot, X.; Wilson, D.J. ClonalFrameML: Efficient Inference of Recombination in Whole Bacterial Genomes. PLoS Comput. Biol. 2015, 11, 1–18. [Google Scholar] [CrossRef]
- Gladstone, R.A.; Lo, S.W.; Goater, R.; Yeats, C.; Taylor, B.; Hadfield, J.; Lees, J.A.; Croucher, N.J.; van Tonder, A.J.; Bentley, L.J.; et al. Visualizing variation within Global Pneumococcal Sequence Clusters (GPSCs) and country population snapshots to contextualize pneumococcal isolates. Microb. Genom. 2020, 6, e000357. [Google Scholar] [CrossRef]
- Lo, S.W.; Gladstone, R.A.; van Tonder, A.J.; Du Plessis, M.; Cornick, J.E.; Hawkins, P.A.; Madhi, S.A.; Nzenze, S.A.; Kandasamy, R.; Ravikumar, K.L.; et al. A mosaic tetracycline resistance gene tet(S/M) detected in an MDR pneumococcal CC230 lineage that underwent capsular switching in South Africa. J. Antimicrob. Chemother. 2020, 75, 512–520. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, X.; Song, Y.; Zeng, Q.; Zhang, Y.; Luo, H. Prochlorococcus have low global mutation rate and small effective population size. Nat. Ecol. Evol. 2021, 6, 183–194. [Google Scholar] [CrossRef]
- Gill, J.L.; Hedge, J.; Wilson, D.J.; MacLean, R.C. Evolutionary Processes Driving the Rise and Fall of Staphylococcus aureus ST239, a Dominant Hybrid Pathogen. mBio 2021, 12, e0216821. [Google Scholar] [CrossRef]
- Chase, A.B.; Sweeney, D.; Muskat, M.N.; Guillén-Matus, D.G.; Jensen, P.R. Vertical Inheritance Facilitates Interspecies Diversification in Biosynthetic Gene Clusters and Specialized Metabolites. mBio 2021, 12, e0270021. [Google Scholar] [CrossRef]
- Wang, J.; Li, Y.; Pinto-Tomás, A.A.; Cheng, K.; Huang, Y. Habitat Adaptation Drives Speciation of a Streptomyces Species with Distinct Habitats and Disparate Geographic Origins. mBio 2022, 13, e0278121. [Google Scholar] [CrossRef]
- Sawhney, S.S.; Ransom, E.M.; Wallace, M.A.; Reich, P.J.; Dantas, G.; Burnham, C.-A.D. Comparative Genomics of Borderline Oxacillin-Resistant Staphylococcus aureus Detected during a Pseudo-outbreak of Methicillin-Resistant S. aureus in a Neonatal Intensive Care Unit. mBio 2022, 13, e0319621. [Google Scholar] [CrossRef]
- Castillo, A.I.; Tsai, C.-W.; Su, C.-C.; Weng, L.-W.; Lin, Y.-C.; Cho, S.-T.; Almeida, R.P.P.; Kuo, C.-H. Genetic differentiation of Xylella fastidiosa following the introduction into Taiwan. Microb. Genom. 2021, 7, 727. [Google Scholar] [CrossRef]
- Sakoparnig, T.; Field, C.; van Nimwegen, E. Whole genome phylogenies reflect the distributions of recombination rates for many bacterial species. eLife 2021, 10, e65366. [Google Scholar] [CrossRef]
- Lerat, E.; Daubin, V.; Moran, N.A. From Gene Trees to Organismal Phylogeny in Prokaryotes: The Case of the γ-Proteobacteria. PLoS Biol. 2003, 1, e19. [Google Scholar] [CrossRef]
- Zhaxybayeva, O.; Gogarten, J.P.; Charlebois, R.L.; Doolittle, W.F.; Papke, R.T. Phylogenetic analyses of cyanobacterial genomes: Quantification of horizontal gene transfer events. Genome Res. 2006, 16, 1099–1108. [Google Scholar] [CrossRef]
- Baroni, M.; Grünewald, S.; Moulton, V.; Semple, C. Bounding the number of hybridisation events for a consistent evolutionary history. J. Math. Biol. 2005, 51, 171–182. [Google Scholar] [CrossRef]
- Szöllosi, G.J.; Boussau, B.; Abby, S.S.; Tannier, E.; Daubin, V. Phylogenetic modeling of lateral gene transfer reconstructs the pattern and relative timing of speciations. Proc. Natl. Acad. Sci. USA 2012, 109, 17513–17518. [Google Scholar] [CrossRef]
