
Atomic-Resolution Structures and Mode of Action of Clinically Relevant Antimicrobial Peptides




Abstract
1. Introduction
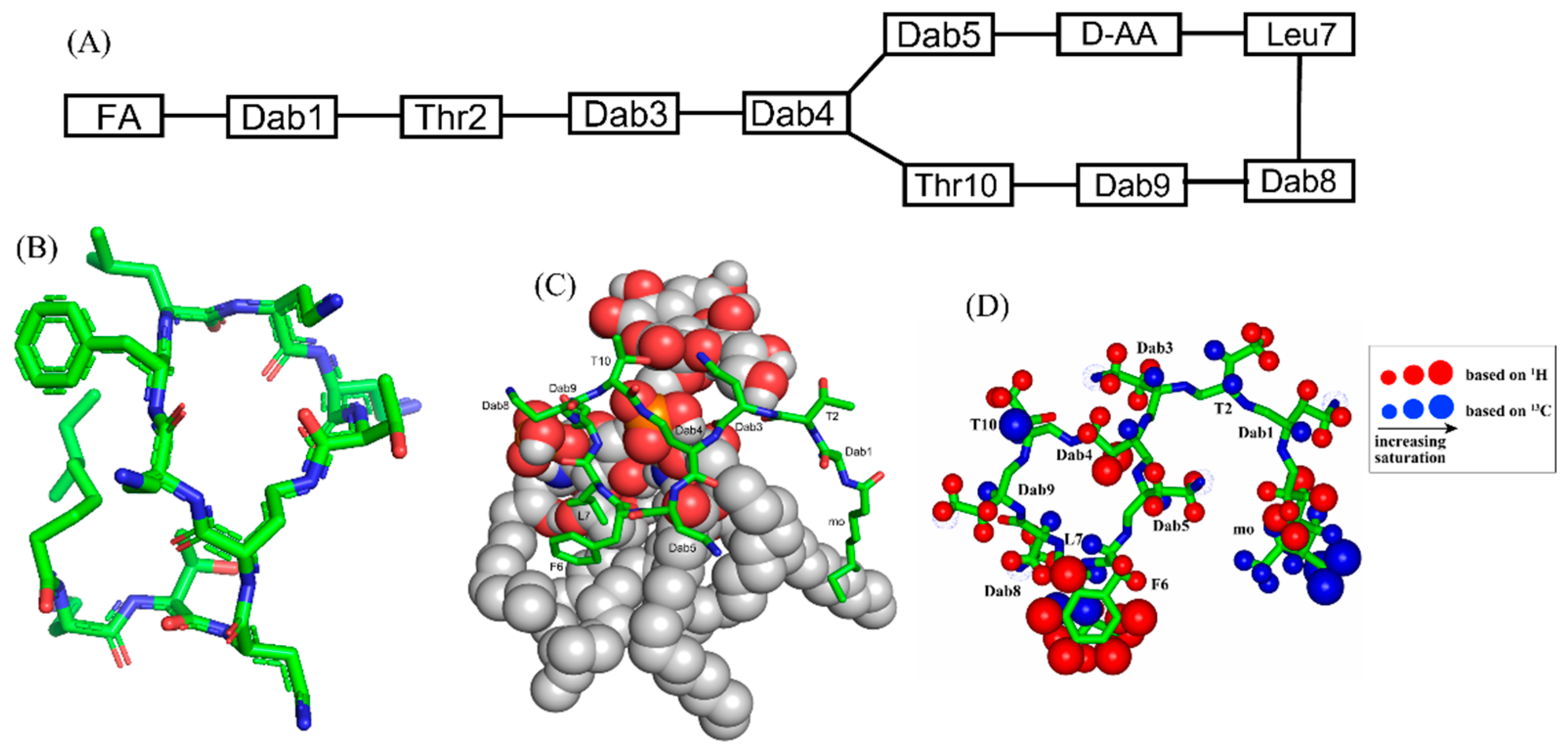
2. Polymyxins
3. β-Sheet AMPs: Protegrins and Thanatin
4. Outer Membrane Protein Targeting Antibiotics (OMPTA)
5. MSI Peptides
6. LL-37
7. De Novo Designed Synthetic Peptides
8. Conclusions and Future Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Taubes, G. The bacteria fight back. Science 2008, 321, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Kupferschmidt, K. Resistance fighters. Science 2016, 352, 758–761. [Google Scholar] [CrossRef] [PubMed]
- Savage, N. Overcoming resistance. Nature 2020, 586, S55–S56. [Google Scholar] [CrossRef]
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations; Government of the United Kingdom: London, UK, 2016.
- Antibiotic Resistance Threats in the United States. 2019. Available online: https://www.cdc.gov/drugresistance/pdf/threats-report/2019-ar-threats-report-508.pdf (accessed on 17 April 2022).
- Antimicrobial Resistance Collaborators. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- Taylor, P.L.; Rossi, L.; De Pascale, G.; Wright, G.D. A forward chemical screen identifies antibiotic adjuvants in Escherichia coli. ACS Chem. Biol. 2012, 7, 1547–1555. [Google Scholar] [CrossRef]
- Theuretzbacher, U. Global antimicrobial resistance in Gram-negative pathogens and clinical need. Curr. Opin. Microbiol. 2017, 39, 106–112. [Google Scholar] [CrossRef]
- Brown, D. Antibiotic resistance breakers: Can repurposed drugs fill the antibiotic discovery void? Nat. Rev. Drug Discov. 2015, 14, 821–832. [Google Scholar] [CrossRef]
- World Health Organization. Antibacterial Agents in Clinical Development: An Analysis of the Antibacterial Clinical Development Pipeline; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Chan, L.W.; Hern, K.E.; Ngambenjawong, C.; Lee, K.; Kwon, E.J.; Hung, D.T.; Bhatia, S.N. Selective Permeabilization of Gram-Negative Bacterial Membranes Using Multivalent Peptide Constructs for Antibiotic Sensitization. ACS Infect. Dis. 2021, 7, 721–732. [Google Scholar] [CrossRef]
- Zgurskaya, H.I.; Löpez, C.A.; Gnanakaran, S. Permeability Barrier of Gram-Negative Cell Envelopes and Approaches to Bypass It. ACS Infect. Dis. 2015, 1, 512–522. [Google Scholar] [CrossRef]
- Nikaido, H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656. [Google Scholar] [CrossRef]
- Lazzaro, B.P.; Zasloff, M.; Rolff, J. Antimicrobial peptides: Application informed by evolution. Science 2020, 368, eaau5480. [Google Scholar] [CrossRef] [PubMed]
- Magana, M.; Pushpanathan, M.; Santos, A.L.; Leanse, L.; Fernandez, M.; Ioannidis, A.; Giulianotti, M.A.; Apidianakis, Y.; Bradfute, S.; Ferguson, A.L.; et al. The value of antimicrobial peptides in the age of resistance. Lancet Infect. Dis. 2020, 20, e216–e230. [Google Scholar] [CrossRef]
- Drayton, M.; Deisinger, J.P.; Ludwig, K.C.; Raheem, N.; Müller, A.; Schneider, T.; Straus, S.K. Host Defense Peptides: Dual Antimicrobial and Immunomodulatory Action. Int. J. Mol. Sci. 2021, 22, 1172. [Google Scholar] [CrossRef] [PubMed]
- Boman, H.G. Antibacterial peptides: Basic facts and emerging concepts. J. Intern. Med. 2003, 254, 197–215. [Google Scholar] [CrossRef] [PubMed]
- Zasloff, M. Antimicrobial Peptides of Multicellular Organisms: My Perspective. Adv. Exp. Med. Biol. 2019, 1117, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuji, T.; Chen, T.H.; Narala, S.; Chun, K.A.; Two, A.M.; Yun, T.; Shafiq, F.; Kotol, P.F.; Bouslimani, A.; Melnik, A.V.; et al. Antimicrobials from human skin commensal bacteria protect against Staphylococcus aureus and are deficient in atopic dermatitis. Sci. Transl. Med. 2017, 9, eaah4680. [Google Scholar] [CrossRef]
- Bhattacharjya, S.; Straus, S.K. Design, Engineering and Discovery of Novel α-Helical and β-Boomerang Antimicrobial Peptides against Drug Resistant Bacteria. Int. J. Mol. Sci. 2020, 21, 5773. [Google Scholar] [CrossRef]
- Haney, E.F.; Straus, S.K.; Hancock, R.E.W. Reassessing the Host Defense Peptide Landscape. Front. Chem. 2019, 7, 43. [Google Scholar] [CrossRef]
- Barreto-Santamaría, A.; Arévalo-Pinzón, G.; Patarroyo, M.A.; Patarroyo, M.E. How to Combat Gram-Negative Bacteria Using Antimicrobial Peptides: A Challenge or an Unattainable Goal? Antibiotics 2021, 10, 1499. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Haney, E.F.; Vogel, H.J. The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol. 2011, 29, 464–472. [Google Scholar] [CrossRef]
- Shai, Y. Mode of action of membrane active antimicrobial peptides. Biopolymers 2002, 66, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K. Why and how are peptide-lipid interactions utilized for self defence? Biochem. Soc. Trans. 2001, 29, 598–601. [Google Scholar] [CrossRef] [PubMed]
- Ebbensgaard, A.; Mordhorst, H.; Aarestrup, F.M.; Hansen, E.B. The Role of Outer Membrane Proteins and Lipopolysaccharides for the Sensitivity of Escherichia coli to Antimicrobial Peptides. Front. Microbiol. 2018, 9, 2153. [Google Scholar] [CrossRef] [PubMed]
- Balhuizen, M.D.; van Dijk, A.; Jansen, J.W.A.; van de Lest, C.H.A.; Veldhuizen, E.J.A.; Haagsman, H.P. Outer Membrane Vesicles Protect Gram-Negative Bacteria against Host Defense Peptides. mSphere 2021, 6, e0052321. [Google Scholar] [CrossRef] [PubMed]
- Bhunia, A.; Domadia, P.N.; Bhattacharjya, S. Structural and thermodynamic analyses of the interaction between melittin and lipopolysaccharide. Biochim. Biophys. Acta BBA Biomembr. 2007, 1768, 3282–3291. [Google Scholar] [CrossRef] [PubMed]
