
Mitochondrial and Neuronal Dysfunctions in L1 Mutant Mice

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
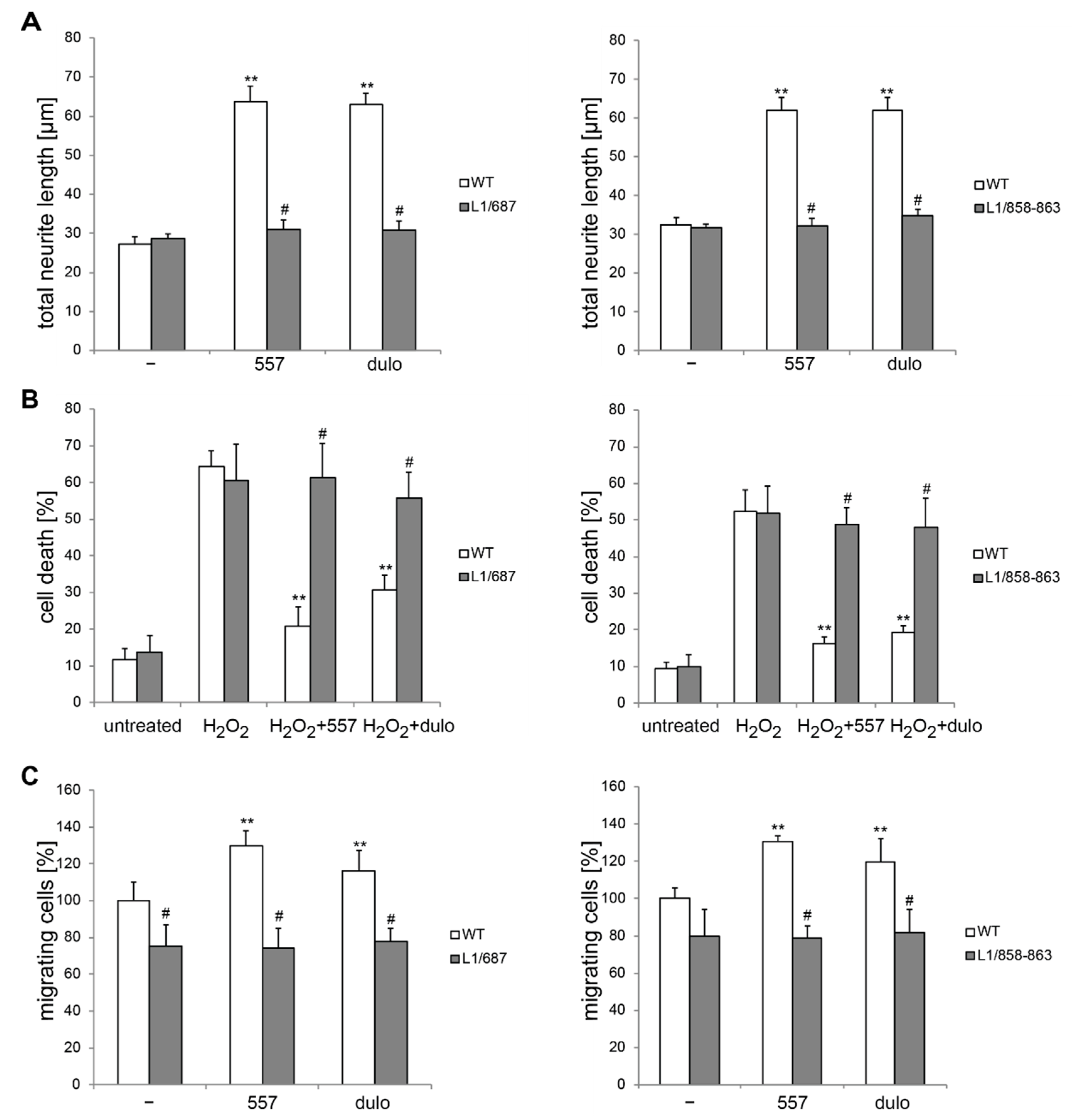
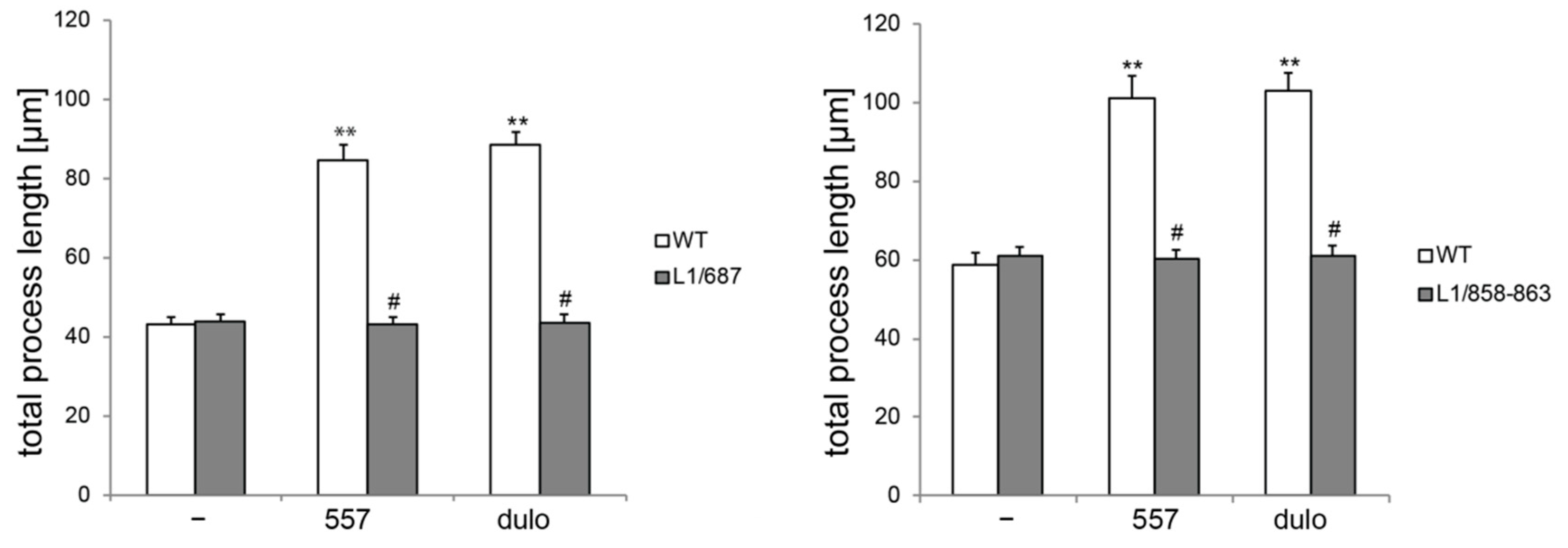
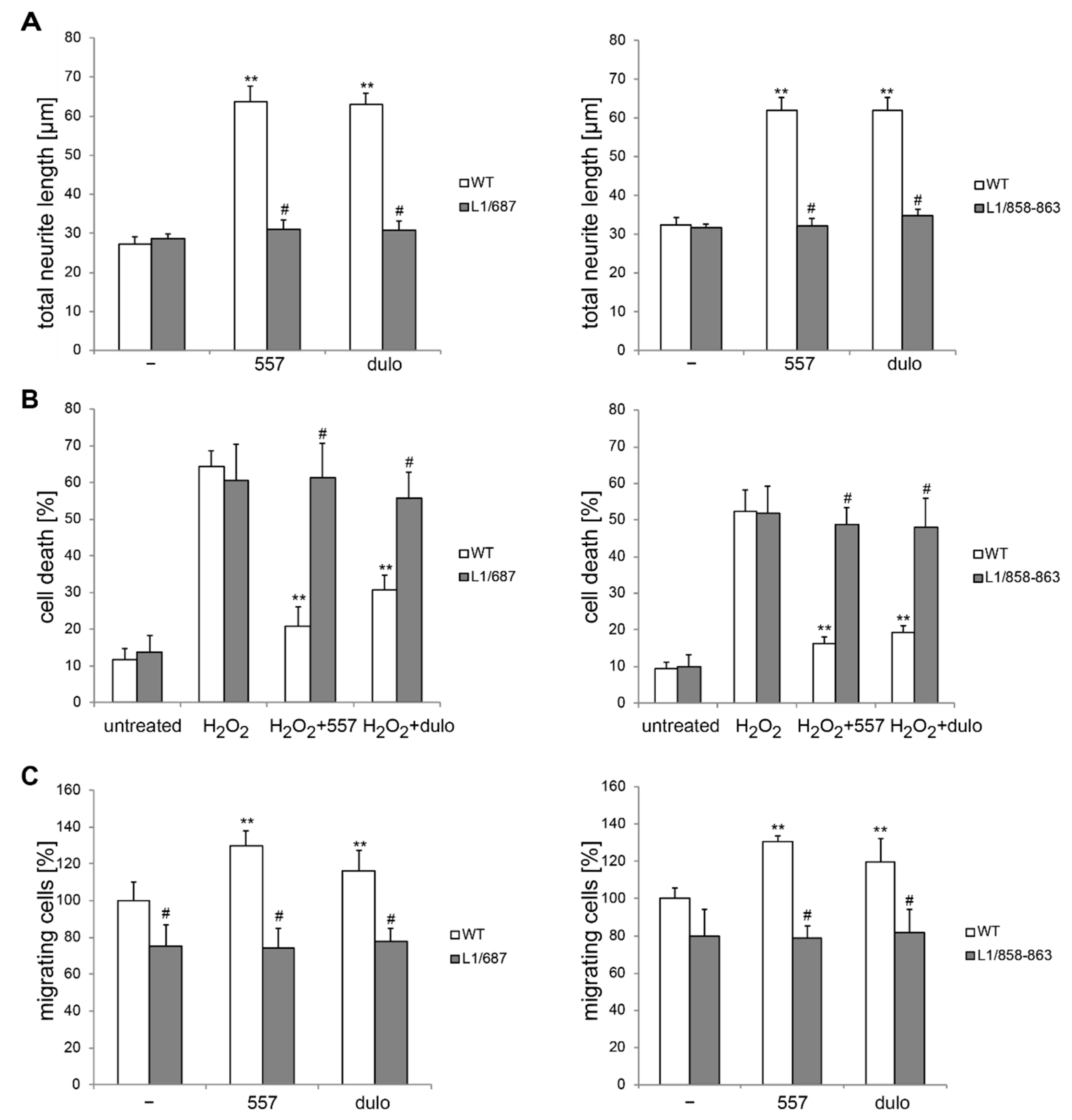
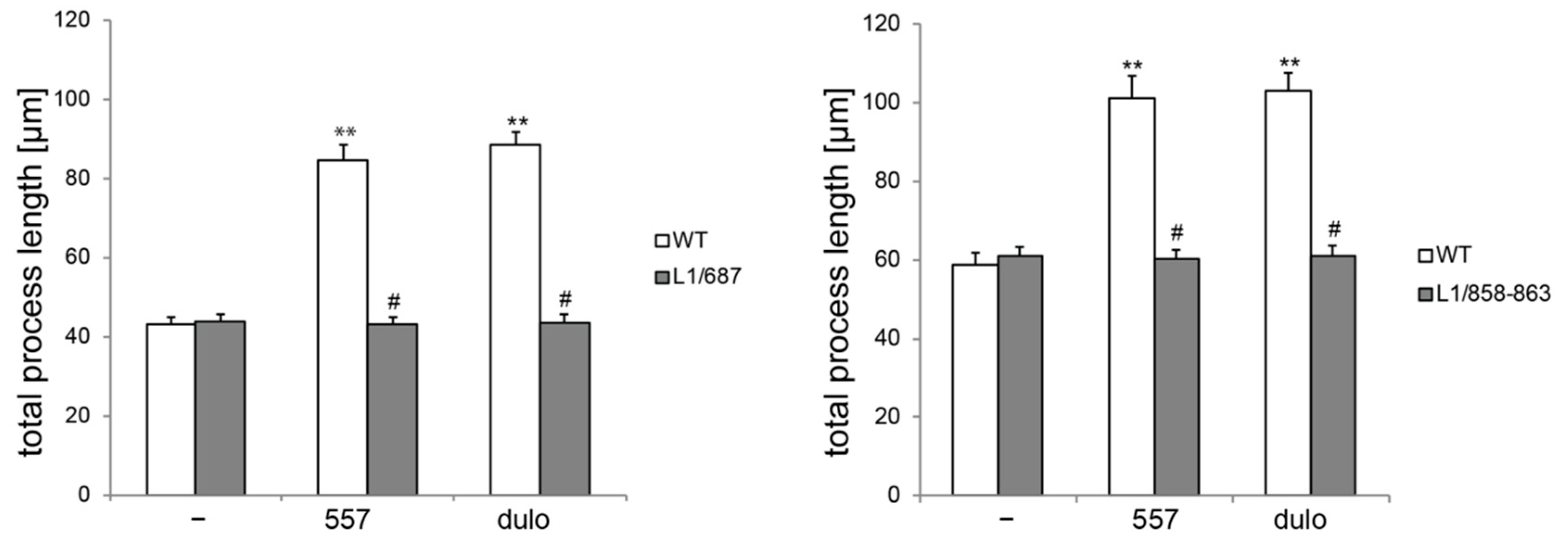
2.1. Survival and Migration of Neurons and Neurite Outgrowth in L1/858-863 and L1/687 Mutant Mice Are Not Enhanced by L1 Function-Triggering Reagents
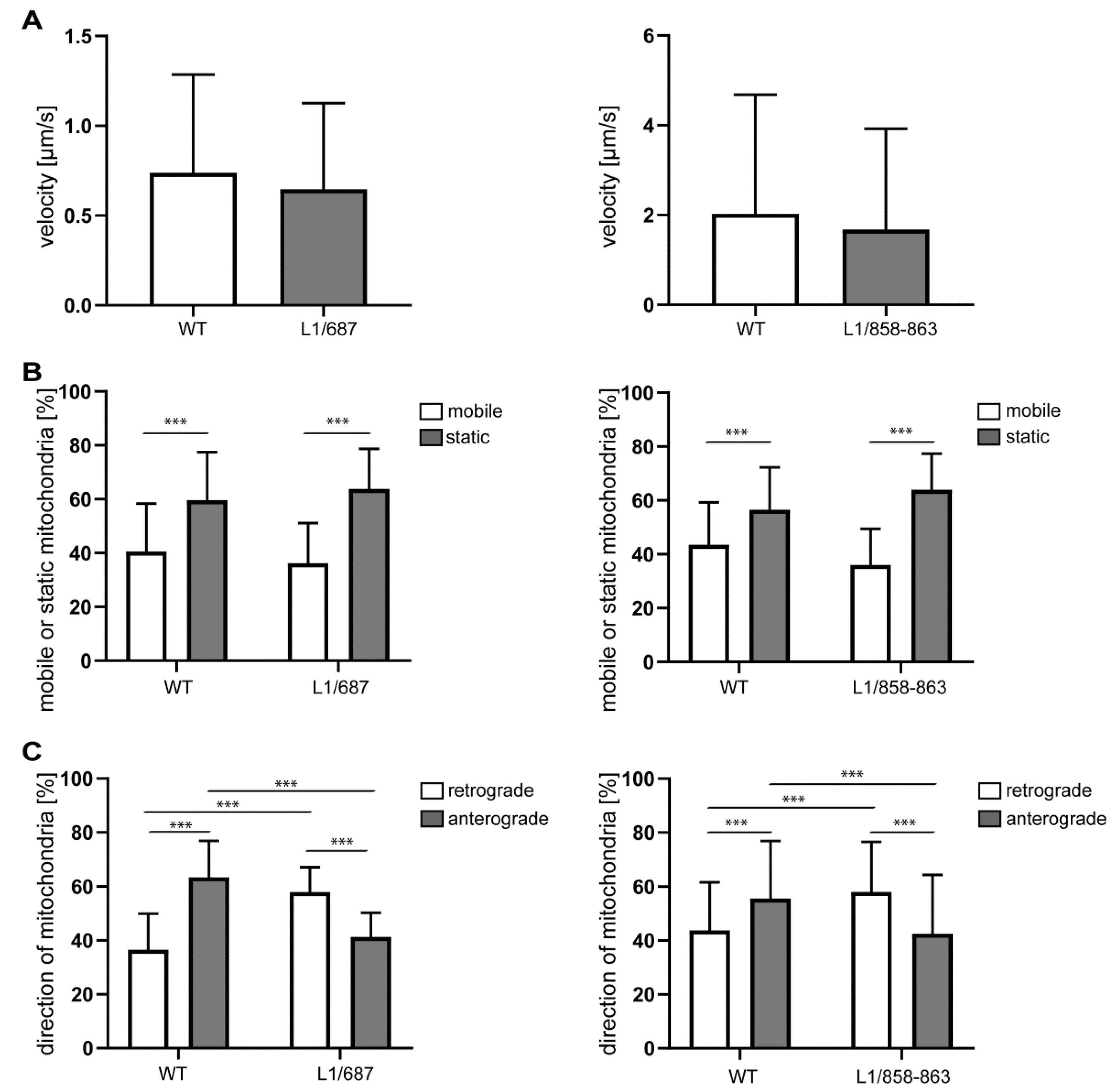
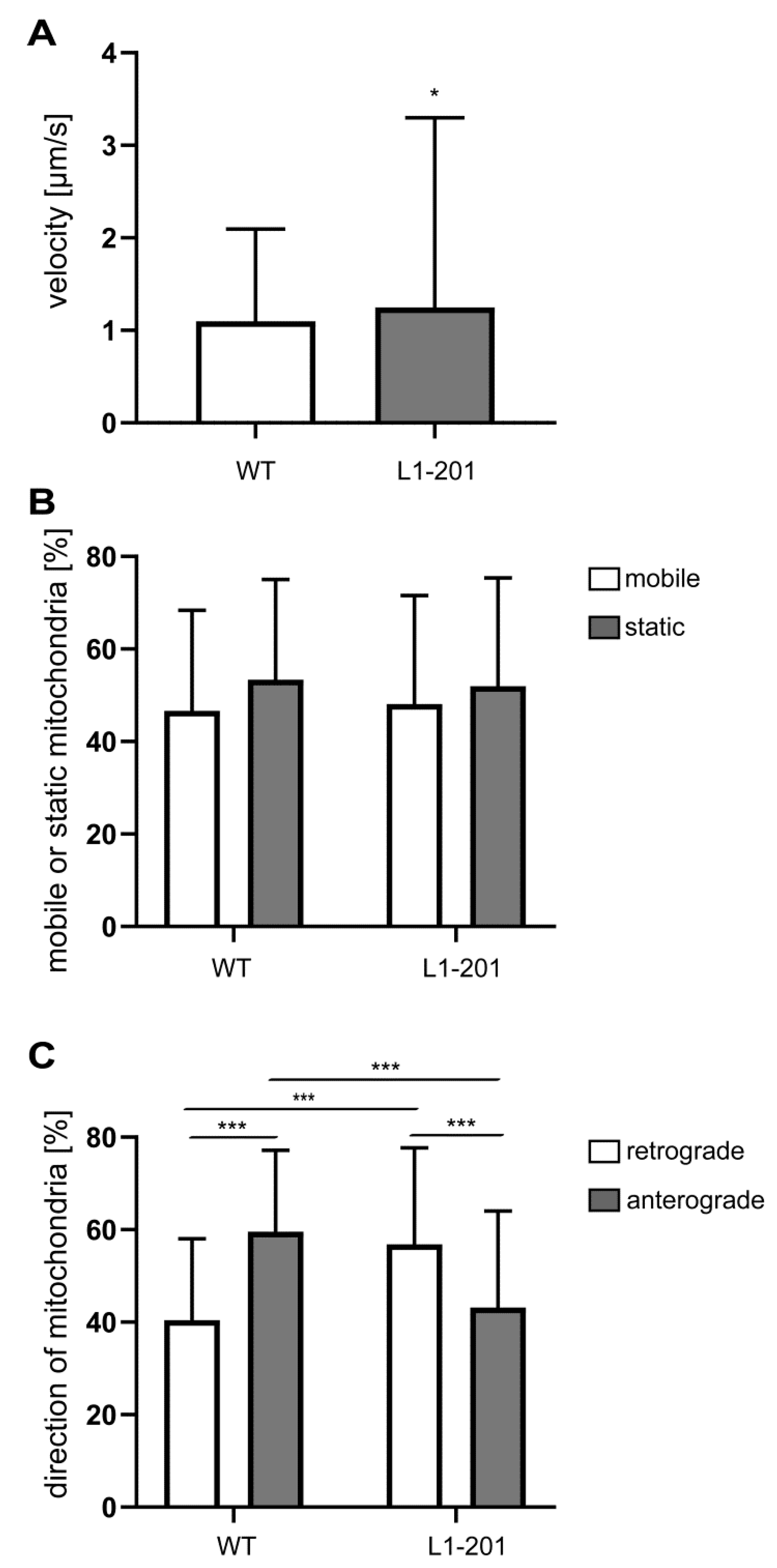
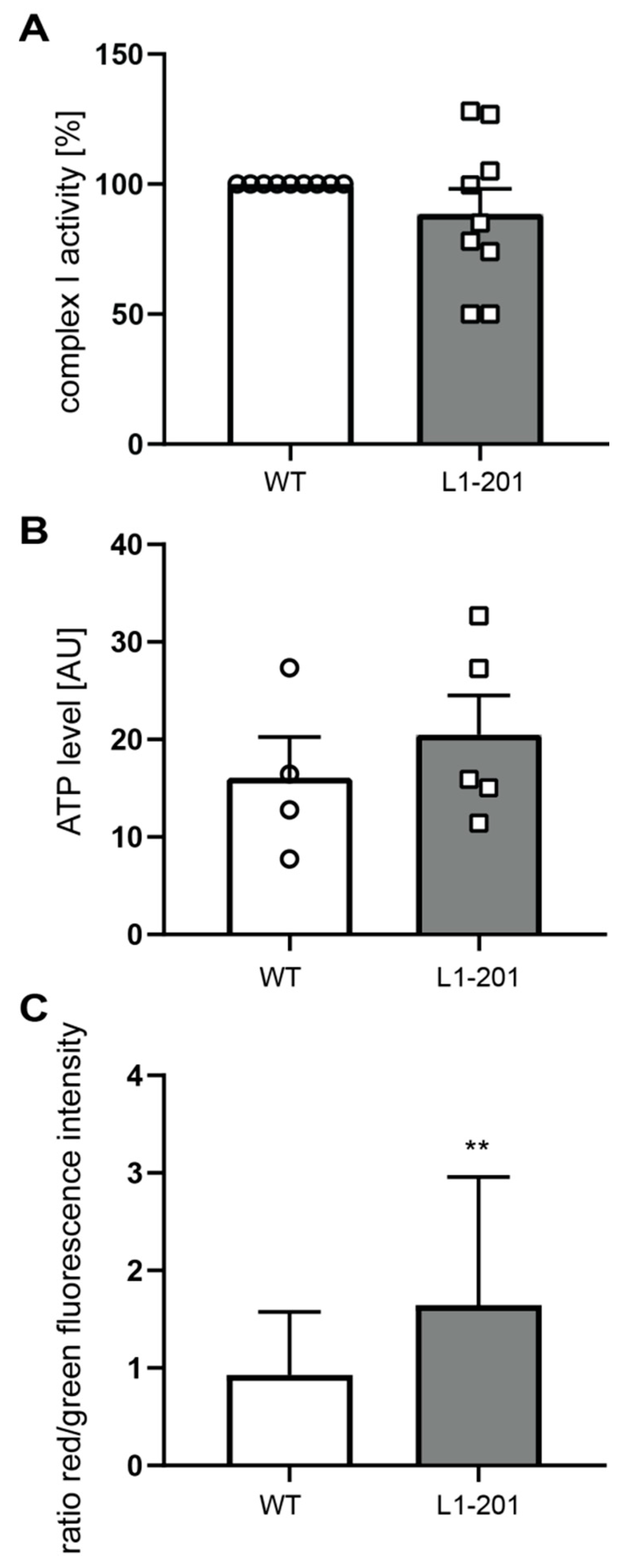
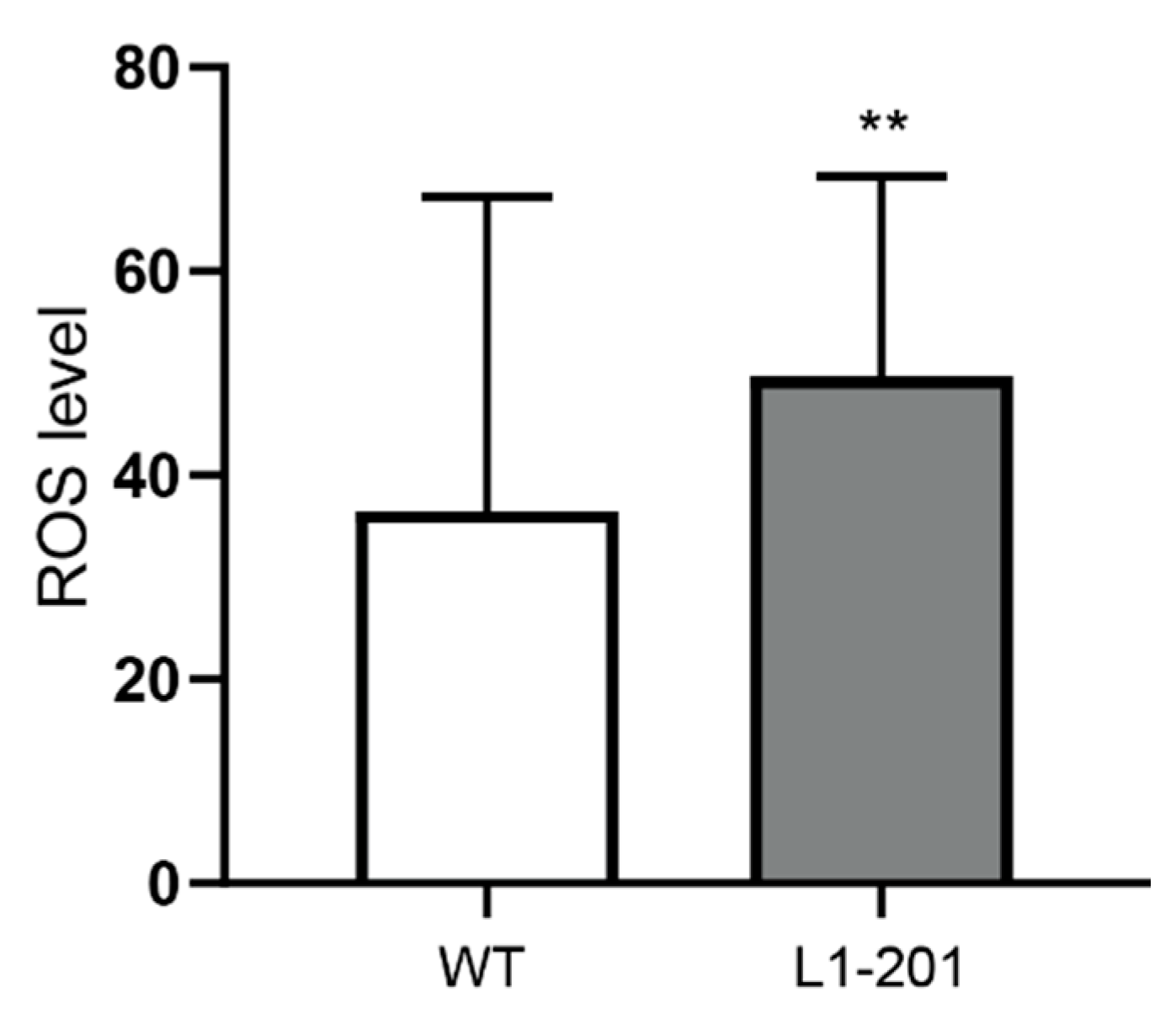
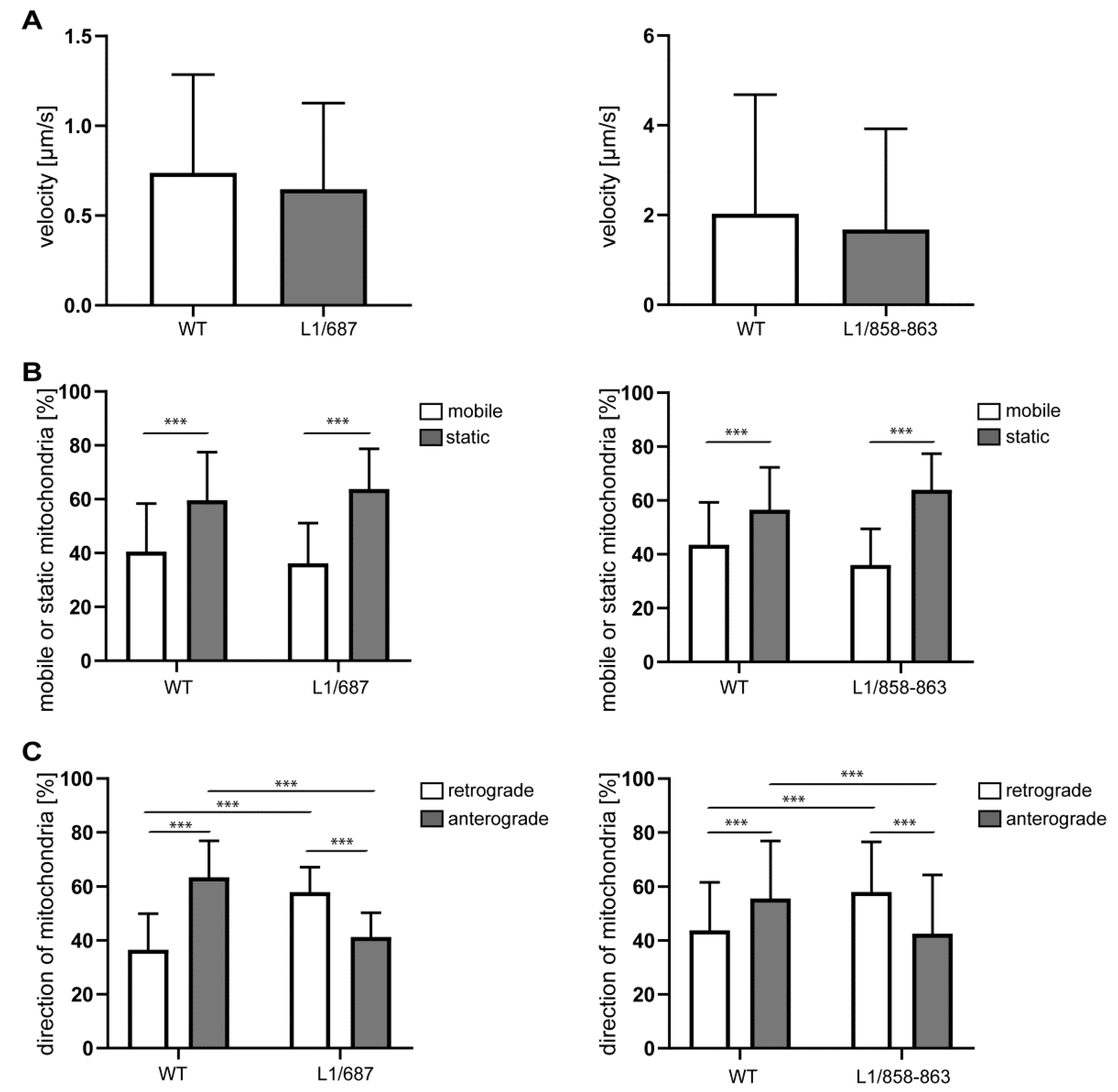
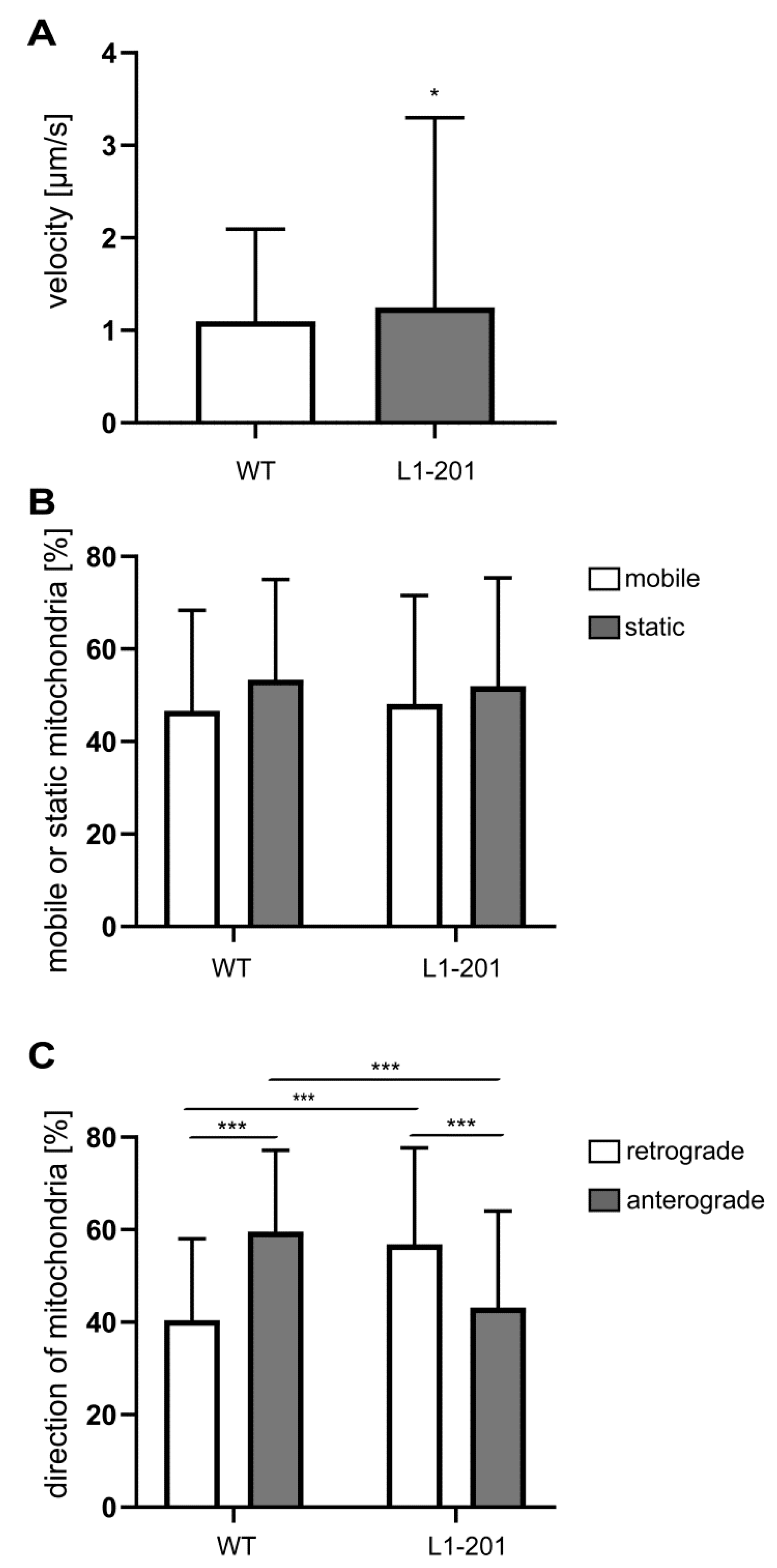
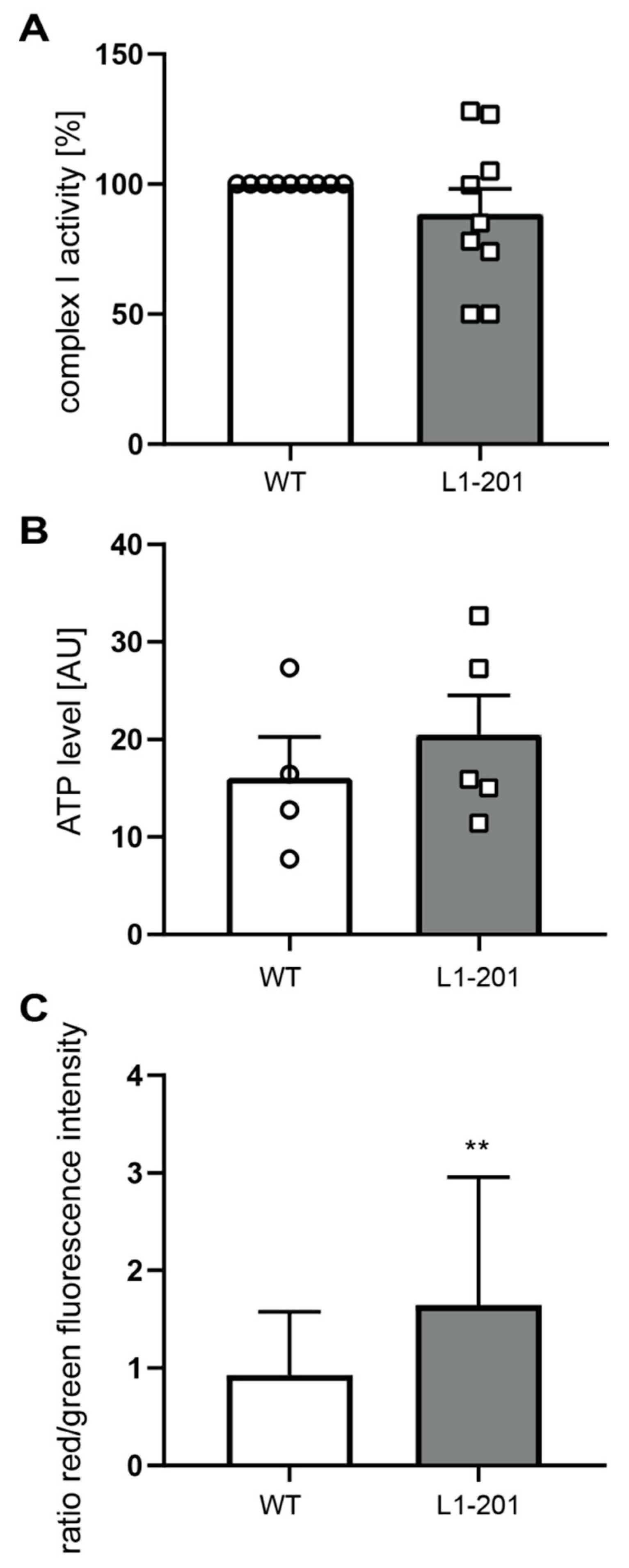
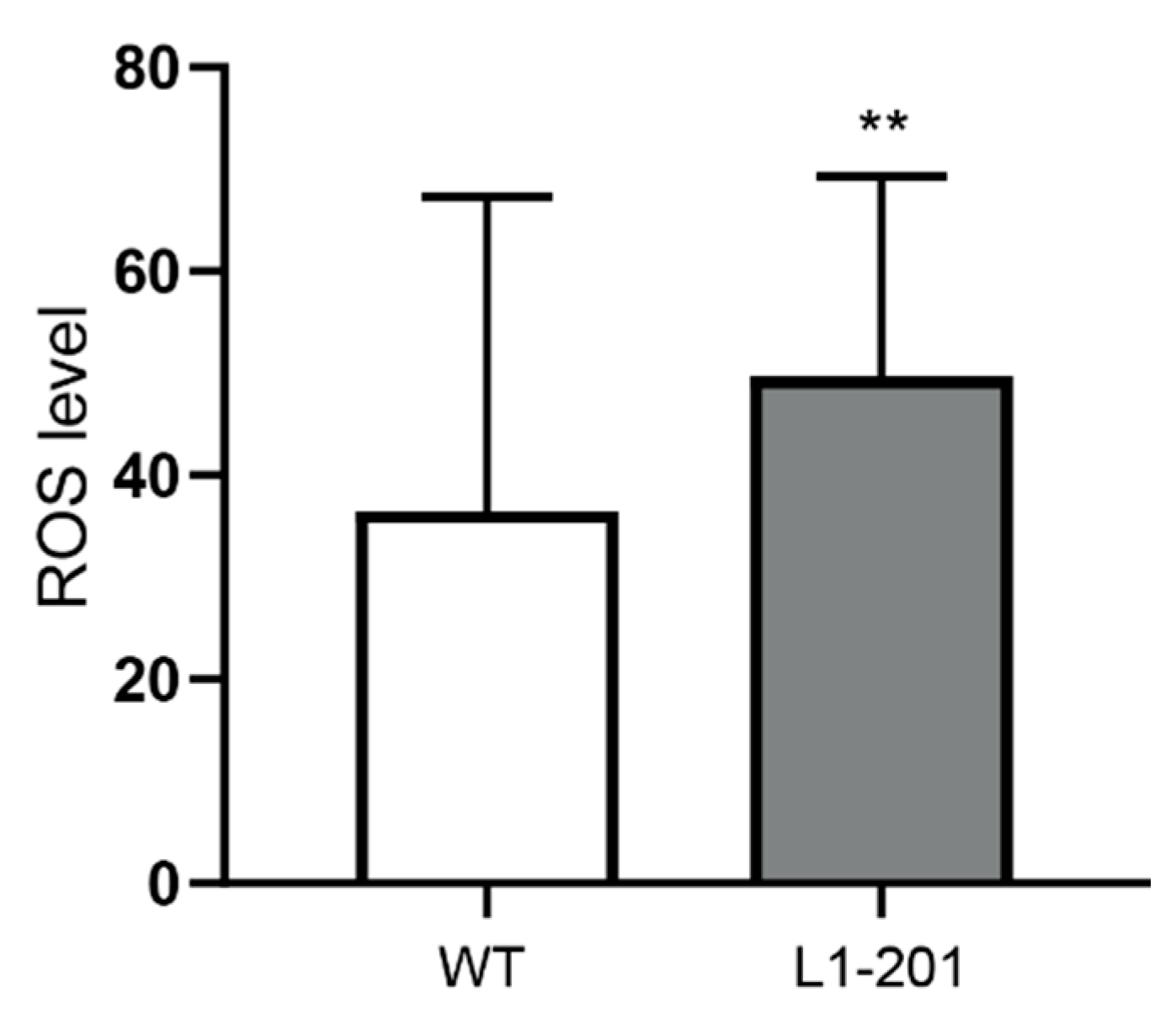
2.2. Impaired Mitochondrial Function in Neurons from L1/858-863, L1/687 and L1-201 Mice
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Cerebellar Neuron Culture, Neurite Outgrowth, Neuronal Migration and Neuronal Survival
4.3. Schwann Cell Process Length
4.4. Mitochondrial Movement
4.5. Mitochondrial Membrane Potential
4.6. Mitochondrial Complex I Activity
4.7. ATP Levels
4.8. Reactive Oxygen Species
4.9. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jung, H.; Kim, S.Y.; Canbakis Cecen, F.S.; Cho, Y.; Kwon, S.K. Dysfunction of mitochondrial Ca(2+) regulatory machineries in brain aging and neurodegenerative diseases. Front. Cell Dev. Biol. 2020, 8, 599792. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y. Mitochondrial support and local translation of mitochondrial proteins in synaptic plasticity and function. Histol. Histopathol. 2021, 36, 1007–1019. [Google Scholar] [PubMed]