- Nelson, K.E.; Clayton, R.A.; Gill, S.R.; Gwinn, M.L.; Dodson, R.J.; Haft, D.H.; Hickey, E.K.; Peterson, J.D.; Nelson, W.C.; Ketchum, K.A.; et al. Evidence for lateral gene transfer between Archaea and bacteria from genome sequence of Thermotoga maritima. Nature 1999, 399, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Clarke, G.D.P.; Beiko, R.G.; Ragan, M.A.; Charlebois, R.L. Inferring genome trees by using a filter to eliminate phylogenetically discordant sequences and a distance matrix based on mean normalized BLASTP scores. J. Bacteriol. 2002, 184, 2072–2080. [Google Scholar] [CrossRef] [PubMed]
- Welch, R.A.; Burland, V.; Plunkett, G.; Redford, P.; Roesch, P.; Rasko, D.; Buckles, E.L.; Liou, S.R.; Boutin, A.; Hackett, J.; et al. Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc. Natl. Acad. Sci. USA 2002, 99, 17020–17024. [Google Scholar] [CrossRef] [PubMed]
- Didelot, X.; Falush, D. Inference of bacterial microevolution using multilocus sequence data. Genetics 2007, 175, 1251–1266. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.; Ekstrom, A.; Li, X.; Yin, Y. HGT-Finder: A New Tool for Horizontal Gene Transfer Finding and Application to Aspergillus genomes. Toxins 2015, 7, 4035–4053. [Google Scholar] [CrossRef]
- Cui, Z.; Wang, S.; Kakar, K.U.; Xie, G.; Li, B.; Chen, G.; Zhu, B. Genome Sequence and Adaptation Analysis of the Human and Rice Pathogenic Strain Burkholderia glumae AU6208. Pathogens 2021, 10, 87. [Google Scholar] [CrossRef]
- Zhu, Q.; Kosoy, M.; Dittmar, K. HGTector: An automated method facilitating genome-wide discovery of putative horizontal gene transfers. BMC Genom. 2014, 15, 717. [Google Scholar] [CrossRef]
- Shimada, S.; Nakai, R.; Aoki, K.; Kudoh, S.; Imura, S.; Shimoeda, N.; Ohno, G.; Watanabe, K.; Miyazaki, Y.; Ishii, Y.; et al. Characterization of the First Cultured Psychrotolerant Representative of Legionella from Antarctica Reveals Its Unique Genome Structure. Microbiol. Spectr. 2021, 9, e0042421. [Google Scholar] [CrossRef]
- Xu, S.; Li, Z.; Huang, Y.; Han, L.; Che, Y.; Hou, X.; Li, D.; Fan, S.; Li, Z. Whole genome sequencing reveals the genomic diversity, taxonomic classification, and evolutionary relationships of the genus Nocardia. PLoS Negl. Trop. Dis. 2021, 15, e0009665. [Google Scholar] [CrossRef]
- Li, X.; Tong, W.; Wang, L.; Rahman, S.U.; Wei, G.; Tao, S. A Novel Strategy for Detecting Recent Horizontal Gene Transfer and Its Application to Rhizobium Strains. Front. Microbiol. 2018, 9, 973. [Google Scholar] [CrossRef]
- Tong, W.; Li, X.; Wang, E.; Cao, Y.; Chen, W.; Tao, S.; Wei, G. Genomic insight into the origins and evolution of symbiosis genes in Phaseolus vulgaris microsymbionts. BMC Genom. 2020, 21, 186. [Google Scholar] [CrossRef]
- Sánchez-Soto, D.; Agüero-Chapin, G.; Armijos-Jaramillo, V.; Perez-Castillo, Y.; Tejera, E.; Antunes, A.; Sánchez-Rodríguez, A. ShadowCaster: Compositional Methods under the Shadow of Phylogenetic Models to Detect Horizontal Gene Transfers in Prokaryotes. Genes 2020, 11, 756. [Google Scholar] [CrossRef]
- Wang, W.B.; Jiang, T.; Gardner, S. Detection of Homologous Recombination Events in Bacterial Genomes. PLoS ONE 2013, 8, e75230. [Google Scholar] [CrossRef]
- Zhang, F.; Hu, Z.; Wu, Z.; Lu, J.; Shi, Y.; Xu, J.; Wang, X.; Wang, J.; Zhang, F.; Wang, M.; et al. Reciprocal adaptation of rice and Xanthomonas oryzae pv. oryzae: Cross-species 2D GWAS reveals the underlying genetics. Plant Cell 2021, 33, 2538–2561. [Google Scholar] [CrossRef]