- Krishnakumari, V.; Binny, T.M.; Adicherla, H.; Nagaraj, R. Lipopolysaccharide Modulates Biological Activities of Human-β-Defensin Analogues but Not Non-Ribosomally Synthesized Peptides. ACS Omega 2020, 5, 6366–6375. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjya, S. NMR Structures and Interactions of Antimicrobial Peptides with Lipopolysaccharide: Connecting Structures to Functions. Curr. Top. Med. Chem. 2016, 16, 4–15. [Google Scholar] [CrossRef]
- Brown, P.; Abdulle, O.; Boakes, S.; Divall, N.; Duperchy, E.; Ganeshwaran, S.; Lester, R.; Moss, S.; Rivers, D.; Simonovic, M.; et al. Influence of Lipophilicity on the Antibacterial Activity of Polymyxin Derivatives and on Their Ability to Act as Potentiators of Rifampicin. ACS Infect. Dis. 2021, 7, 894–905. [Google Scholar] [CrossRef]
- Baker, K.R.; Jana, B.; Hansen, A.M.; Nielsen, H.M.; Franzyk, H.; Guardabassi, L. Repurposing Azithromycin and Rifampicin Against Gram-Negative Pathogens by Combination with Peptidomimetics. Front. Cell. Infect. Microbiol. 2019, 9, 236. [Google Scholar] [CrossRef]
- Bernal, P.; Molina-Santiago, C.; Daddaoua, A.; Llamas, M.A. Antibiotic adjuvants: Identification and clinical use. Microb. Biotechnol. 2013, 6, 445–449. [Google Scholar] [CrossRef]
- Wright, G.D. Antibiotic Adjuvants: Rescuing Antibiotics from Resistance. Trends Microbiol. 2016, 24, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Swarbrick, J.D.; Karas, J.A.; Li, J.; Velkov, T. Structure of micelle bound cationic peptides by NMR spectroscopy using a lanthanide shift reagent. Chem. Commun. 2020, 56, 2897–2900. [Google Scholar] [CrossRef] [PubMed]
- Ilyas, H.; Kim, J.; Lee, D.; Malmsten, M.; Bhunia, A. Structural insights into the combinatorial effects of antimicrobial peptides reveal a role of aromatic-aromatic interactions in antibacterial synergism. J. Biol. Chem. 2019, 294, 14615–14633. [Google Scholar] [CrossRef] [PubMed]
- Saravanan, R.; Holdbrook, D.A.; Petrlova, J.; Singh, S.; Berglund, N.A.; Choong, Y.K.; Kjellström, S.; Bond, P.J.; Malmsten, M.; Schmidtchen, A. Structural basis for endotoxin neutralisation and anti-inflammatory activity of thrombin-derived C-terminal peptides. Nat. Commun. 2018, 9, 2762. [Google Scholar] [CrossRef]
- Yu, H.Y.; Chen, Y.A.; Yip, B.S.; Wang, S.Y.; Wei, H.J.; Chih, Y.H.; Chen, K.H.; Cheng, J.W. Role of β-naphthylalanine end-tags in the enhancement of antiendotoxin activities: Solution structure of the antimicrobial peptide S1-Nal-Nal in complex with lipopolysaccharide. Biochim. Biophys. Acta BBA Biomembr. 2017, 1859, 1114–1123. [Google Scholar] [CrossRef]
- Kushibiki, T.; Kamiya, M.; Aizawa, T.; Kumaki, Y.; Kikukawa, T.; Mizuguchi, M.; Demura, M.; Kawabata, S.; Kawano, K. Interaction between tachyplesin I, an antimicrobial peptide derived from horseshoe crab, and lipopolysaccharide. Biochim. Biophys. Acta BBA Proteins Proteom. 2014, 1844, 527–534. [Google Scholar] [CrossRef]
- Bai, Y.; Liu, S.; Li, J.; Lakshminarayanan, R.; Sarawathi, P.; Tang, C.; Ho, D.; Verma, C.; Beuerman, R.W.; Pervushin, K. Progressive structuring of a branched antimicrobial peptide on the path to the inner membrane target. J. Biol. Chem. 2012, 287, 26606–26617. [Google Scholar] [CrossRef]
- De Breij, A.; Riool, M.; Cordfunke, R.A.; Malanovic, N.; de Boer, L.; Koning, R.I.; Ravensbergen, E.; Franken, M.; van der Heijde, T.; Boekema, B.K.; et al. The antimicrobial peptide SAAP-148 combats drug-resistant bacteria and biofilms. Sci. Transl. Med. 2018, 10, eaan4044. [Google Scholar] [CrossRef]
- Luther, A.; Urfer, M.; Zahn, M.; Müller, M.; Wang, S.Y.; Mondal, M.; Vitale, A.; Hartmann, J.B.; Sharpe, T.; Monte, F.L.; et al. Chimeric peptidomimetic antibiotics against Gram-negative bacteria. Nature 2019, 576, 452–458. [Google Scholar] [CrossRef]
- Nicolas, I.; Bordeau, V.; Bondon, A.; Baudy-Floc’h, M.; Felden, B. Novel antibiotics effective against gram-positive and -negative multi-resistant bacteria with limited resistance. PLoS Biol. 2019, 17, e3000337. [Google Scholar] [CrossRef]
- Mishra, B.; Lakshmaiah Narayana, J.; Lushnikova, T.; Wang, X.; Wang, G. Low cationicity is important for systemic in vivo efficacy of database-derived peptides against drug-resistant Gram-positive pathogens. Proc. Natl. Acad. Sci. USA 2019, 116, 13517–13522. [Google Scholar] [CrossRef] [PubMed]
- Mwangi, J.; Yin, Y.; Wang, G.; Yang, M.; Li, Y.; Zhang, Z.; Lai, R. The antimicrobial peptide ZY4 combats multidrug-resistant. Proc. Natl. Acad. Sci. USA 2019, 116, 26516–26522. [Google Scholar] [CrossRef] [PubMed]
- Spohn, R.; Daruka, L.; Lázár, V.; Martins, A.; Vidovics, F.; Grézal, G.; Méhi, O.; Kintses, B.; Számel, M.; Jangir, P.K.; et al. Integrated evolutionary analysis reveals antimicrobial peptides with limited resistance. Nat. Commun. 2019, 10, 4538. [Google Scholar] [CrossRef] [PubMed]
- Lázár, V.; Martins, A.; Spohn, R.; Daruka, L.; Grézal, G.; Fekete, G.; Számel, M.; Jangir, P.K.; Kintses, B.; Csörgő, B.; et al. Antibiotic-resistant bacteria show widespread collateral sensitivity to antimicrobial peptides. Nat. Microbiol. 2018, 3, 718–731. [Google Scholar] [CrossRef]
- Silva, A.R.P.; Guimarães, M.S.; Rabelo, J.; Belén, L.H.; Perecin, C.J.; Farías, J.G.; Santos, J.H.P.M.; Rangel-Yagui, C.O. Recent advances in the design of antimicrobial peptide conjugates. J. Mater. Chem. B 2022. [Google Scholar] [CrossRef]
- Kuang, M.; Yu, H.; Qiao, S.; Huang, T.; Zhang, J.; Sun, M.; Shi, X.; Chen, H. A Novel Nano-Antimicrobial Polymer Engineered with Chitosan Nanoparticles and Bioactive Peptides as Promising Food Biopreservative Effective against Foodborne Pathogen. Int. J. Mol. Sci. 2021, 22, 3580. [Google Scholar] [CrossRef]
- Si, Z.; Zheng, W.; Prananty, D.; Li, J.; Koh, C.H.; Kang, E.T.; Pethe, K.; Chan-Park, M.B. Polymers as advanced antibacterial and antibiofilm agents for direct and combination therapies. Chem. Sci. 2022, 13, 345–364. [Google Scholar] [CrossRef]