- Stanga, S.; Caretto, A.; Boido, M.; Vercelli, A. Mitochondrial dysfunctions: A red thread across neurodegenerative diseases. Int. J. Mol. Sci. 2020, 21, 3719. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Z.; Ristow, M. Mitochondria and metabolic homeostasis. Antioxid. Redox Signal. 2013, 19, 240–242. [Google Scholar] [CrossRef]
- Madreiter-Sokolowski, C.T.; Ramadani-Muja, J.; Ziomek, G.; Burgstaller, S.; Bischof, H.; Koshenov, Z.; Gottschalk, B.; Malli, R.; Graier, W.F. Tracking intra- and inter-organelle signaling of mitochondria. FEBS J. 2019, 286, 4378–4401. [Google Scholar] [CrossRef] [Green Version]
- Protasoni, M.; Zeviani, M. Mitochondrial structure and bioenergetics in normal and disease conditions. Int. J. Mol. Sci. 2021, 22, 586. [Google Scholar] [CrossRef]
- Brunetti, D.; Dykstra, W.; Le, S.; Zink, A.; Prigione, A. Mitochondria in neurogenesis: Implications for mitochondrial diseases. Stem Cells 2021, 39, 1289–1297. [Google Scholar] [CrossRef]
- Kawamata, H.; Manfredi, G. Proteinopathies and OXPHOS dysfunction in neurodegenerative diseases. J. Cell Biol. 2017, 216, 3917–3929. [Google Scholar] [CrossRef]
- McAvoy, K.; Kawamata, H. Glial mitochondrial function and dysfunction in health and neurodegeneration. Mol. Cell. Neurosci. 2019, 101, 103417. [Google Scholar] [CrossRef]
- Dionisio, P.A.; Amaral, J.D.; Rodrigues, C.M.P. Oxidative stress and regulated cell death in Parkinson’s disease. Ageing Res. Rev. 2021, 67, 101263. [Google Scholar] [CrossRef] [PubMed]
