
Multi-Cohort Transcriptomic Subtyping of B-Cell Acute Lymphoblastic Leukemia

 , , ,
, , , 
Abstract
:1. Introduction
2. Results
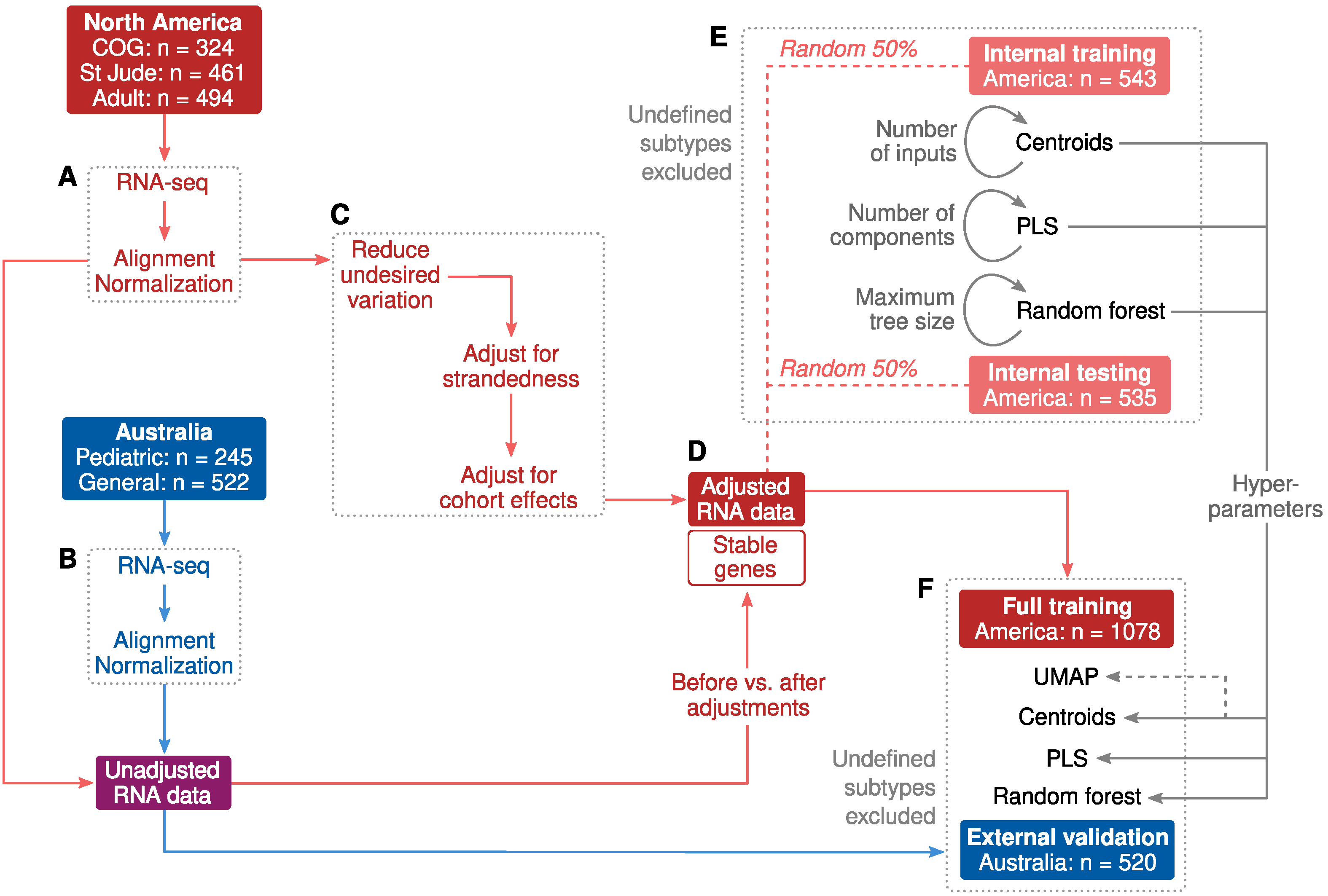
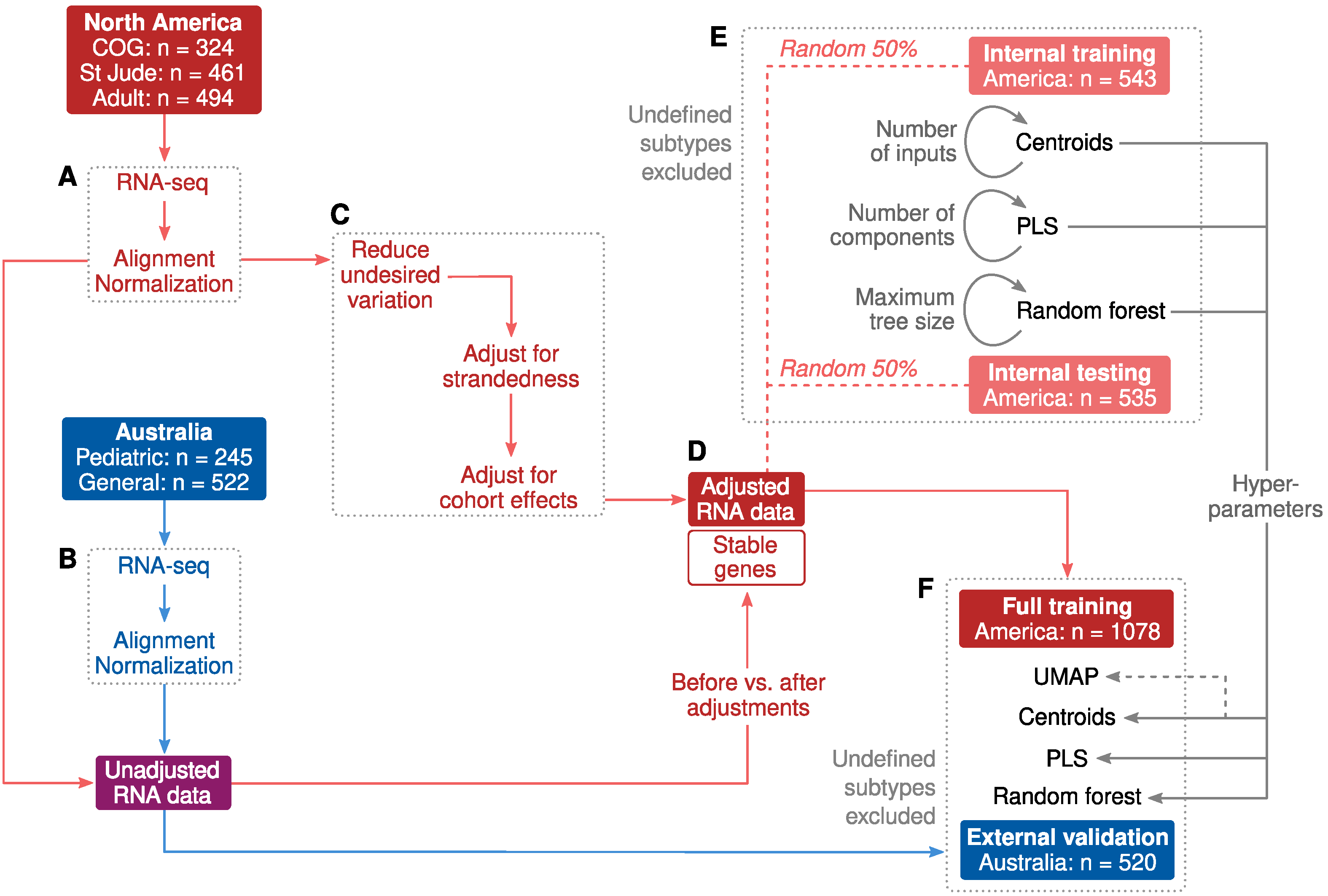
2.1. Cohort Characteristics and Study Design
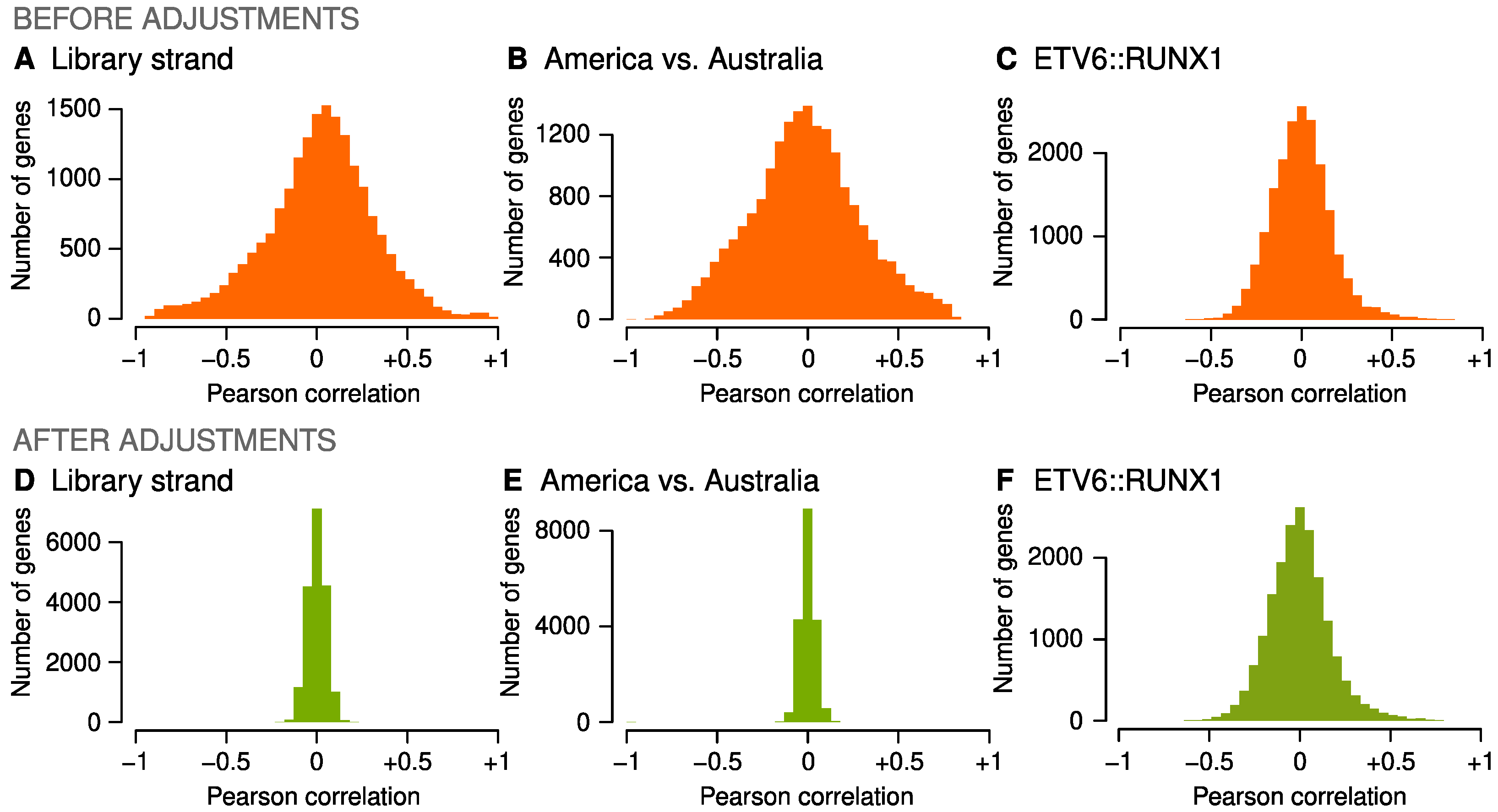
2.2. Adjustments for Confounders
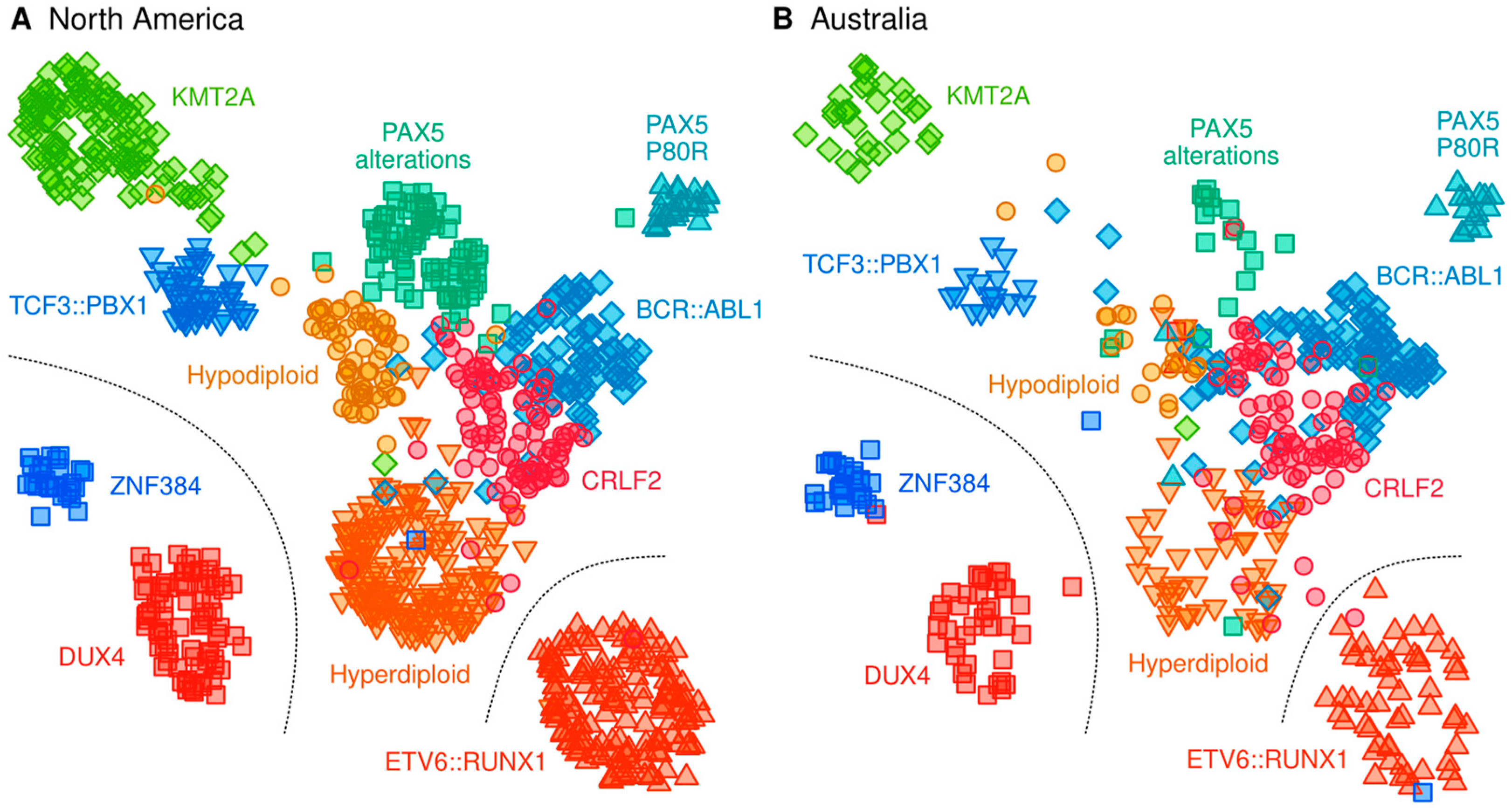
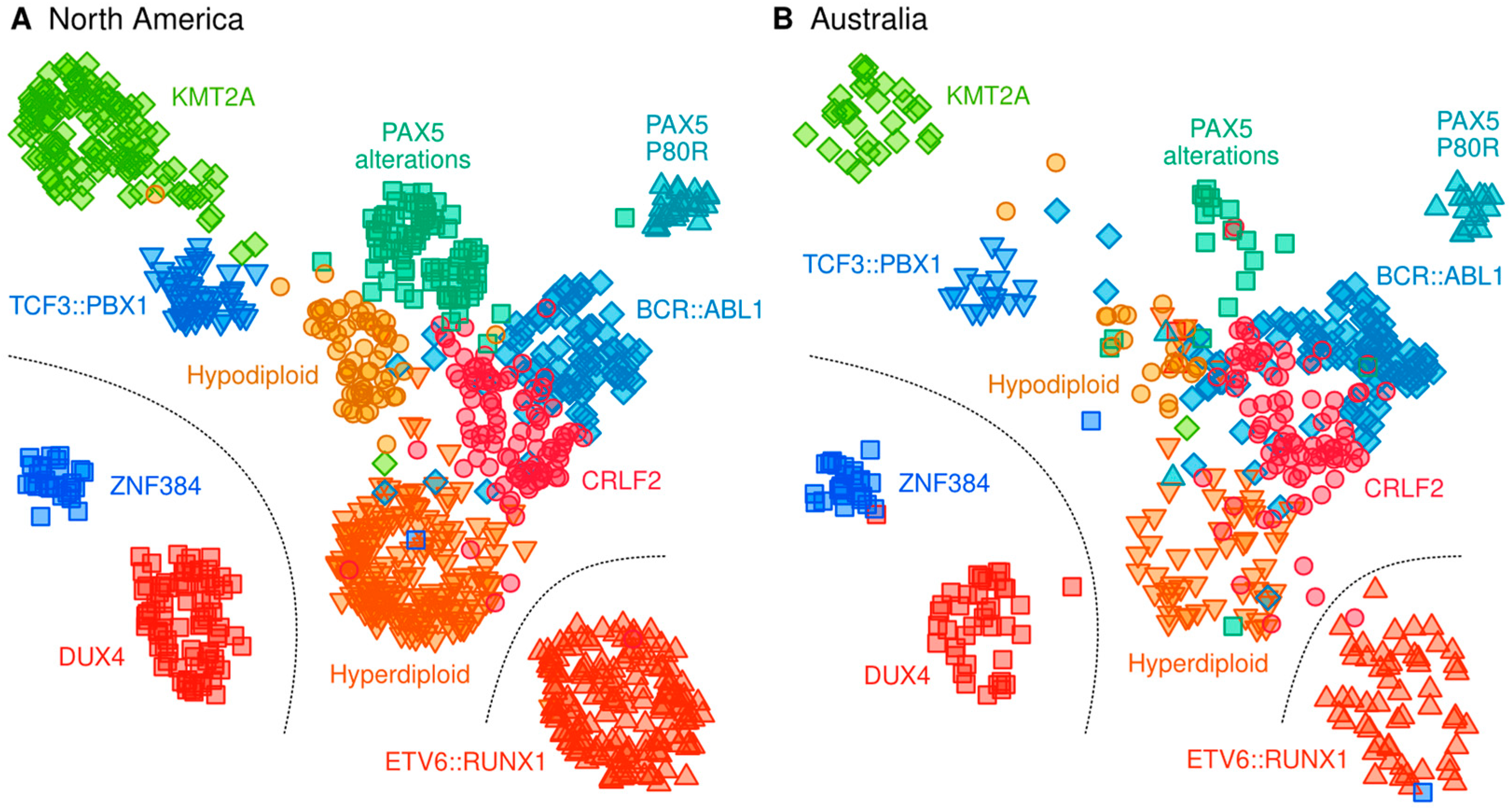
2.3. Classification of B-Cell ALL Subtypes Based on RNA-Seq Profiling
2.4. Allspice Classifier
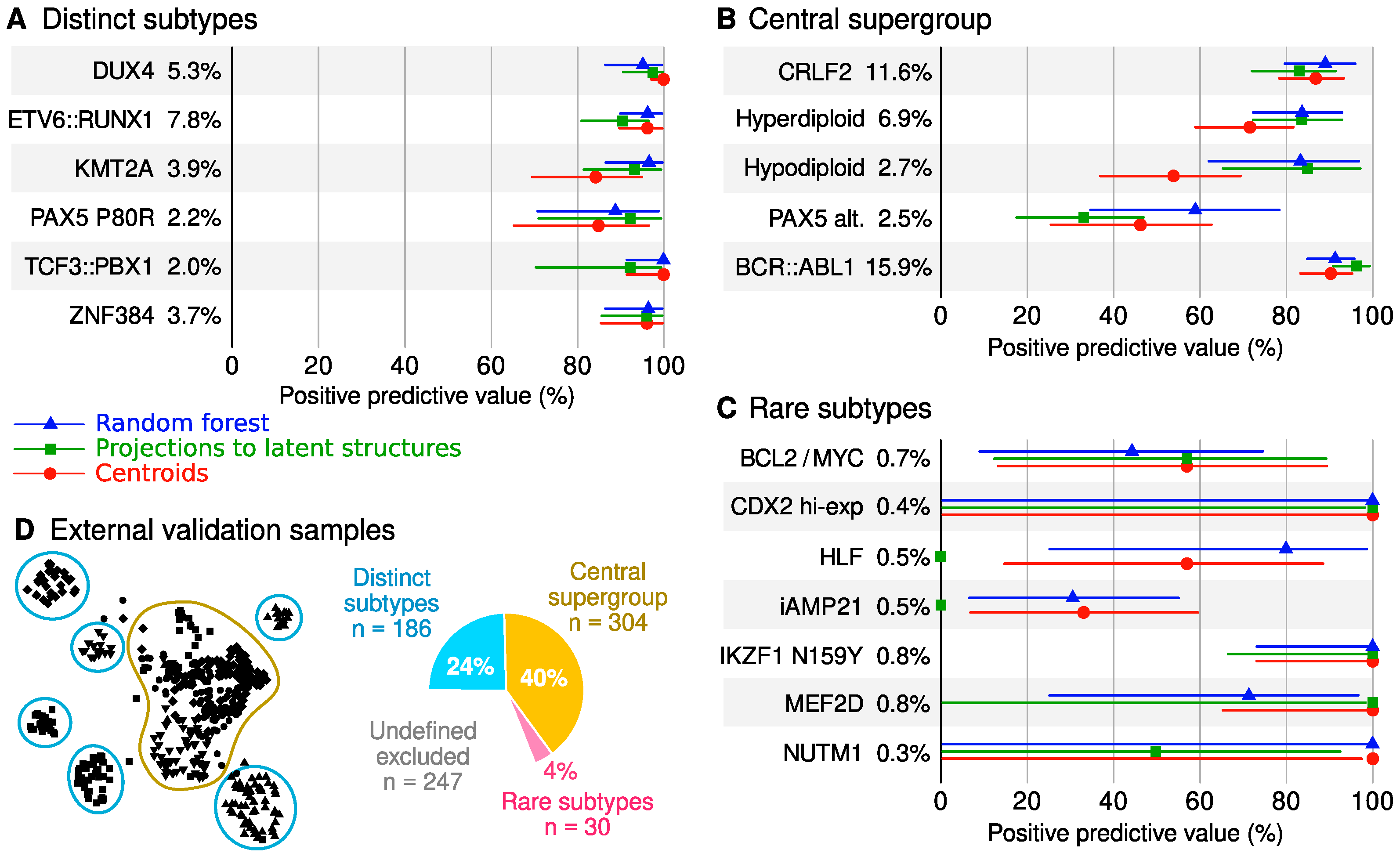
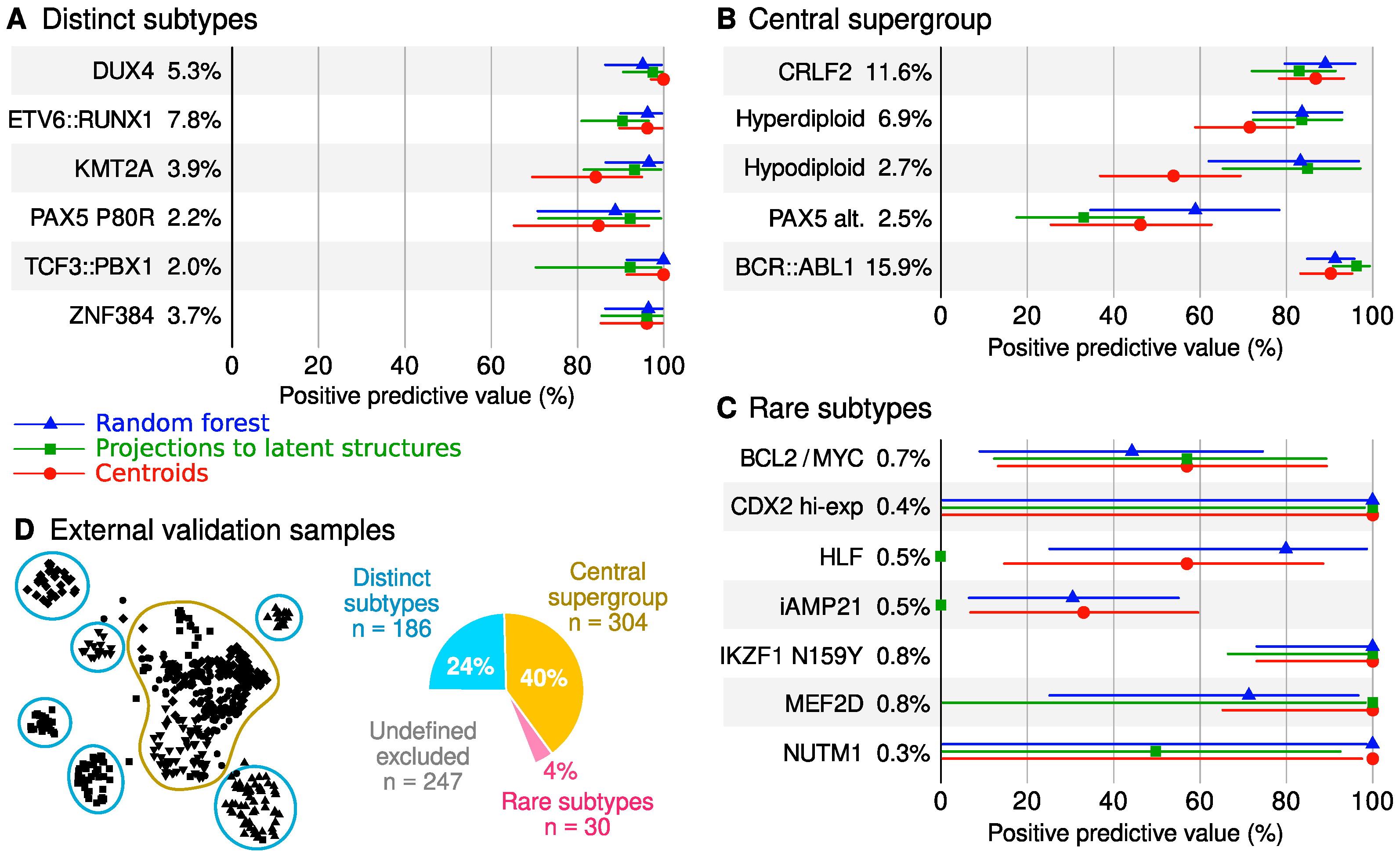
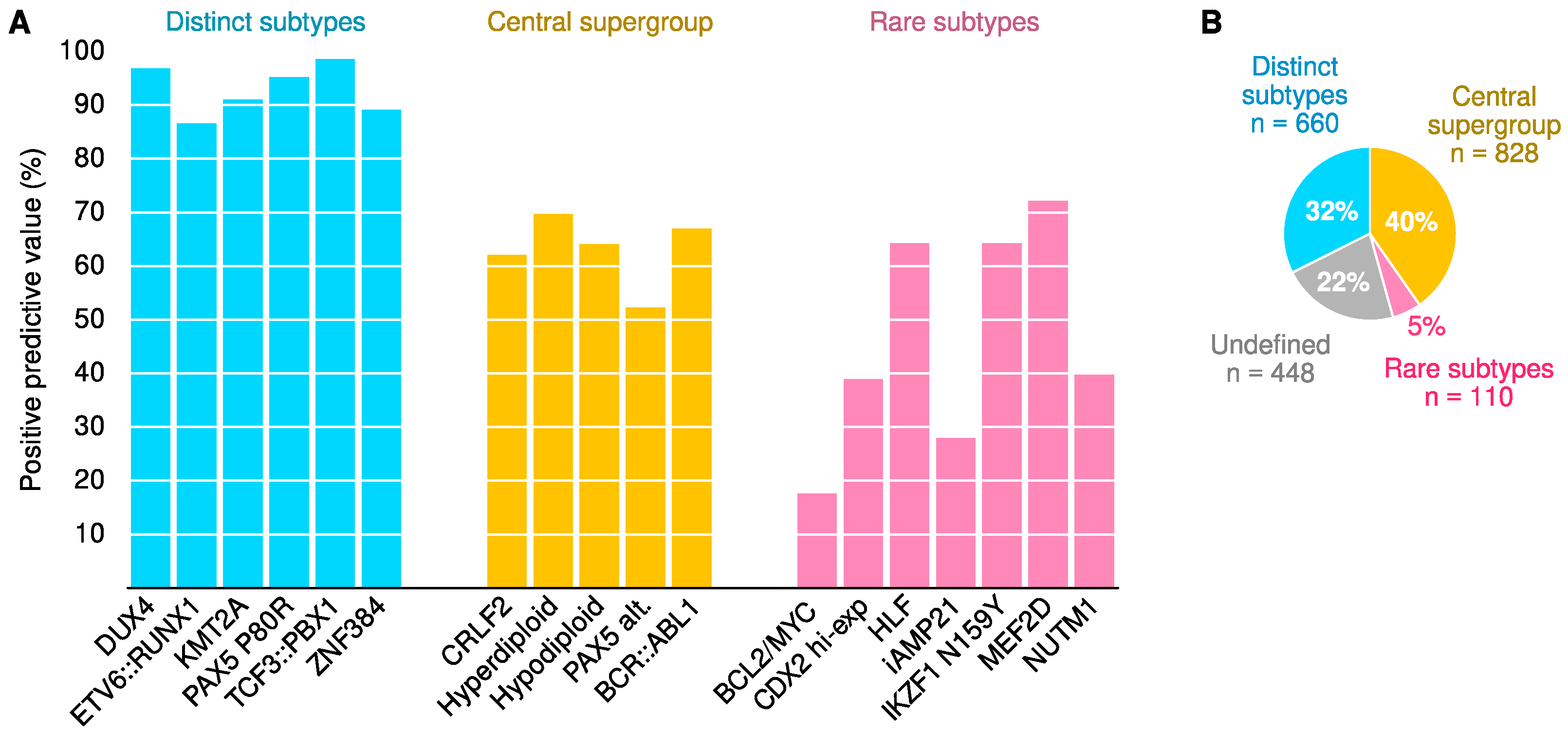
2.5. Classifier Performance
3. Discussion
3.1. Definition of ALL Subtypes
3.2. Classification Performance and Utility
3.3. Strengths and Weaknesses
3.4. Practical Considerations
4. Materials and Methods
4.1. North American Participants
4.2. Australian Participants
4.3. Supporting RNA Data
4.4. RNA Sequencing and Pre-Processing
4.5. Genomic Subtyping
4.6. Adjustments for Confounders
4.7. Machine Learning
4.8. Pruning of Correlated Input Features
4.9. Training, Testing, External Validation and Performance Metrics
4.10. Proximity and Exclusivity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Terwilliger, T.; Abdul-Hay, M. Acute lymphoblastic leukemia: A comprehensive review and 2017 update. Blood Cancer J. 2017, 7, e577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunger, S.P.; Mullighan, C.G. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, M.; Zhou, L.; Li, A.; Luo, S.; Wu, K. Global burden and trend of acute lymphoblastic leukemia from 1990 to 2017. Aging 2020, 12, 22869–22891. [Google Scholar] [CrossRef] [PubMed]
- Cario, G.; Leoni, V.; Conter, V.; Attarbaschi, A.; Zaliova, M.; Sramkova, L.; Cazzaniga, G.; Fazio, G.; Sutton, R.; Elitzur, S.; et al. Relapses and treatment-related events contributed equally to poor prognosis in children with ABL-class fusion positive B-cell acute lymphoblastic leukemia treated according to AIEOP-BFM protocols. Haematologica 2020, 105, 1887–1894. [Google Scholar] [CrossRef]