- Vaara, M. Polymyxins and Their Potential Next Generation as Therapeutic Antibiotics. Front. Microbiol. 2019, 10, 1689. [Google Scholar] [CrossRef]
- Yu, Z.; Qin, W.; Lin, J.; Fang, S.; Qiu, J. Antibacterial mechanisms of polymyxin and bacterial resistance. Biomed. Res. Int. 2015, 2015, 679109. [Google Scholar] [CrossRef]
- Velkov, T.; Thompson, P.E.; Nation, R.L.; Li, J. Structure–activity relationships of polymyxin antibiotics. J. Med. Chem. 2010, 53, 1898–1916. [Google Scholar] [CrossRef]
- Bhunia, A.; Bhattacharjya, S. Mapping residue-specific contacts of polymyxin B with lipopolysaccharide by saturation transfer difference NMR: Insights into outer-membrane disruption and endotoxin neutralization. Biopolymers 2011, 96, 273–287. [Google Scholar] [CrossRef] [PubMed]
- Koch-Weser, J.; Sidel, V.W.; Federman, E.B.; Kanarek, P.; Finer, D.C.; Eaton, A.E. Adverse effects of sodium colistimethate. Manifestations and specific reaction rates during 317 courses of therapy. Ann. Intern. Med. 1970, 72, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Gallardo-Godoy, A.; Hansford, K.A.; Muldoon, C.; Becker, B.; Elliott, A.G.; Huang, J.X.; Pelingon, R.; Butler, M.S.; Blaskovich, M.A.T.; Cooper, M.A. Structure-Function Studies of Polymyxin B Lipononapeptides. Molecules 2019, 24, 553. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, B.T.; Pogue, J.M.; Zavascki, A.P.; Paul, M.; Daikos, G.L.; Forrest, A.; Giacobbe, D.R.; Viscoli, C.; Giamarellou, H.; Karaiskos, I.; et al. International Consensus Guidelines for the Optimal Use of the Polymyxins: Endorsed by the American College of Clinical Pharmacy (ACCP), European Society of Clinical Microbiology and Infectious Diseases (ESCMID), Infectious Diseases Society of America (IDSA), International Society for Anti-infective Pharmacology (ISAP), Society of Critical Care Medicine (SCCM), and Society of Infectious Diseases Pharmacists (SIDP). Pharmacotherapy 2019, 39, 10–39. [Google Scholar] [CrossRef] [PubMed]
- Vaara, M.; Vaara, T.; Vingsbo Lundberg, C. Polymyxin derivatives NAB739 and NAB815 are more effective than polymyxin B in murine Escherichia coli pyelonephritis. J. Antimicrob. Chemother. 2018, 73, 452–455. [Google Scholar] [CrossRef]
- Corbett, D.; Wise, A.; Langley, T.; Skinner, K.; Trimby, E.; Birchall, S.; Dorali, A.; Sandiford, S.; Williams, J.; Warn, P.; et al. Potentiation of Antibiotic Activity by a Novel Cationic Peptide: Potency and Spectrum of Activity of SPR741. Antimicrob. Agents Chemother. 2017, 61, e00200-17. [Google Scholar] [CrossRef]
- Bian, X.; Liu, X.; Hu, F.; Feng, M.; Chen, Y.; Bergen, P.J.; Li, J.; Li, X.; Guo, Y.; Zhang, J. Pharmacokinetic/Pharmacodynamic Based Breakpoints of Polymyxin B for Bloodstream Infections Caused by Multidrug-Resistant Gram-Negative Pathogens. Front. Pharmacol. 2021, 12, 785893. [Google Scholar] [CrossRef]
- Vaara, M.; Vaara, T.; Tyrrell, J.M. Structure-activity studies on polymyxin derivatives carrying three positive charges only reveal a new class of compounds with strong antibacterial activity. Peptides 2017, 91, 8–12. [Google Scholar] [CrossRef]
- Newton, B.A. The properties and mode of action of the polymyxins. Bacteriol. Rev. 1956, 20, 14–27. [Google Scholar] [CrossRef]
- Hancock, R.E. Alterations in outer membrane permeability. Annu. Rev. Microbiol. 1984, 38, 237–264. [Google Scholar] [CrossRef]
- Mohapatra, S.S.; Dwibedy, S.K.; Padhy, I. Polymyxins, the last-resort antibiotics: Mode of action, resistance emergence, and potential solutions. J. Biosci. 2021, 46, 85. [Google Scholar] [CrossRef]
- Bhattacharjya, S.; David, S.A.; Mathan, D.V.I.; Balaram, P. Polymyxin B Nonapeptide: Conformations in Water and in the Lipopolysaccharide-Bound State Determined by Two-Dimensional NMR and Molecular Dynamics. Biopolym. Orig. Res. Biomol. 1997, 41, 251–256. [Google Scholar] [CrossRef]
- Pristovsek, P.; Kidric, J. Solution structure of polymyxins B and E and effect of binding to lipopolysaccharide: An NMR and molecular modeling study. J. Med. Chem. 1999, 42, 4604–4613. [Google Scholar] [CrossRef] [PubMed]
- Mares, J.; Kumaran, S.; Gobbo, M.; Zerbe, O. Interactions of lipopolysaccharide and polymyxin studied by NMR spectroscopy. J. Biol. Chem. 2009, 284, 11498–11506. [Google Scholar] [CrossRef] [PubMed]
- Gennaro, R.; Zanetti, M. Structural features and biological activities of the cathelicidin-derived antimicrobial peptides. Pept. Sci. 2000, 55, 31–49. [Google Scholar] [CrossRef]
- Dimarcq, J.-L.; Bulet, P.; Hetru, C.; Hoffmann, J. Cysteine-rich antimicrobial peptides in invertebrates. Pept. Sci. 1998, 47, 465–477. [Google Scholar] [CrossRef]
- Kokryakov, V.N.; Harwig, S.S.; Panyutich, E.A.; Shevchenko, A.A.; Aleshina, G.M.; Shamova, O.V.; Korneva, H.A.; Lehrer, R.I. Protegrins: Leukocyte antimicrobial peptides that combine features of corticostatic defensins and tachyplesins. FEBS Lett. 1993, 327, 231–236. [Google Scholar] [CrossRef]
- Hancock, R.E. Resistance mechanisms in Pseudomonas aeruginosa and other nonfermentative gram-negative bacteria. Clin. Infect. Dis. 1998, 27 (Suppl. S1), S93–S99. [Google Scholar] [CrossRef]
- Chen, H.; Gordon, D.; Heinemann, S.H. Modulation of cloned skeletal muscle sodium channels by the scorpion toxins Lqh II, Lqh III, and Lqh alphaIT. Pflüg. Arch. 2000, 439, 423–432. [Google Scholar] [CrossRef]
- Mosca, D.A.; Hurst, M.A.; So, W.; Viajar, B.S.; Fujii, C.A.; Falla, T.J. IB-367, a protegrin peptide with in vitro and in vivo activities against the microflora associated with oral mucositis. Antimicrob. Agents Chemother. 2000, 44, 1803–1808. [Google Scholar] [CrossRef]
- Kollef, M.; Pittet, D.; Sánchez García, M.; Chastre, J.; Fagon, J.Y.; Bonten, M.; Hyzy, R.; Fleming, T.R.; Fuchs, H.; Bellm, L.; et al. A randomized double-blind trial of iseganan in prevention of ventilator-associated pneumonia. Am. J. Respir. Crit. Care Med. 2006, 173, 91–97. [Google Scholar] [CrossRef]