- Grimm, A.; Mensah-Nyagan, A.G.; Eckert, A. Alzheimer, mitochondria and gender. Neurosci. Biobehav. Rev. 2016, 67, 89–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, M.; Zhao, X.; Song, F. Aging-dependent mitophagy dysfunction in Alzheimer’s disease. Mol. Neurobiol. 2021, 58, 2362–2378. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Boyd-Kimball, D. Mitochondrial oxidative and nitrosative stress and Alzheimer disease. Antioxidants 2020, 9, 818. [Google Scholar] [CrossRef]
- Obrador, E.; Salvador, R.; Lopez-Blanch, R.; Jihad-Jebbar, A.; Valles, S.L.; Estrela, J.M. Oxidative stress, neuroinflammation and mitochondria in the pathophysiology of amyotrophic lateral sclerosis. Antioxidants 2020, 9, 901. [Google Scholar] [CrossRef]
- Park, H.; Kim, J.; Shin, C.; Lee, S. Intersection between redox homeostasis and autophagy: Valuable insights into neurodegeneration. Antioxidants 2021, 10, 694. [Google Scholar] [CrossRef]
- Khacho, M.; Harris, R.; Slack, R.S. Mitochondria as central regulators of neural stem cell fate and cognitive function. Nat. Rev. Neurosci. 2019, 20, 34–48. [Google Scholar] [CrossRef]
- Adebayo, M.; Singh, S.; Singh, A.P.; Dasgupta, S. Mitochondrial fusion and fission: The fine-tune balance for cellular homeostasis. FASEB J. 2021, 35, e21620. [Google Scholar] [CrossRef]