- Passet, M.; Boissel, N.; Sigaux, F.; Saillard, C.; Bargetzi, M.; Ba, I.; Thomas, X.; Graux, C.; Chalandon, Y.; Leguay, T.; et al. PAX5 P80R mutation identifies a novel subtype of B-cell precursor acute lymphoblastic leukemia with favorable outcome. Blood 2019, 133, 280–284. [Google Scholar] [CrossRef]
- Samra, B.; Jabbour, E.; Ravandi, F.; Kantarjian, H.; Short, N.J. Evolving therapy of adult acute lymphoblastic leukemia: State-of-the-art treatment and future directions. J. Hematol. Oncol. 2020, 13, 70. [Google Scholar] [CrossRef]
- Karol, S.E.; Pui, C.-H. Personalized therapy in pediatric high-risk B-cell acute lymphoblastic leukemia. Ther. Adv. Hematol. 2020, 11, 204062072092757. [Google Scholar] [CrossRef]
- Moorman, A.V.; Chilton, L.; Wilkinson, J.; Ensor, H.M.; Bown, N.; Proctor, S.J. A population-based cytogenetic study of adults with acute lymphoblastic leukemia. Blood 2010, 115, 206–214. [Google Scholar] [CrossRef]
- Chiaretti, S.; Zini, G.; Bassan, R. Diagnosis and subclassification of acute lymphoblastic leukemia. Mediterr. J. Hematol. Infect. Dis. 2014, 6, e2014073. [Google Scholar] [CrossRef] [Green Version]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Brown, L.M.; Lonsdale, A.; Zhu, A.; Davidson, N.M.; Schmidt, B.; Hawkins, A.; Wallach, E.; Martin, M.; Mechinaud, F.M.; Khaw, S.L.; et al. The application of RNA sequencing for the diagnosis and genomic classification of pediatric acute lymphoblastic leukemia. Blood Adv. 2020, 4, 930–942. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Churchman, M.L.; Roberts, K.G.; Moore, I.; Zhou, X.; Nakitandwe, J.; Hagiwara, K.; Pelletier, S.; Gingras, S.; Berns, H.; et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat. Genet. 