- Gidalevitz, D.; Ishitsuka, Y.; Muresan, A.S.; Konovalov, O.; Waring, A.J.; Lehrer, R.I.; Lee, K.Y. Interaction of antimicrobial peptide protegrin with biomembranes. Proc. Natl. Acad. Sci. USA 2003, 100, 6302–6307. [Google Scholar] [CrossRef] [PubMed]
- Mani, R.; Cady, S.D.; Tang, M.; Waring, A.J.; Lehrer, R.I.; Hong, M. Membrane-dependent oligomeric structure and pore formation of a beta-hairpin antimicrobial peptide in lipid bilayers from solid-state NMR. Proc. Natl. Acad. Sci. USA 2006, 103, 16242–16247. [Google Scholar] [CrossRef] [PubMed]
- Fahrner, R.L.; Dieckmann, T.; Harwig, S.S.; Lehrer, R.I.; Eisenberg, D.; Feigon, J. Solution structure of protegrin-1, a broad-spectrum antimicrobial peptide from porcine leukocytes. Chem. Biol. 1996, 3, 543–550. [Google Scholar] [CrossRef]
- Su, Y.; Waring, A.J.; Ruchala, P.; Hong, M. Structures of β-hairpin antimicrobial protegrin peptides in lipopolysaccharide membranes: Mechanism of gram selectivity obtained from solid-state nuclear magnetic resonance. Biochemistry 2011, 50, 2072–2083. [Google Scholar] [CrossRef]
- Roumestand, C.; Louis, V.; Aumelas, A.; Grassy, G.; Calas, B.; Chavanieu, A. Oligomerization of protegrin-1 in the presence of DPC micelles. A proton high-resolution NMR study. FEBS Lett. 1998, 421, 263–267. [Google Scholar] [CrossRef]
- Tang, M.; Waring, A.J.; Hong, M. Phosphate-mediated arginine insertion into lipid membranes and pore formation by a cationic membrane peptide from solid-state NMR. J. Am. Chem. Soc. 2007, 129, 11438–11446. [Google Scholar] [CrossRef]
- Mohanram, H.; Bhattacharjya, S. Cysteine deleted protegrin-1 (CDP-1): Anti-bacterial activity, outer-membrane disruption and selectivity. Biochim. Biophys. Acta BBA Gen. Subj. 2014, 1840, 3006–3016. [Google Scholar] [CrossRef]
- Fehlbaum, P.; Bulet, P.; Chernysh, S.; Briand, J.P.; Roussel, J.P.; Letellier, L.; Hetru, C.; Hoffmann, J.A. Structure-activity analysis of thanatin, a 21-residue inducible insect defense peptide with sequence homology to frog skin antimicrobial peptides. Proc. Natl. Acad. Sci. USA 1996, 93, 1221–1225. [Google Scholar] [CrossRef]
- Dash, R.; Bhattacharjya, S. Thanatin: An Emerging Host Defense Antimicrobial Peptide with Multiple Modes of Action. Int. J. Mol. Sci. 2021, 22, 1522. [Google Scholar] [CrossRef]
- Edwards, I.A.; Elliott, A.G.; Kavanagh, A.M.; Zuegg, J.; Blaskovich, M.A.; Cooper, M.A. Contribution of Amphipathicity and Hydrophobicity to the Antimicrobial Activity and Cytotoxicity of β-Hairpin Peptides. ACS Infect. Dis. 2016, 2, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Fang, C.; Lu, L.; Wang, M.; Xue, X.; Zhou, Y.; Li, M.; Hu, Y.; Luo, X.; Hou, Z. The antimicrobial peptide thanatin disrupts the bacterial outer membrane and inactivates the NDM-1 metallo-β-lactamase. Nat. Commun. 2019, 10, 3517. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Fan, X.; Li, L.; Wang, H.; Ding, J.; Hongbin, W.; Zhao, R.; Gou, L.; Shen, Z.; Xi, T. Interaction of antimicrobial peptide s-thanatin with lipopolysaccharide in vitro and in an experimental mouse model of septic shock caused by a multidrug-resistant clinical isolate of Escherichia coli. Int. J. Antimicrob. Agents 2010, 35, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Zheng, L.; Mu, Y.; Ng, W.J.; Bhattacharjya, S. Structure and Interactions of a Host Defense Antimicrobial Peptide Thanatin in Lipopolysaccharide Micelles Reveal Mechanism of Bacterial Cell Agglutination. Sci. Rep. 2017, 7, 17795. [Google Scholar] [CrossRef]
- Fiorentino, F.; Sauer, J.B.; Qiu, X.; Corey, R.A.; Cassidy, C.K.; Mynors-Wallis, B.; Mehmood, S.; Bolla, J.R.; Stansfeld, P.J.; Robinson, C.V. Dynamics of an LPS translocon induced by substrate and an antimicrobial peptide. Nat. Chem. Biol. 2021, 17, 187–195. [Google Scholar] [CrossRef]
- Vetterli, S.U.; Zerbe, K.; Müller, M.; Urfer, M.; Mondal, M.; Wang, S.Y.; Moehle, K.; Zerbe, O.; Vitale, A.; Pessi, G.; et al. Thanatin targets the intermembrane protein complex required for lipopolysaccharide transport in Escherichia coli. Sci. Adv. 2018, 4, eaau2634. [Google Scholar] [CrossRef]
- Moura, E.C.C.M.; Baeta, T.; Romanelli, A.; Laguri, C.; Martorana, A.M.; Erba, E.; Simorre, J.P.; Sperandeo, P.; Polissi, A. Thanatin Impairs Lipopolysaccharide Transport Complex Assembly by Targeting LptC-LptA Interaction and Decreasing LptA Stability. Front. Microbiol. 2020, 11, 909. [Google Scholar] [CrossRef]
- Sinha, S.; Ng, W.J.; Bhattacharjya, S. NMR structure and localization of the host defense antimicrobial peptide thanatin in zwitterionic dodecylphosphocholine micelle: Implications in antimicrobial activity. Biochim. Biophys. Acta BBA Biomembr. 2020, 1862, 183432. [Google Scholar] [CrossRef]
- Sinha, S.; Dhanabal, V.B.; Sperandeo, P.; Polissi, A.; Bhattacharjya, S. Linking dual mode of action of host defense antimicrobial peptide thanatin: Structures, lipopolysaccharide and LptA. Biochim. Biophys. Acta BBA Biomembr. 2022, 1864, 183839. [Google Scholar] [CrossRef]
- Upert, G.; Luther, A.; Obrecht, D.; Ermert, P. Emerging peptide antibiotics with therapeutic potential. Med. Drug Discov. 2021, 9, 100078. [Google Scholar] [CrossRef]
- Luther, A.; Bisang, C.; Obrecht, D. Advances in macrocyclic peptide-based antibiotics. Bioorg. Med. Chem. 2018, 26, 2850–2858. [Google Scholar] [CrossRef] [PubMed]
- Luther, A.; Moehle, K.; Chevalier, E.; Dale, G.; Obrecht, D. Protein epitope mimetic macrocycles as biopharmaceuticals. Curr. Opin. Chem. Biol. 2017, 38, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, N.; Jetter, P.; Ueberbacher, B.J.; Werneburg, M.; Zerbe, K.; Steinmann, J.; Van der Meijden, B.; Bernardini, F.; Lederer, A.; Dias, R.L.; et al. Peptidomimetic antibiotics target outer-membrane biogenesis in Pseudomonas aeruginosa. Science 2010, 327, 1010–1013. [Google Scholar] [CrossRef] [PubMed]