- Kim, I.; Rodriguez-Enriquez, S.; Lemasters, J.J. Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 2007, 462, 245–253. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Ying, J.; Wang, X.; Zhao, T.; Yoon, S.; Fang, Y.; Zheng, Q.; Liu, X.; Yu, W.; Hua, F. Mitochondrial dynamics: A key role in neurodegeneration and a potential target for neurodegenerative disease. Front. Neurosci. 2021, 15, 654785. [Google Scholar] [CrossRef]
- Castellanos, E.; Lanning, N.J. Phosphorylation of OXPHOS machinery subunits: Functional implications in cell biology and disease. Yale J. Biol. Med. 2019, 92, 523–531. [Google Scholar] [PubMed]
- Maness, P.F.; Schachner, M. Neural recognition molecules of the immunoglobulin superfamily: Signaling transducers of axon guidance and neuronal migration. Nat. Neurosci. 2007, 10, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Sytnyk, V.; Leshchyns’ka, I.; Schachner, M. Neural cell adhesion molecules of the immunoglobulin superfamily regulate synapse formation, maintenance, and function. Trends Neurosci. 2017, 40, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, K.; Mualla, R.; Hortsch, M. The L1 family of cell adhesion molecules: A sickening number of mutations and protein functions. Adv. Neurobiol. 2014, 8, 195–229. [Google Scholar]
- Stumpel, C.; Vos, Y.J. L1 Syndrome. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2004. [Google Scholar]
- Schäfer, M.; Altevogt, P. L1CAM malfunction in the nervous system and human carcinomas. Cell. Mol. Life Sci. 2010, 67, 2425–2437. [Google Scholar] [CrossRef]
- Schäfer, M.K.; Nam, Y.C.; Moumen, A.; Keglowich, L.; Bouche, E.; Kuffner, M.; Bock, H.H.; Rathjen, F.G.; Raoul, C.; Frotscher, M. L1 syndrome mutations impair neuronal L1 function at different levels by divergent mechanisms. Neurobiol. Dis. 2010, 40, 222–237. [Google Scholar] [CrossRef]