- Lin, M.; Kussell, E. Inferring bacterial recombination rates from large-scale sequencing datasets. Nat. Methods 2019, 16, 199–204. [Google Scholar] [CrossRef]
- Crits-Christoph, A.; Olm, M.R.; Diamond, S.; Bouma-Gregson, K.; Banfield, J.F. Soil bacterial populations are shaped by recombination and gene-specific selection across a grassland meadow. ISME J. 2020, 14, 1834–1846. [Google Scholar] [CrossRef]
- Cámara, P.G.; Levine, A.J.; Rabadán, R. Inference of Ancestral Recombination Graphs through Topological Data Analysis. PLoS Comput. Biol. 2016, 12, e1005071. [Google Scholar] [CrossRef]
- Koonin, E.V.; Makarova, K.S.; Aravind, L. Horizontal gene transfer in prokaryotes: Quantification and classification. Annu. Rev. Microbiol. 2001, 55, 709–742. [Google Scholar] [CrossRef]
- Zamani-Dahaj, S.A.; Okasha, M.; Kosakowski, J.; Higgs, P.G. Estimating the Frequency of Horizontal Gene Transfer Using Phylogenetic Models of Gene Gain and Loss. Mol. Biol. Evol. 2016, 33, 1843–1857. [Google Scholar] [CrossRef]
- Jeong, H.; Nasir, A. A Preliminary List of Horizontally Transferred Genes in Prokaryotes Determined by Tree Reconstruction and Reconciliation. Front. Genet. 2017, 8, 112. [Google Scholar] [CrossRef]
- Vogan, A.A.; Higgs, P.G. The advantages and disadvantages of horizontal gene transfer and the emergence of the first species. Biol. Direct 2011, 6, 1. [Google Scholar] [CrossRef]
- Narechania, A.; Baker, R.; DeSalle, R.; Mathema, B.; Kolokotronis, S.O.; Kreiswirth, B.; Planet, P.J. Clusterflock: A flocking algorithm for isolating congruent phylogenomic datasets. Gigascience 2016, 5, s13742-016-0152-3. [Google Scholar] [CrossRef]
- Domazet-Lošo, M.; Domazet-Lošo, T. gmos: Rapid Detection of Genome Mosaicism over Short Evolutionary Distances. PLoS ONE 2016, 11, e0166602. [Google Scholar] [CrossRef]
- Wan, Y.; Wick, R.R.; Zobel, J.; Ingle, D.J.; Inouye, M.; Holt, K.E. GeneMates: An R package for detecting horizontal gene co-transfer between bacteria using gene-gene associations controlled for population structure. BMC Genom. 2020, 21, 658. [Google Scholar] [CrossRef] [PubMed]
- Trappe, K.; Marschall, T.; Renard, B.Y. Detecting horizontal gene transfer by mapping sequencing reads across species boundaries. Bioinformatics 2016, 32, i595–i604. [Google Scholar] [CrossRef] [PubMed]
- Bay, R.A.; Bielawski, J.P. Recombination detection under evolutionary scenarios relevant to functional divergence. J. Mol. Evol. 2011, 73, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, Y.J.K.; Johansson, M.; Norberg, P. Revisiting Recombination Signal in the Tick-Borne Encephalitis Virus: A Simulation Approach. PLoS ONE 2016, 11, e0164435. [Google Scholar] [CrossRef]
- Reed, F.A.; Tishkoff, S.A. Positive selection can create false hotspots of recombination. Genetics 2006, 172, 2011–2014. [Google Scholar] [CrossRef][Green Version]
- Sun, S.; Evans, B.J.; Golding, G.B. “Patchy-tachy” leads to false positives for recombination. Mol. Biol. Evol. 2011, 28, 2549–2559. [Google Scholar] [CrossRef]
- Friedman, R.; Ely, B. Codon usage methods for horizontal gene transfer detection generate an abundance of false positive and false negative results. Curr. Microbiol. 2012, 65, 639–642. [Google Scholar] [CrossRef]
- Godfroid, M.; Dagan, T.; Kupczok, A. Recombination Signal in Mycobacterium tuberculosis Stems from Reference-guided Assemblies and Alignment Artefacts. Genome Biol. Evol. 2018, 10, 1920–1926. [Google Scholar] [CrossRef]