- Díez-Aguilar, M.; Hernández-García, M.; Morosini, M.I.; Fluit, A.; Tunney, M.M.; Huertas, N.; Del Campo, R.; Obrecht, D.; Bernardini, F.; Ekkelenkamp, M.; et al. Murepavadin antimicrobial activity against and resistance development in cystic fibrosis Pseudomonas aeruginosa isolates. J. Antimicrob. Chemother. 2021, 76, 984–992. [Google Scholar] [CrossRef]
- Dong, H.; Xiang, Q.; Gu, Y.; Wang, Z.; Paterson, N.G.; Stansfeld, P.J.; He, C.; Zhang, Y.; Wang, W.; Dong, C. Structural basis for outer membrane lipopolysaccharide insertion. Nature 2014, 511, 52–56. [Google Scholar] [CrossRef]
- Dale, G.E.; Halabi, A.; Petersen-Sylla, M.; Wach, A.; Zwingelstein, C. Pharmacokinetics, Tolerability, and Safety of Murepavadin, a Novel Antipseudomonal Antibiotic, in Subjects with Mild, Moderate, or Severe Renal Function Impairment. Antimicrob. Agents Chemother. 2018, 62, e0049018. [Google Scholar] [CrossRef]
- Imai, Y.; Meyer, K.J.; Iinishi, A.; Favre-Godal, Q.; Green, R.; Manuse, S.; Caboni, M.; Mori, M.; Niles, S.; Ghiglieri, M.; et al. A new antibiotic selectively kills Gram-negative pathogens. Nature 2019, 576, 459–464. [Google Scholar] [CrossRef]
- Kaur, H.; Jakob, R.P.; Marzinek, J.K.; Green, R.; Imai, Y.; Bolla, J.R.; Agustoni, E.; Robinson, C.V.; Bond, P.J.; Lewis, K.; et al. The antibiotic darobactin mimics a β-strand to inhibit outer membrane insertase. Nature 2021, 593, 125–129. [Google Scholar] [CrossRef]
- Zasloff, M. Magainins, a class of antimicrobial peptides from Xenopus skin: Isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc. Natl. Acad. Sci. USA 1987, 84, 5449–5453. [Google Scholar] [CrossRef]
- Zasloff, M.; Martin, B.; Chen, H.C. Antimicrobial activity of synthetic magainin peptides and several analogues. Proc. Natl. Acad. Sci. USA 1988, 85, 910–913. [Google Scholar] [CrossRef]
- Porcelli, F.; Buck-Koehntop, B.A.; Thennarasu, S.; Ramamoorthy, A.; Veglia, G. Structures of the dimeric and monomeric variants of magainin antimicrobial peptides (MSI-78 and MSI-594) in micelles and bilayers, determined by NMR spectroscopy. Biochemistry 2006, 45, 5793–5799. [Google Scholar] [CrossRef] [PubMed]
- Bechinger, B.; Juhl, D.W.; Glattard, E.; Aisenbrey, C. Revealing the Mechanisms of Synergistic Action of Two Magainin Antimicrobial Peptides. Front. Med. Technol. 2020, 2, 615494. [Google Scholar] [CrossRef] [PubMed]
- Islam, K.; Hawser, S.P. MSI-78 Magainin Pharmaceuticals. Idrugs Investig. Drugs J. 1998, 1, 605–609. [Google Scholar]
- Flamm, R.K.; Rhomberg, P.R.; Simpson, K.M.; Farrell, D.J.; Sader, H.S.; Jones, R.N. In vitro spectrum of pexiganan activity when tested against pathogens from diabetic foot infections and with selected resistance mechanisms. Antimicrob. Agents Chemother. 2015, 59, 1751–1754. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gottler, L.M.; Ramamoorthy, A. Structure, membrane orientation, mechanism, and function of pexiganan—A highly potent antimicrobial peptide designed from magainin. Biochim. Biophys. Acta BBA Biomembr. 2009, 1788, 1680–1686. [Google Scholar] [CrossRef]
- Gomes, D.; Santos, R.; Soares, R.S.; Reis, S.; Carvalho, S.; Rego, P.; Peleteiro, M.C.; Tavares, L.; Oliveira, M. Pexiganan in Combination with Nisin to Control Polymicrobial Diabetic Foot Infections. Antibiotics 2020, 9, 128. [Google Scholar] [CrossRef]
- Leontiadou, H.; Mark, A.E.; Marrink, S.J. Antimicrobial peptides in action. J. Am. Chem. Soc. 2006, 128, 12156–12161. [Google Scholar] [CrossRef]
- Takeshima, K.; Chikushi, A.; Lee, K.K.; Yonehara, S.; Matsuzaki, K. Translocation of analogues of the antimicrobial peptides magainin and buforin across human cell membranes. J. Biol. Chem. 2003, 278, 1310–1315. [Google Scholar] [CrossRef]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef]
- Mukai, Y.; Matsushita, Y.; Niidome, T.; Hatekeyama, T.; Aoyag, H. Parallel and antiparallel dimers of magainin 2: Their interaction with phospholipid membrane and antibacterial activity. J. Pept. Sci. Off. Publ. Eur. Pept. Soc. 2002, 8, 570–577. [Google Scholar] [CrossRef]
- Papo, N.; Shai, Y. Can we predict biological activity of antimicrobial peptides from their interactions with model phospholipid membranes? Peptides 2003, 24, 1693–1703. [Google Scholar] [CrossRef] [PubMed]
- Hayouka, Z.; Mortenson, D.E.; Kreitler, D.F.; Weisblum, B.; Forest, K.T.; Gellman, S.H. Evidence for phenylalanine zipper-mediated dimerization in the X-ray crystal structure of a magainin 2 analogue. J. Am. Chem. Soc. 2013, 135, 15738–15741. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bhunia, A.; Domadia, P.N.; Torres, J.; Hallock, K.J.; Ramamoorthy, A.; Bhattacharjya, S. NMR structure of pardaxin, a pore-forming antimicrobial peptide, in lipopolysaccharide micelles: Mechanism of outer membrane permeabilization. J. Biol. Chem. 2010, 285, 3883–3895. [Google Scholar] [CrossRef] [PubMed]
- Ramamoorthy, A.; Thennarasu, S.; Lee, D.K.; Tan, A.; Maloy, L. Solid-state NMR investigation of the membrane-disrupting mechanism of antimicrobial peptides MSI-78 and MSI-594 derived from magainin 2 and melittin. Biophys. J. 2006, 91, 206–216. [Google Scholar] [CrossRef]
- Hallock, K.J.; Lee, D.K.; Ramamoorthy, A. MSI-78, an analogue of the magainin antimicrobial peptides, disrupts lipid bilayer structure via positive curvature strain. Biophys. J. 2003, 84, 3052–3060. [Google Scholar] [CrossRef]
- Strandberg, E.; Tremouilhac, P.; Wadhwani, P.; Ulrich, A.S. Synergistic transmembrane insertion of the heterodimeric PGLa/magainin 2 complex studied by solid-state NMR. Biochim. Biophys. Acta BBA Biomembr. 2009, 1788, 1667–1679. [Google Scholar] [CrossRef]