2019, 51, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, R.; Ribera, J.; Morgades, M.; Ayala, R.; Onecha, E.; Ruiz-Heredia, Y.; Juárez-Rufián, A.; de Nicolás, R.; Sánchez-Pina, J.; Vives, S.; et al. A novel targeted RNA-Seq panel identifies a subset of adult patients with acute lymphoblastic leukemia with BCR-ABL1-like characteristics. Blood Cancer J. 2020, 10, 43. [Google Scholar] [CrossRef] [PubMed]
- Marincevic-Zuniga, Y.; Dahlberg, J.; Nilsson, S.; Raine, A.; Nystedt, S.; Lindqvist, C.M.; Berglund, E.C.; Abrahamsson, J.; Cavelier, L.; Forestier, E.; et al. Transcriptome sequencing in pediatric acute lymphoblastic leukemia identifies fusion genes associated with distinct DNA methylation profiles. J. Hematol. Oncol. 2017, 10, 148. [Google Scholar] [CrossRef] [Green Version]
- Li, J.-F.; Dai, Y.-T.; Lilljebjörn, H.; Shen, S.-H.; Cui, B.-W.; Bai, L.; Liu, Y.-F.; Qian, M.-X.; Kubota, Y.; Kiyoi, H.; et al. Transcriptional landscape of B cell precursor acute lymphoblastic leukemia based on an international study of 1223 cases. Proc. Natl. Acad. Sci. USA 2018, 115, E11711–E11720. [Google Scholar] [CrossRef] [Green Version]
- Braoudaki, M.; Tzortzatou-Stathopoulou, F. Clinical Cytogenetics in Pediatric Acute Leukemia: An Update. Clin. Lymphoma Myeloma Leuk. 2012, 12, 230–237. [Google Scholar] [CrossRef]
- Rehn, J.A.; O’Connor, M.J.; White, D.L.; Yeung, D.T. DUX Hunting-Clinical Features and Diagnostic Challenges Associated with DUX4-Rearranged Leukaemia. Cancers 2020, 12, 2815. [Google Scholar] [CrossRef]
- Lee, J.; Cho, S.; Hong, S.-E.; Kang, D.; Choi, H.; Lee, J.-M.; Yoon, J.-H.; Cho, B.-S.; Lee, S.; Kim, H.-J.; et al. Integrative Analysis of Gene Expression Data by RNA Sequencing for Differential Diagnosis of Acute Leukemia: Potential Application of Machine Learning. Front. Oncol. 2021, 11, 717616. [Google Scholar] [CrossRef]