- Kraus, K.; Kleene, R.; Braren, I.; Loers, G.; Lutz, D.; Schachner, M. A fragment of adhesion molecule L1 is imported into mitochondria, and regulates mitochondrial metabolism and trafficking. J. Cell Sci. 2018, 131, jcs210500. [Google Scholar] [CrossRef] [Green Version]
- Loers, G.; Makhina, T.; Bork, U.; Dorner, A.; Schachner, M.; Kleene, R. The interaction between cell adhesion molecule L1, matrix metalloproteinase 14, and adenine nucleotide translocator at the plasma membrane regulates L1-mediated neurite outgrowth of murine cerebellar neurons. J. Neurosci. 2012, 32, 3917–3930. [Google Scholar] [CrossRef] [Green Version]
- Kleene, R.; Lutz, D.; Loers, G.; Bork, U.; Borgmeyer, U.; Hermans-Borgmeyer, I.; Schachner, M. Revisiting the proteolytic processing of cell adhesion molecule L1. J. Neurochem. 2021, 157, 1102–1117. [Google Scholar] [CrossRef]
- Loers, G.; Appel, D.; Lutz, D.; Congiu, L.; Kleene, R.; Hermans-Borgmeyer, I.; Schafer, M.K.E.; Schachner, M. Amelioration of the abnormal phenotype of a new L1 syndrome mouse mutation with L1 mimetics. FASEB J. 2021, 35, e21329. [Google Scholar] [CrossRef]
- Kataria, H.; Lutz, D.; Chaudhary, H.; Schachner, M.; Loers, G. Small Molecule Agonists of Cell Adhesion Molecule L1 Mimic L1 Functions In Vivo. Mol. Neurobiol. 2016, 53, 4461–4483. [Google Scholar] [CrossRef] [PubMed]