- Marassi, F.M.; Opella, S.J. A solid-state NMR index of helical membrane protein structure and topology. J. Magn. Reson. 2000, 144, 150–155. [Google Scholar] [CrossRef]
- Domadia, P.N.; Bhunia, A.; Ramamoorthy, A.; Bhattacharjya, S. Structure, interactions, and antibacterial activities of MSI-594 derived mutant peptide MSI-594F5A in lipopolysaccharide micelles: Role of the helical hairpin conformation in outer-membrane permeabilization. J. Am. Chem. Soc. 2010, 132, 18417–18428. [Google Scholar] [CrossRef]
- Bhunia, A.; Ramamoorthy, A.; Bhattacharjya, S. Helical hairpin structure of a potent antimicrobial peptide MSI-594 in lipopolysaccharide micelles by NMR spectroscopy. Chemistry 2009, 15, 2036–2040. [Google Scholar] [CrossRef]
- Lee, D.K.; Bhunia, A.; Kotler, S.A.; Ramamoorthy, A. Detergent-type membrane fragmentation by MSI-78, MSI-367, MSI-594, and MSI-843 antimicrobial peptides and inhibition by cholesterol: A solid-state nuclear magnetic resonance study. Biochemistry 2015, 54, 1897–1907. [Google Scholar] [CrossRef]
- Bhattacharjya, S.; Ramamoorthy, A. Multifunctional host defense peptides: Functional and mechanistic insights from NMR structures of potent antimicrobial peptides. FEBS J. 2009, 276, 6465–6473. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Kar, R.K.; Nanga, R.P.R.; Mroue, K.H.; Ramamoorthy, A.; Bhunia, A. Accelerated molecular dynamics simulation analysis of MSI-594 in a lipid bilayer. Phys. Chem. Chem. Phys. 2017, 19, 19289–19299. [Google Scholar] [CrossRef] [PubMed]
- Nijnik, A.; Hancock, R.E. The roles of cathelicidin LL-37 in immune defences and novel clinical applications. Curr. Opin. Hematol. 2009, 16, 41–47. [Google Scholar] [CrossRef]
- Wang, G.; Narayana, J.L.; Mishra, B.; Zhang, Y.; Wang, F.; Wang, C.; Zarena, D.; Lushnikova, T.; Wang, X. Design of Antimicrobial Peptides: Progress Made with Human Cathelicidin LL-37. Adv. Exp. Med. Biol. 2019, 1117, 215–240. [Google Scholar] [CrossRef] [PubMed]
- Thennarasu, S.; Tan, A.; Penumatchu, R.; Shelburne, C.E.; Heyl, D.L.; Ramamoorthy, A. Antimicrobial and membrane disrupting activities of a peptide derived from the human cathelicidin antimicrobial peptide LL37. Biophys. J. 2010, 98, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Sharma, N.; Mishra, P.; Joseph, J.; Mishra, D.K.; Garg, P.; Roy, S. Differential Expression of Antimicrobial Peptides in Streptococcus pneumoniae Keratitis and STAT3-Dependent Expression of LL-37 by Streptococcus pneumoniae in Human Corneal Epithelial Cells. Pathogens 2019, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Zanetti, M.; Gennaro, R.; Romeo, D. Cathelicidins: A novel protein family with a common proregion and a variable C-terminal antimicrobial domain. FEBS Lett. 1995, 374, 1–5. [Google Scholar] [CrossRef]
- Malmsten, M.; Davoudi, M.; Walse, B.; Rydengård, V.; Pasupuleti, M.; Mörgelin, M.; Schmidtchen, A. Antimicrobial peptides derived from growth factors. Growth Factors 2007, 25, 60–70. [Google Scholar] [CrossRef]
- Dürr, U.H.; Sudheendra, U.S.; Ramamoorthy, A. LL-37, the only human member of the cathelicidin family of antimicrobial peptides. Biochim. Biophys. Acta BBA Biomembr. 2006, 1758, 1408–1425. [Google Scholar] [CrossRef]
- Koo, H.B.; Seo, J. Antimicrobial peptides under clinical investigation. Pept. Sci. 2019, 111, e24122. [Google Scholar] [CrossRef]
- Wang, G. Structures of human host defense cathelicidin LL-37 and its smallest antimicrobial peptide KR-12 in lipid micelles. J. Biol. Chem. 2008, 283, 32637–32643. [Google Scholar] [CrossRef] [PubMed]
- Sancho-Vaello, E.; François, P.; Bonetti, E.J.; Lilie, H.; Finger, S.; Gil-Ortiz, F.; Gil-Carton, D.; Zeth, K. Structural remodeling and oligomerization of human cathelicidin on membranes suggest fibril-like structures as active species. Sci. Rep. 2017, 7, 15371. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, Y.; Han, H.; Miller, D.W.; Wang, G. Solution structures of human LL-37 fragments and NMR-based identification of a minimal membrane-targeting antimicrobial and anticancer region. J. Am. Chem. Soc. 2006, 128, 5776–5785. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, I.; Tamura, H.; Reich, J. Therapeutic Potential of Cathelicidin Peptide LL-37, an Antimicrobial Agent, in a Murine Sepsis Model. Int. J. Mol. Sci. 2020, 21, 5973. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zou, X.; Qi, G.; Tang, Y.; Guo, Y.; Si, J.; Liang, L. Roles and Mechanisms of Human Cathelicidin LL-37 in Cancer. Cell. Physiol. Biochem. 2018, 47, 1060–1073. [Google Scholar] [CrossRef]
- Ridyard, K.E.; Overhage, J. The Potential of Human Peptide LL-37 as an Antimicrobial and Anti-Biofilm Agent. Antibiotics 2021, 10, 650. [Google Scholar] [CrossRef]
- Zeth, K.; Sancho-Vaello, E. Structural Plasticity of LL-37 Indicates Elaborate Functional Adaptation Mechanisms to Bacterial Target Structures. Int. J. Mol. Sci. 2021, 22, 5200. [Google Scholar] [CrossRef]
- Porcelli, F.; Verardi, R.; Shi, L.; Henzler-Wildman, K.A.; Ramamoorthy, A.; Veglia, G. NMR structure of the cathelicidin-derived human antimicrobial peptide LL-37 in dodecylphosphocholine micelles. Biochemistry 2008, 47, 5565–5572. [Google Scholar] [CrossRef]
- Chen, K.; Gong, W.; Huang, J.; Yoshimura, T.; Wang, J.M. The potentials of short fragments of human anti-microbial peptide LL-37 as a novel therapeutic modality for diseases. Front. Biosci. Landmark Ed. 2021, 26, 1362–1372. [Google Scholar] [CrossRef]
- Wang, G.; Mishra, B.; Epand, R.F.; Epand, R.M. High-quality 3D structures shine light on antibacterial, anti-biofilm and antiviral activities of human cathelicidin LL-37 and its fragments. Biochim. Biophys. Acta BBA Biomembr. 2014, 1838, 2160–2172. [Google Scholar] [CrossRef]