- Walter, W.; Shahswar, R.; Stengel, A.; Meggendorfer, M.; Kern, W.; Haferlach, T.; Haferlach, C. Clinical application of whole transcriptome sequencing for the classification of patients with acute lymphoblastic leukemia. BMC Cancer 2021, 21, 886. [Google Scholar] [CrossRef]
- Pui, C.-H. Precision medicine in acute lymphoblastic leukemia. Front. Med. 2020, 14, 689–700. [Google Scholar] [CrossRef]
- Schmidt, B.; Brown, L.M.; Ryland, G.L.; Lonsdale, A.; Kosasih, H.J.; Ludlow, L.E.; Majewski, I.J.; Blombery, P.; Ekert, P.G.; Davidson, N.M.; et al. ALLSorts: A RNA-Seq Classifier for B-Cell Acute Lymphoblastic Leukemia; Bioinformatics: Bethesda, MD, USA, 2021. [Google Scholar]
- Gianfrancesco, M.A.; Tamang, S.; Yazdany, J.; Schmajuk, G. Potential Biases in Machine Learning Algorithms Using Electronic Health Record Data. JAMA Intern. Med. 2018, 178, 1544. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, E.; Retico, A.; Bacciu, D. Measuring the effects of confounders in medical supervised classification problems: The Confounding Index (CI). Artif. Intell. Med. 2020, 103, 101804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skelly, A.C.; Dettori, J.R.; Brodt, E.D. Assessing bias: The importance of considering confounding. Evid.-Based Spine-Care J. 2012, 3, 9–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boer, J.M.; Marchante, J.R.M.; Evans, W.E.; Horstmann, M.A.; Escherich, G.; Pieters, R.; Den Boer, M.L. BCR-ABL1-like cases in pediatric acute lymphoblastic leukemia: A comparison between DCOG/Erasmus MC and COG/St. Jude signatures. Haematologica 2015, 100, e354–e357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, K.G. The biology of Philadelphia chromosome-like ALL. Best Pract. Res. Clin. Haematol. 2017, 30, 212–221. [Google Scholar] [CrossRef]
- Harvey, R.C.; Kang, H.; Roberts, K.G.; Chen, I.-M.L.; Atlas, S.R.; Bedrick, E.J.; Gastier-Foster, J.M.; Zhang, J.; Gerhard, D.S.; Smith, M.A.; et al. Development and Validation Of a Highly Sensitive and Specific Gene Expression Classifier To Prospectively Screen and Identify B-Precursor Acute Lymphoblastic Leukemia (ALL) Patients With a Philadelphia Chromosome-Like (“Ph-like” or “BCR-ABL1-Like”) Signature For Therapeutic Targeting and Clinical Intervention. Blood 2013, 122, 826. [Google Scholar] [CrossRef]