- Meyerhans, A.; Vartanian, J.P.; Wain-Hobson, S. DNA recombination during PCR. Nucleic Acids Res. 1990, 18, 1687–1691. [Google Scholar] [CrossRef]
- Zagordi, O.; Klein, R.; Däumer, M.; Beerenwinkel, N. Error correction of next-generation sequencing data and reliable estimation of HIV quasispecies. Nucleic Acids Res. 2010, 38, 7400–7409. [Google Scholar] [CrossRef]
- Posada, D. Evaluation of Methods for Detecting Recombination from DNA Sequences: Empirical Data. Mol. Biol. Evol. 2002, 19, 708–717. [Google Scholar] [CrossRef]
- Brown, T.; Didelot, X.; Wilson, D.J.; Maio, N. De SimBac: Simulation of whole bacterial genomes with homologous recombination. Microb. Genom. 2016, 2, e000044. [Google Scholar] [CrossRef]
- Sipola, A.; Marttinen, P.; Corander, J. Bacmeta: Simulator for genomic evolution in bacterial metapopulations. Bioinformatics 2018, 34, 2308–2310. [Google Scholar] [CrossRef]
- Bobay, L.M. CoreSimul: A forward-in-time simulator of genome evolution for prokaryotes modeling homologous recombination. BMC Bioinform. 2020, 21, 1–7. [Google Scholar] [CrossRef]
- Song, W.; Steensen, K.; Thomas, T. HgtSIM: A simulator for horizontal gene transfer (HGT) in microbial communities. PeerJ 2017, 5, e4015. [Google Scholar] [CrossRef]
- Cury, J.; Haller, B.C.; Achaz, G.; Jay, F. Simulation of bacterial populations with SLiM. Peer Community J. 2022, 2, e7. [Google Scholar] [CrossRef]
- Marttinen, P.; Hanage, W.P.; Croucher, N.J.; Connor, T.R.; Harris, S.R.; Bentley, S.D.; Corander, J. Detection of recombination events in bacterial genomes from large population samples. Nucleic Acids Res. 2012, 40, 1–12. [Google Scholar] [CrossRef]
- Lehtinen, S.; Lehtinen, S.; Chewapreecha, C.; Chewapreecha, C.; Chewapreecha, C.; Lees, J.; Hanage, W.P.; Lipsitch, M.; Croucher, N.J.; Bentley, S.D.; et al. Horizontal gene transfer rate is not the primary determinant of observed antibiotic resistance frequencies in Streptococcus pneumonia. Sci. Adv. 2020, 6, eaaz6137. [Google Scholar] [CrossRef]


{kind=link}
| Tool | Applied Approach | Method’s Class | Input | Output | Detected Events | Advantages | Limitations | References |
|---|---|---|---|---|---|---|---|---|
| Homologous Recombination (HR) Identification | ||||||||
| RDP4/RDP5 | Combination of phylogenetic and distance methods | Phylogenetic and distance-based | Alignments in FASTA format | Recombination events with phylogenetic relationships and breakpoints coordinates for chimeric sequences in tabular format | Recent | Robustness and providing the information on the direction of exchanges | Inability to reveal distant events and high computational costs | [66,67] |
| Gubbins | Revealing increased substitution rate among ML-tree branches | Phylogenetic | Alignments in FASTA format | Coordinates of recombination events tabular format and their visualization on the genome alignment | Recent and ancestral | Precise reconstruction of ancestral state | High computational costs and possible false-positive results when analyzing trees with short branches (theoretically) | [44] |
| ClonalFrameML | Maximal likelihood-based clonal model | Phylogenetic | Alignments in FASTA format and guiding tree | Phylogeny regarding recombination and visualization of events’ coordinates on the genome alignment in tabular format | Recent and ancestral | Computational effectiveness | Underestimation of recombination rate in datasets with intensive recombination | [73] |