- Jacob, B.; Park, I.S.; Bang, J.K.; Shin, S.Y. Short KR-12 analogs designed from human cathelicidin LL-37 possessing both antimicrobial and antiendotoxic activities without mammalian cell toxicity. J. Pept. Sci. 2013, 19, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; McLean, D.T.; Linden, G.J.; McAuley, D.F.; McMullan, R.; Lundy, F.T. The Naturally Occurring Host Defense Peptide, LL-37, and Its Truncated Mimetics KE-18 and KR-12 Have Selected Biocidal and Antibiofilm Activities Against. Front. Microbiol. 2017, 8, 544. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Elliott, M.; Cogen, A.L.; Ezell, E.L.; Gallo, R.L.; Hancock, R.E. Structure, dynamics, and antimicrobial and immune modulatory activities of human LL-23 and its single-residue variants mutated on the basis of homologous primate cathelicidins. Biochemistry 2012, 51, 653–664. [Google Scholar] [CrossRef]
- Mohammed, I.; Said, D.G.; Nubile, M.; Mastropasqua, L.; Dua, H.S. Cathelicidin-Derived Synthetic Peptide Improves Therapeutic Potential of Vancomycin Against. Front. Microbiol. 2019, 10, 2190. [Google Scholar] [CrossRef] [PubMed]
- Nell, M.J.; Tjabringa, G.S.; Wafelman, A.R.; Verrijk, R.; Hiemstra, P.S.; Drijfhout, J.W.; Grote, J.J. Development of novel LL-37 derived antimicrobial peptides with LPS and LTA neutralizing and antimicrobial activities for therapeutic application. Peptides 2006, 27, 649–660. [Google Scholar] [CrossRef]
- Ming, L.; Huang, J.A. The Antibacterial Effects of Antimicrobial Peptides OP-145 against Clinically Isolated Multi-Resistant Strains. Jpn. J. Infect. Dis. 2017, 70, 601–603. [Google Scholar] [CrossRef]
- Singh, S.; Datta, A.; Borro, B.C.; Davoudi, M.; Schmidtchen, A.; Bhunia, A.; Malmsten, M. Conformational Aspects of High Content Packing of Antimicrobial Peptides in Polymer Microgels. ACS Appl. Mater. Interfaces 2017, 9, 40094–40106. [Google Scholar] [CrossRef]
- Bhunia, A.; Mohanram, H.; Bhattacharjya, S. Lipopolysaccharide bound structures of the active fragments of fowlicidin-1, a cathelicidin family of antimicrobial and antiendotoxic peptide from chicken, determined by transferred nuclear Overhauser effect spectroscopy. Biopolymers 2009, 92, 9–22. [Google Scholar] [CrossRef]
- Browne, K.; Chakraborty, S.; Chen, R.; Willcox, M.D.; Black, D.S.; Walsh, W.R.; Kumar, N. A New Era of Antibiotics: The Clinical Potential of Antimicrobial Peptides. Int. J. Mol. Sci. 2020, 21, 7047. [Google Scholar] [CrossRef]
- Ting, D.S.J.; Beuerman, R.W.; Dua, H.S.; Lakshminarayanan, R.; Mohammed, I. Strategies in Translating the Therapeutic Potentials of Host Defense Peptides. Front. Immunol. 2020, 11, 983. [Google Scholar] [CrossRef]
- Nordström, R.; Malmsten, M. Delivery systems for antimicrobial peptides. Adv. Colloid Interface Sci. 2017, 242, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Mercer, D.K.; Robertson, J.C.; Miller, L.; Stewart, C.S.; O’Neil, D.A. NP213 (Novexatin®): A unique therapy candidate for onychomycosis with a differentiated safety and efficacy profile. Med. Mycol. 2020, 58, 1064–1072. [Google Scholar] [CrossRef] [PubMed]
- Ryder, M.P.; Wu, X.; McKelvey, G.R.; McGuire, J.; Schilke, K.F. Binding interactions of bacterial lipopolysaccharide and the cationic amphiphilic peptides polymyxin B and WLBU2. Colloids Surf. B Biointerfaces 2014, 120, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Swedan, S.; Shubair, Z.; Almaaytah, A. Synergism of cationic antimicrobial peptide WLBU2 with antibacterial agents against biofilms of multi-drug resistant. Infect. Drug Resist. 2019, 12, 2019–2030. [Google Scholar] [CrossRef] [PubMed]
- Golbek, T.W.; Franz, J.; Elliott Fowler, J.; Schilke, K.F.; Weidner, T.; Baio, J.E. Identifying the selectivity of antimicrobial peptides to cell membranes by sum frequency generation spectroscopy. Biointerphases 2017, 12, 02D406. [Google Scholar] [CrossRef] [PubMed]
- Elsalem, L.; Al Sheboul, S.; Khasawneh, A. Synergism between WLBU2 peptide and antibiotics against methicillin-resistant Staphylococcus aureus and extended-spectrum beta-lactamase-producing Enterobacter cloacae. J. Appl. Biomed. 2021, 19, 14–25. [Google Scholar] [CrossRef]
- Saravolatz, L.D.; Pawlak, J.; Johnson, L.; Bonilla, H.; Fakih, M.G.; Fugelli, A.; Olsen, W.M. In vitro activities of LTX-109, a synthetic antimicrobial peptide, against methicillin-resistant, vancomycin-intermediate, vancomycin-resistant, daptomycin-nonsusceptible, and linezolid-nonsusceptible Staphylococcus aureus. Antimicrob. Agents Chemother. 2012, 56, 4478–4482. [Google Scholar] [CrossRef]
- Isaksson, J.; Brandsdal, B.O.; Engqvist, M.; Flaten, G.E.; Svendsen, J.S.; Stensen, W. A synthetic antimicrobial peptidomimetic (LTX 109): Stereochemical impact on membrane disruption. J. Med. Chem. 2011, 54, 5786–5795. [Google Scholar] [CrossRef]
- Javia, A.; Amrutiya, J.; Lalani, R.; Patel, V.; Bhatt, P.; Misra, A. Antimicrobial peptide delivery: An emerging therapeutic for the treatment of burn and wounds. Ther. Deliv. 2018, 9, 375–386. [Google Scholar] [CrossRef]
- Sierra, J.M.; Fusté, E.; Rabanal, F.; Vinuesa, T.; Viñas, M. An overview of antimicrobial peptides and the latest advances in their development. Expert Opin. Biol. Ther. 2017, 17, 663–676. [Google Scholar] [CrossRef]
- Boto, A.; Pérez de la Lastra, J.M.; González, C.C. The Road from Host-Defense Peptides to a New Generation of Antimicrobial Drugs. Molecules 2018, 23, 311. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Jordan, O.; Hanawa, T.; Borchard, G.; Patrulea, V. Are Antimicrobial Peptide Dendrimers an Escape from ESKAPE? Adv. Wound Care 2020, 9, 378–395. [Google Scholar] [CrossRef] [PubMed]
- Datta, A.; Ghosh, A.; Airoldi, C.; Sperandeo, P.; Mroue, K.H.; Jiménez-Barbero, J.; Kundu, P.; Ramamoorthy, A.; Bhunia, A. Antimicrobial Peptides: Insights into Membrane Permeabilization, Lipopolysaccharide Fragmentation and Application in Plant Disease Control. Sci. Rep. 2015, 5, 11951. [Google Scholar] [CrossRef]