- Appel, F.; Holm, J.; Conscience, J.F.; von Bohlen, F.; Faissner, A.; James, P.; Schachner, M. Identification of the border between fibronectin type III homologous repeats 2 and 3 of the neural cell adhesion molecule L1 as a neurite outgrowth promoting and signal transducing domain. J. Neurobiol. 1995, 28, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Fransen, E.; Van Camp, G.; Vits, L.; Willems, P.J. L1-associated diseases: Clinical geneticists divide, molecular geneticists unite. Hum. Mol. Genet. 1997, 6, 1625–1632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, Y.; Murakami, Y.; Asou, H.; Uyemura, K. The roles of cell adhesion molecules on the formation of peripheral myelin. Keio J. Med. 2001, 50, 240–248. [Google Scholar] [CrossRef]
- Kraus, K.; Kleene, R.; Henis, M.; Braren, I.; Kataria, H.; Sharaf, A.; Loers, G.; Schachner, M.; Lutz, D. A fragment of adhesion molecule L1 binds to nuclear receptors to regulate synaptic plasticity and motor coordination. Mol. Neurobiol. 2018, 55, 7164–7178. [Google Scholar] [CrossRef]
- Lutz, D.; Wolters-Eisfeld, G.; Joshi, G.; Djogo, N.; Jakovcevski, I.; Schachner, M.; Kleene, R. Generation and nuclear translocation of sumoylated transmembrane fragment of cell adhesion molecule L1. J. Biol. Chem. 2012, 287, 17161–17175. [Google Scholar] [CrossRef] [Green Version]
- Morris, R.L.; Hollenbeck, P.J. The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. J. Cell Sci. 1993, 104 Pt 3, 917–927. [Google Scholar] [CrossRef]
- Vakifahmetoglu-Norberg, H.; Ouchida, A.T.; Norberg, E. The role of mitochondria in metabolism and cell death. Biochem. Biophys. Res. Commun. 2017, 482, 426–431. [Google Scholar] [CrossRef]