- Kashef, A.; Khatibi, T.; Mehrvar, A. Treatment outcome classification of pediatric Acute Lymphoblastic Leukemia patients with clinical and medical data using machine learning: A case study at MAHAK hospital. Inform. Med. Unlocked 2020, 20, 100399. [Google Scholar] [CrossRef]
- Pan, L.; Liu, G.; Lin, F.; Zhong, S.; Xia, H.; Sun, X.; Liang, H. Machine learning applications for prediction of relapse in childhood acute lymphoblastic leukemia. Sci. Rep. 2017, 7, 7402. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Wang, L.; Liu, Q.; Tárnok, A.; Su, X. Deep learning-based light scattering microfluidic cytometry for label-free acute lymphocytic leukemia classification. Biomed. Opt. Express 2020, 11, 6674. [Google Scholar] [CrossRef]
- Reddy, S.; Rogers, W.; Makinen, V.-P.; Coiera, E.; Brown, P.; Wenzel, M.; Weicken, E.; Ansari, S.; Mathur, P.; Casey, A.; et al. Evaluation framework to guide implementation of AI systems into healthcare settings. BMJ Health Care Inform. 2021, 28, e100444. [Google Scholar] [CrossRef]
- Van der Maaten, L.; Hinton, G. Viualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- The Geuvadis Consortium; Lappalainen, T.; Sammeth, M.; Friedländer, M.R.; Hoen, P.A.C.; Monlong, J.; Rivas, M.A.; Gonzàlez-Porta, M.; Kurbatova, N.; Griebel, T.; et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature 2013, 501, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Gingeras, T.R. Mapping RNA-seq Reads with STAR. Curr. Protoc. Bioinform. 2015, 51, 11.14.1–11.14.19. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinforma. Oxf. Engl. 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, T.; Sanada, M.; Kawazu, M.; Kojima, S.; Tsuzuki, S.; Ueno, H.; Iwamoto, E.; Iijima-Yamashita, Y.; Yamada, T.; Kanamori, T.; et al. Two novel high-risk adult B-cell acute lymphoblastic leukemia subtypes with high expression of CDX2 and IDH1/2 mutations. Blood 2021, 139, 1850–1862. [Google Scholar] [CrossRef]