- Biswas, K.; Ilyas, H.; Datta, A.; Bhunia, A. NMR Assisted Antimicrobial Peptide Designing: Structure Based Modifications and Functional Correlation of a Designed Peptide VG16KRKP. Curr. Med. Chem. 2020, 27, 1387–1404. [Google Scholar] [CrossRef] [PubMed]
- Datta, A.; Bhattacharyya, D.; Singh, S.; Ghosh, A.; Schmidtchen, A.; Malmsten, M.; Bhunia, A. Role of Aromatic Amino Acids in Lipopolysaccharide and Membrane Interactions of Antimicrobial Peptides for Use in Plant Disease Control. J. Biol. Chem. 2016, 291, 13301–13317. [Google Scholar] [CrossRef] [PubMed]
- Datta, A.; Yadav, V.; Ghosh, A.; Choi, J.; Bhattacharyya, D.; Kar, R.K.; Ilyas, H.; Dutta, A.; An, E.; Mukhopadhyay, J.; et al. Mode of Action of a Designed Antimicrobial Peptide: High Potency against Cryptococcus neoformans. Biophys. J. 2016, 111, 1724–1737. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.; Ilyas, H.; Ghosh, A.; Ali, H.; Ghorai, A.; Midya, A.; Jana, N.R.; Das, S.; Bhunia, A. Multivalent gold nanoparticle-peptide conjugates for targeting intracellular bacterial infections. Nanoscale 2017, 9, 14074–14093. [Google Scholar] [CrossRef]
- Mohid, S.A.; Ghorai, A.; Ilyas, H.; Mroue, K.H.; Narayanan, G.; Sarkar, A.; Ray, S.K.; Biswas, K.; Bera, A.K.; Malmsten, M.; et al. Application of tungsten disulfide quantum dot-conjugated antimicrobial peptides in bio-imaging and antimicrobial therapy. Colloids Surf. B Biointerfaces 2019, 176, 360–370. [Google Scholar] [CrossRef]
- Mohid, S.A.; Sharma, P.; Alghalayini, A.; Saini, T.; Datta, D.; Willcox, M.D.P.; Ali, H.; Raha, S.; Singha, A.; Lee, D.; et al. A rationally designed synthetic antimicrobial peptide against Pseudomonas-associated corneal keratitis: Structure-function correlation. Biophys. Chem. 2022, 106802. [Google Scholar] [CrossRef]
- Bhattacharjya, S.; Domadia, P.N.; Bhunia, A.; Malladi, S.; David, S.A. High-resolution solution structure of a designed peptide bound to lipopolysaccharide: Transferred nuclear Overhauser effects, micelle selectivity, and anti-endotoxic activity. Biochemistry 2007, 46, 5864–5874. [Google Scholar] [CrossRef]
- Liu, F.; Mu, J.; Wu, X.; Bhattacharjya, S.; Yeow, E.K.; Xing, B. Peptide-perylene diimide functionalized magnetic nano-platforms for fluorescence turn-on detection and clearance of bacterial lipopolysaccharides. Chem. Commun. 2014, 50, 6200–6203. [Google Scholar] [CrossRef] [PubMed]
- Bhunia, A.; Mohanram, H.; Domadia, P.N.; Torres, J.; Bhattacharjya, S. Designed beta-boomerang antiendotoxic and antimicrobial peptides: Structures and activities in lipopolysaccharide. J. Biol. Chem. 2009, 284, 21991–22004. [Google Scholar] [CrossRef] [PubMed]
- Mohanram, H.; Bhattacharjya, S. β-Boomerang Antimicrobial and Antiendotoxic Peptides: Lipidation and Disulfide Bond Effects on Activity and Structure. Pharmaceuticals 2014, 7, 482–501. [Google Scholar] [CrossRef] [PubMed]
- Mohanram, H.; Bhattacharjya, S. Resurrecting inactive antimicrobial peptides from the lipopolysaccharide trap. Antimicrob. Agents Chemother. 2014, 58, 1987–1996. [Google Scholar] [CrossRef]
- Liu, F.; Soh Yan Ni, A.; Lim, Y.; Mohanram, H.; Bhattacharjya, S.; Xing, B. Lipopolysaccharide neutralizing peptide-porphyrin conjugates for effective photoinactivation and intracellular imaging of gram-negative bacteria strains. Bioconjug. Chem. 2012, 23, 1639–1647. [Google Scholar] [CrossRef]
- Lu, S.; Bi, W.; Du, Q.; Sinha, S.; Wu, X.; Subrata, A.; Bhattacharjya, S.; Xing, B.; Yeow, E.K.L. Lipopolysaccharide-affinity copolymer senses the rapid motility of swarmer bacteria to trigger antimicrobial drug release. Nat. Commun. 2018, 9, 4277. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | FA | Sequence |
|---|---|---|
| Polymyxin B (PMB) | Methyloctanoyl/methylheptanoyl | Dab1-Thr2-Dab3-cy[Dab4-Dab5-DPhe6-Leu7-Dab8-Dab8-Thr10] |
| Polymyxin E (PME, Colistin) | Methyloctanoyl/methylheptanoyl | Dab1-Thr2-Dab3-cy[Dab4-Dab5-DLeu6-Leu7-Dab8-Dab8-Thr10] |
| FADD002 | Octanoyl | Dab1-Thr2-Dab3-cy[Dab4-Dab5-DAda6-Leu7-Dab8-Dab8-Thr10] |
| FADD287 | Octanoyl | Dab1-Thr2-Dap3-cy[Dab4-Dab5-DLeu6-Abu7-Dab8-Dab8-Thr10] |
| CA284 | (S)-1-(2-methylpropyl)-piperazine-2-carbonyl+ | Thr1-Dab2-cy[Dab3-Dab4-DPhe-Leu6-Dab7-Dab8-Thr9] |
| SPR206 | (3S)-4-amino-3-(3-chlorophenyl)butanoyl | Thr1-Dab2-cy[Dab3-Dab4-DPhe-Leu6-Dab7-Dab8-Thr9] |
| MicuRx-12 | 3-(2,2-dimethyl-butanoyloxy)-propanoyl (ester bond) | Dab1-Thr2-Dab3-cy[Dab4-Dab5-DPhe6-Leu7-Dab8-Dab8-Thr10] |
| NAB379 | Octanoyl | Thr1-DSer2-cy[Dab3-Dab4-DPhe5-Leu6-Dab7-Dab8-Thr9] |
| NAB815 | Octanoyl | Dab1-Thr2-DThr3-cy[Dab4-Dab5-DPhe6-Leu7-Abu8-Dab8-Thr10] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhattacharjya, S.; Mohid, S.A.; Bhunia, A. Atomic-Resolution Structures and Mode of Action of Clinically Relevant Antimicrobial Peptides. Int. J. Mol. Sci. 2022, 23, 4558. https://doi.org/10.3390/ijms23094558
Bhattacharjya S, Mohid SA, Bhunia A. Atomic-Resolution Structures and Mode of Action of Clinically Relevant Antimicrobial Peptides. International Journal of Molecular Sciences. 2022; 23(9):4558. https://doi.org/10.3390/ijms23094558
Chicago/Turabian StyleBhattacharjya, Surajit, Sk Abdul Mohid, and Anirban Bhunia. 2022. "Atomic-Resolution Structures and Mode of Action of Clinically Relevant Antimicrobial Peptides" International Journal of Molecular Sciences 23, no. 9: 4558. https://doi.org/10.3390/ijms23094558
APA StyleBhattacharjya, S., Mohid, S. A., & Bhunia, A. (2022). Atomic-Resolution Structures and Mode of Action of Clinically Relevant Antimicrobial Peptides. International Journal of Molecular Sciences, 23(9), 4558. https://doi.org/10.3390/ijms23094558