- Vinokurov, A.Y.; Stelmashuk, O.A.; Ukolova, P.A.; Zherebtsov, E.A.; Abramov, A.Y. Brain region specificity in reactive oxygen species production and maintenance of redox balance. Free Radic. Biol. Med. 2021, 174, 195–201. [Google Scholar] [CrossRef]
- Lin-Hendel, E.G.; McManus, M.J.; Wallace, D.C.; Anderson, S.A.; Golden, J.A. Differential mitochondrial requirements for radially and non-radially migrating cortical neurons: Implications for mitochondrial disorders. Cell Rep. 2016, 15, 229–237. [Google Scholar] [CrossRef] [Green Version]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biol. 2010, 8, e1000412. [Google Scholar] [CrossRef] [PubMed]
- Loers, G.; Chen, S.; Grumet, M.; Schachner, M. Signal transduction pathways implicated in neural recognition molecule L1 triggered neuroprotection and neuritogenesis. J. Neurochem. 2005, 92, 1463–1476. [Google Scholar] [CrossRef] [PubMed]
- Jakovcevski, I.; Siering, J.; Hargus, G.; Karl, N.; Hoelters, L.; Djogo, N.; Yin, S.; Zecevic, N.; Schachner, M.; Irintchev, A. Close homologue of adhesion molecule L1 promotes survival of Purkinje and granule cells and granule cell migration during murine cerebellar development. J. Comp. Neurol. 2009, 513, 496–510. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehanna, A.; Mishra, B.; Kurschat, N.; Schulze, C.; Bian, S.; Loers, G.; Irintchev, A.; Schachner, M. Polysialic acid glycomimetics promote myelination and functional recovery after peripheral nerve injury in mice. Brain 2009, 