- Nicorici, D.; Satalan, M.; Edgren, H.; Kangaspeska, S.; Murumagi, A.; Kallioniemi, O.; Virtanen, S.; Kilkku, O. FusionCatcher—A Tool for Finding Somatic Fusion Genes in Paired-End RNA-Sequencing Data; Bioinformatics: Bethesda, MD, USA, 2014. [Google Scholar]
- Jia, W.; Qiu, K.; He, M.; Song, P.; Zhou, Q.; Zhou, F.; Yu, Y.; Zhu, D.; Nickerson, M.L.; Wan, S.; et al. SOAPfuse: An algorithm for identifying fusion transcripts from paired-end RNA-Seq data. Genome Biol. 2013, 14, R12. [Google Scholar] [CrossRef] [Green Version]
- Davidson, N.M.; Majewski, I.J.; Oshlack, A. JAFFA: High sensitivity transcriptome-focused fusion gene detection. Genome Med. 2015, 7, 43. [Google Scholar] [CrossRef] [Green Version]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [Green Version]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.J.; Carneiro, M.O.; Van der Auwera, G.A.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling Accurate Genetic Variant Discovery to Tens of Thousands of Samples; Genomics: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Leek, J.T.; Storey, J.D. Capturing Heterogeneity in Gene Expression Studies by Surrogate Variable Analysis. PLoS Genet. 2007, 3, e161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, S.; Mutter, S.; Casey, A.; Mäkinen, V.-P. Numero: A statistical framework to define multivariable subgroups in complex population-based datasets. Int. J. Epidemiol. 2019, 48, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Trygg, J.; Wold, S. Orthogonal projections to latent structures (O-PLS). J. Chemom. 2002, 16, 119–128. [Google Scholar] [CrossRef]