| BratNextGen | Bayesian modeling | Substitution distribution | Alignments in FASTA format | Coordinates of the events in tabular format and visualization of transmitted regions on the genome alignment | Recent and ancestral | Computational effectiveness | False-negative results in the case of mosaic sequences with multiple recombination events | [126] |
| fastGEAR | HMM algorithms coupled with Bayesian clustering | Substitution distribution | Alignments in FASTA format | Coordinates of ancestral and recent recombination events in tabular format | Recent and ancestral | Computational effectiveness, high sensitivity, and handling of missing data | Missing events between closely related species | [10] |
| ptACR | Genome-wise average SNP compatibility calculation | Compatibility | Gap-free alignments in PHYLIP format | Genomic coordinates of recombination events in tabular format | Recent | High accuracy and robustness to false-positive results | Inability to process alignments with gaps and high false-negative rate when processing divergent sequences | [40] |
| HREfinder | Genome partitioning into SNP-flanked blocks | Substitution distribution | Genomes in FASTA format, tree in Newick format, and SNP list in tabular format | List of sequences subjected to recombination in tabular format | Recent | High accuracy | High false-negative rate when processing divergent sequences | [99] |
| mcorr | Building correlation profile of synonymous substitution | Parametric | Alignments in XMFA or BAM formats | Tables and figures depicting the average recombination rate | The total rate of recent/ancient events | The ability to process raw reads and metagenomic data | Has not been compared to conventional r/m rate calculating tools | [101] |
| Bacter | Markov chain Monte Carlo (MCMC) | ARG | Alignments in FASTA format | Ancestral recombination graph (ARG) in Newick format | Recent | Improved detection of the events in the case of poor phylogenetic signal | Dependence on predetermined parameters and high computational costs | [35] |
| TARGet | Topological data analysis (TDA) | ARG | Alignments in FASTA format without gaps or segregating sites denoted by 1 and 0 | Ancestral recombination graph (ARG) in XML format and positions of reticulate events | Recent | Computational effectiveness | Inability to process alignments with gaps | [103] |
| Horizontal Gene Transfer (HGT) Detection | ||||||||
| Clusterflock | Self-organizing flock algorithm | Parametric | Sequences and a distance matrix | Clusters of sequences in tabular format | Recent | Applicability to any distance metrics and resilience to missing data | Has not been compared to the existing tools | [108] |
| gmos | Pairwise local alignments with subsequent regions overlapping | Parametric | Query and subject genomes in FASTA format | Structural variants in FASTA format | Recent | Computational effectiveness and the ability to reveal both HR and HGT | Depends heavily on the high similarity between transferred regions | [109] |
| GeneMates | Association tests with the linear-mixed model accounting for population structure | Parametric | Genome assemblies in FASTA format and raw reads in FASTQ format | The linkage network of horizontally co-transferred alleles in tabular format | Recent | Resolving co-occurred HGT events | Reduced sensitivity due to the dependence on core SNPs | [110] |