132 Pt 6, 1449–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marra, M.H.; Tobias, Z.J.; Cohen, H.R.; Glover, G.; Weissman, T.A. In vivo time-lapse imaging in the zebrafish lateral line: A flexible, open-ended research project for an undergraduate neurobiology laboratory course. J. Undergrad. Neurosci. Educ. 2015, 13, A215–A224. [Google Scholar]
- Menke, T.; Gille, G.; Reber, F.; Janetzky, B.; Andler, W.; Funk, R.H.; Reichmann, H. Coenzyme Q10 reduces the toxicity of rotenone in neuronal cultures by preserving the mitochondrial membrane potential. Biofactors 2003, 18, 65–72. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Congiu, L.; Granato, V.; Loers, G.; Kleene, R.; Schachner, M. Mitochondrial and Neuronal Dysfunctions in L1 Mutant Mice. Int. J. Mol. Sci. 2022, 23, 4337. https://doi.org/10.3390/ijms23084337
Congiu L, Granato V, Loers G, Kleene R, Schachner M. Mitochondrial and Neuronal Dysfunctions in L1 Mutant Mice. International Journal of Molecular Sciences. 2022; 23(8):4337. https://doi.org/10.3390/ijms23084337
Chicago/Turabian StyleCongiu, Ludovica, Viviana Granato, Gabriele Loers, Ralf Kleene, and Melitta Schachner. 2022. "Mitochondrial and Neuronal Dysfunctions in L1 Mutant Mice" International Journal of Molecular Sciences 23, no. 8: 4337. https://doi.org/10.3390/ijms23084337
APA StyleCongiu, L., Granato, V., Loers, G., Kleene, R., & Schachner, M. (2022). Mitochondrial and Neuronal Dysfunctions in L1 Mutant Mice. International Journal of Molecular Sciences, 23(8), 4337. https://doi.org/10.3390/ijms23084337