- McInnes, L.; Healy, J.; Saul, N.; Großberger, L. UMAP: Uniform Manifold Approximation and Projection. J. Open Source Softw. 2018, 3, 861. [Google Scholar] [CrossRef]
- Mäkinen, V.-P.; Civelek, M.; Meng, Q.; Zhang, B.; Zhu, J.; Levian, C.; Huan, T.; Segrè, A.V.; Ghosh, S.; Vivar, J.; et al. Integrative Genomics Reveals Novel Molecular Pathways and Gene Networks for Coronary Artery Disease. PLoS Genet. 2014, 10, e1004502. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pediatric Cohorts | Adult and General Cohorts | ||||||
|---|---|---|---|---|---|---|---|
| COG | St Jude | Australia | p-Value | Multiple * | Australia | p-Value | |
| Male | 177 | 221 | 83 | 4.4 × 10−6 | 251 | 304 | 0.021 |
| Female | 120 | 188 | 73 | 0.016 | 233 | 205 | 0.013 |
| Unknown | 27 | 52 | 89 | 1.8 × 10−21 | 10 | 13 | 0.77 |
| Age (years) | 9.2 ± 5.7 | 6.4 ± 4.4 | 7.4 ± 4.0 | 7.6 × 10−11 | 45.1 ± 14.8 | 35.7 ± 22.6 | 3.6 × 10−13 |
| BCL2/MYC | 0.0 | 0.4 | 0.4 | 0.50 | 2.8 | 0.8 | 0.024 |
| CDX2 hi-exp | 0.3 | 0.0 | 0.0 | 0.34 | 0.6 | 0.6 | 1.0 |
| CRLF2 | 7.4 | 3.0 | 18.4 | 8.4 × 10−12 | 13.2 | 8.4 | 0.020 |
| DUX4 | 7.4 | 7.4 | 2.0 | 0.0096 | 4.9 | 6.9 | 0.21 |
| ETV6::RUNX1 | 9.9 | 26.7 | 17.6 | 2.3 × 10−8 | 1.0 | 2.3 | 0.18 |
| HLF | 0.0 | 0.4 | 0.4 | 0.50 | 0.6 | 0.6 | 1.0 |
| Hyperdiploid | 21.0 | 21.3 | 9.8 | 0.00034 | 4.0 | 5.6 | 0.27 |
| Hypodiploid | 1.2 | 0.7 | 0.8 | 0.68 | 11.5 | 3.6 | 3.1 × 10−6 |
| iAMP21 | 6.5 | 0.4 | 0.8 | 0.72 × 10−8 | 0.2 | 0.4 | 1.0 |
| IKZF1 N159Y | 0.3 | 0.0 | 1.2 | 0.043 | 0.4 | 0.6 | 1.0 |
| KMT2A | 2.2 | 10.8 | 2.9 | 2.0 × 10−7 | 13.8 | 4.4 | 3.2 × 10−7 |
| MEF2D | 2.2 | 1.1 | 0.0 | 0.058 | 2.0 | 1.1 | 0.39 |
| NUTM1 | 0.3 | 1.1 | 0.4 | 0.36 | 0.0 | 0.2 | 1.0 |
| PAX5 Alt | 4.0 | 5.9 | 3.7 | 0.32 | 8.3 | 1.9 | 6.4 × 10−6 |
| PAX5 P80R | 0.9 | 1.1 | 0.8 | 0.94 | 3.4 | 2.9 | 0.74 |
| BCR::ABL1 | 5.6 | 2.4 | 4.1 | 0.070 | 12.3 | 21.5 | 1.6 × 10−4 |
| TCF3::PBX1 | 2.2 | 6.7 | 1.2 | 0.00023 | 3.0 | 2.3 | 0.59 |
| ZNF384 | 1.9 | 2.0 | 2.0 | 0.99 | 2.8 | 4.4 | 0.24 |