| ShadowCaster | Support vector machine-based hybrid approach | Implicit phylogenetic and parametric | A query genome and proteome and list of related proteomes in FASTA format | The list of HGT candidates with corresponding likelihood calculations in tabular format | Recent and ancestral | High sensitivity when reveling both recent and ancient events and reduced false-positive rate | Does not determine the directions of transfers and processes only a single genome | [98] |
| nearHGT | Calculating synteny index (SI) followed by constant relative mutability (CRM) measurement | Synteny-based and parametric | Reference and putatively transferred sequences in FASTA format | Chi-square-based p-value denoting the probability of HGT | Recent | High sensitivity | No ready-made application is available | [49] |
| HGT-Finder | Similarity ratio evaluation for proteins according to BLAST hits and taxonomic distance calculation based on the NCBI Taxonomy annotation | Implicit phylogenetic | The BLAST search result and the NCBI Taxonomy database | Tabular format file with the transfer index value for a protein | Recent | Detecting mostly true events | High reliance on the taxonomic nomenclature and low sensitivity | [91] |
| HGTector | Analyzing BLAST hit distribution patterns according to predefined evolutionary categories | Implicit phylogenetic | FASTA files of amino acid sequences for each analyzed genome | List of candidate HGT-derived genes with the respective silhouette scores in tabular format | Recent | Insensitive to gene loss, rate variations, and database errors | High reliance on the taxonomic nomenclature and low sensitivity | [93] |
| RecentHGT | The expectation-maximization algorithm based on the sequence-similarity distribution of orthologous genes | Implicit phylogenetic | Tabular file with strains information and RAST-annotated GenBank file | Putative HGT events in chromosomal and plasmid regions in tabular format | Recent | Reduced false-positive rate when processing conserved genes | Missing events when analyzing divergent sequences | [96] |
| Daisy | Mapping-based detection relying on short read pairs and coverage information | Parametric | Reads from the analyzed organism and poposed acceptor and donor genomes in FASTA format | A variant call format (VCF) file reporting HGT candidates meeting the predefined threshold and tabular format file with all potential events | Recent | Outperforms reference genome-based approaches if short reads are available | Requires short reads only and explicit specifying recipient and donor genomes | [11] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shikov, A.E.; Malovichko, Y.V.; Nizhnikov, A.A.; Antonets, K.S. Current Methods for Recombination Detection in Bacteria. Int. J. Mol. Sci. 2022, 23, 6257. https://doi.org/10.3390/ijms23116257
Shikov AE, Malovichko YV, Nizhnikov AA, Antonets KS. Current Methods for Recombination Detection in Bacteria. International Journal of Molecular Sciences. 2022; 23(11):6257. https://doi.org/10.3390/ijms23116257
Chicago/Turabian StyleShikov, Anton E., Yury V. Malovichko, Anton A. Nizhnikov, and Kirill S. Antonets. 2022. "Current Methods for Recombination Detection in Bacteria" International Journal of Molecular Sciences 23, no. 11: 6257. https://doi.org/10.3390/ijms23116257
APA StyleShikov, A. E., Malovichko, Y. V., Nizhnikov, A. A., & Antonets, K. S. (2022). Current Methods for Recombination Detection in Bacteria. International Journal of Molecular Sciences, 23(11), 6257. https://doi.org/10.3390/ijms23116257