| Undefined | 26.9 | 8.7 | 33.5 | 1.7 × 10−16 | 15.0 | 31.6 | 7.0 × 10−9 |
| All Samples | Defined Subtypes | Defined Subtypes and QC Pass | |
|---|---|---|---|
| Number of samples | 2046 | 1598 | 1292 |
| DUX4 (%) | 96.8 | 100.0 | 100.0 |
| ETV6::RUNX1 (%) | 86.6 | 97.7 | 98.6 |
| KMT2A (%) | 91.1 | 96.6 | 100.0 |
| PAX5 P80R (%) | 95.2 | 97.6 | 100.0 |
| TCF3::PBX1 (%) | 98.5 | 98.5 | 100.0 |
| ZNF384 (%) | 89.1 | 96.6 | 100.0 |
| CRLF2 (%) | 62.0 | 91.2 | 97.6 |
| Hyperdiploid (%) | 69.8 | 92.1 | 99.0 |
| Hypodiploid (%) | 64.2 | 79.0 | 89.2 |
| PAX5 Alt (%) | 52.1 | 74.8 | 92.7 |
| BCR::ABL1 (%) | 67.2 | 84.8 | 96.6 |
| BCL2/MYC (%) | 17.5 | 54.1 | 66.7 |
| CDX2 hi-exp (%) | 38.9 | 77.8 | 100.0 |
| HLF (%) | 64.3 | 75.0 | 90.0 |
| iAMP21 (%) | 27.9 | 66.7 | 100.0 |
| IKZF1 N159Y (%) | 64.3 | 90.0 | 100.0 |
| MEF2D (%) | 72.2 | 78.8 | 100.0 |
| NUTM1 (%) | 40.0 | 66.7 | 100.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mäkinen, V.-P.; Rehn, J.; Breen, J.; Yeung, D.; White, D.L. Multi-Cohort Transcriptomic Subtyping of B-Cell Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2022, 23, 4574. https://doi.org/10.3390/ijms23094574
Mäkinen V-P, Rehn J, Breen J, Yeung D, White DL. Multi-Cohort Transcriptomic Subtyping of B-Cell Acute Lymphoblastic Leukemia. International Journal of Molecular Sciences. 2022; 23(9):4574. https://doi.org/10.3390/ijms23094574
Chicago/Turabian StyleMäkinen, Ville-Petteri, Jacqueline Rehn, James Breen, David Yeung, and Deborah L. White. 2022. "Multi-Cohort Transcriptomic Subtyping of B-Cell Acute Lymphoblastic Leukemia" International Journal of Molecular Sciences 23, no. 9: 4574. https://doi.org/10.3390/ijms23094574
APA StyleMäkinen, V.-P., Rehn, J., Breen, J., Yeung, D., & White, D. L. (2022). Multi-Cohort Transcriptomic Subtyping of B-Cell Acute Lymphoblastic Leukemia. International Journal of Molecular Sciences, 23(9), 4574. https://doi.org/10.3390/ijms23094574