
Template-Free Preparation of a Mesopore-Rich Hierarchically Porous Carbon Monolith from a Thermally Rearrangeable Polyurea Network


Abstract
: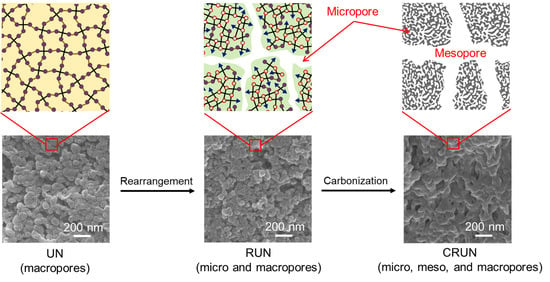
{kind=link}
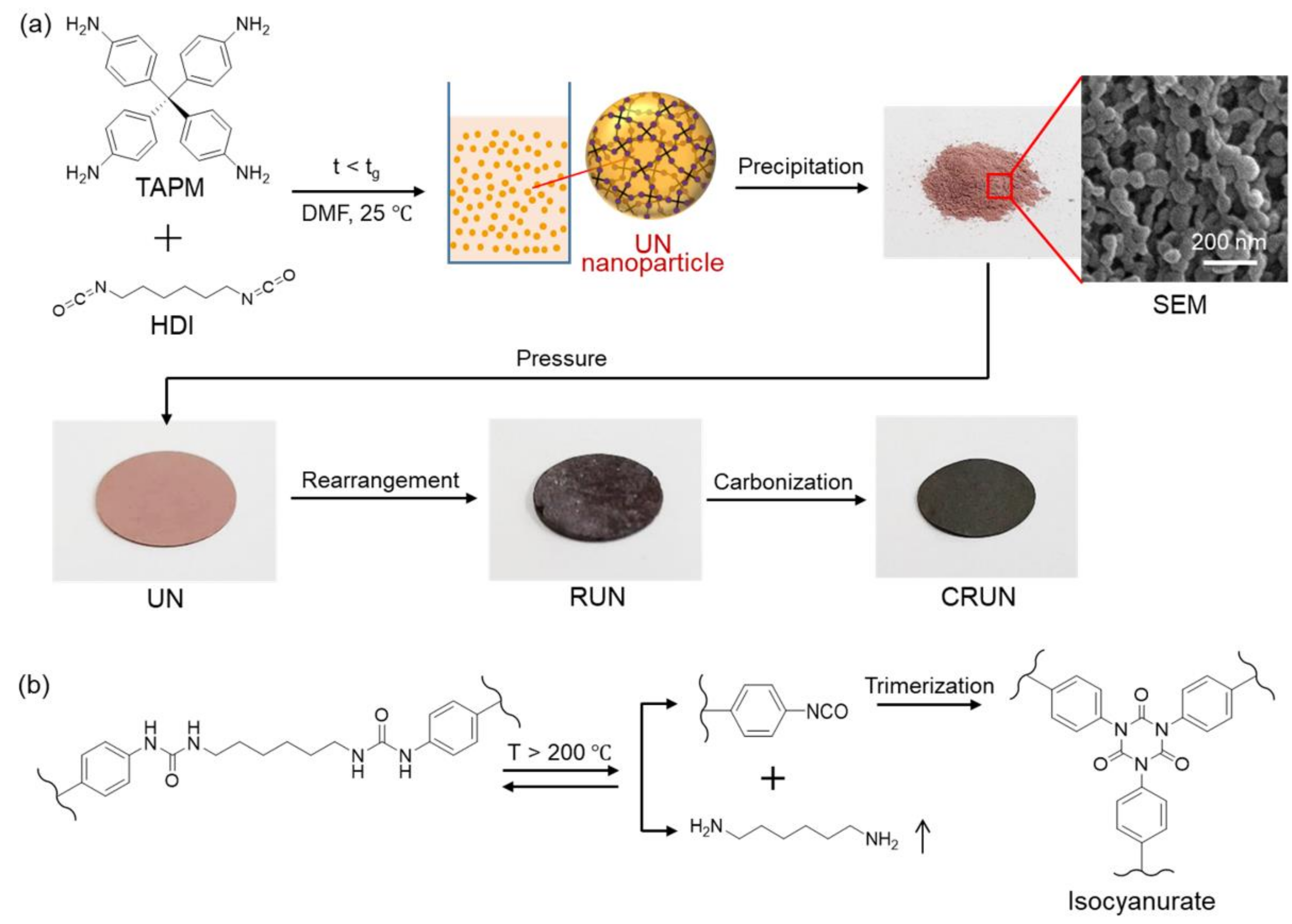{kind=link}
{kind=link}
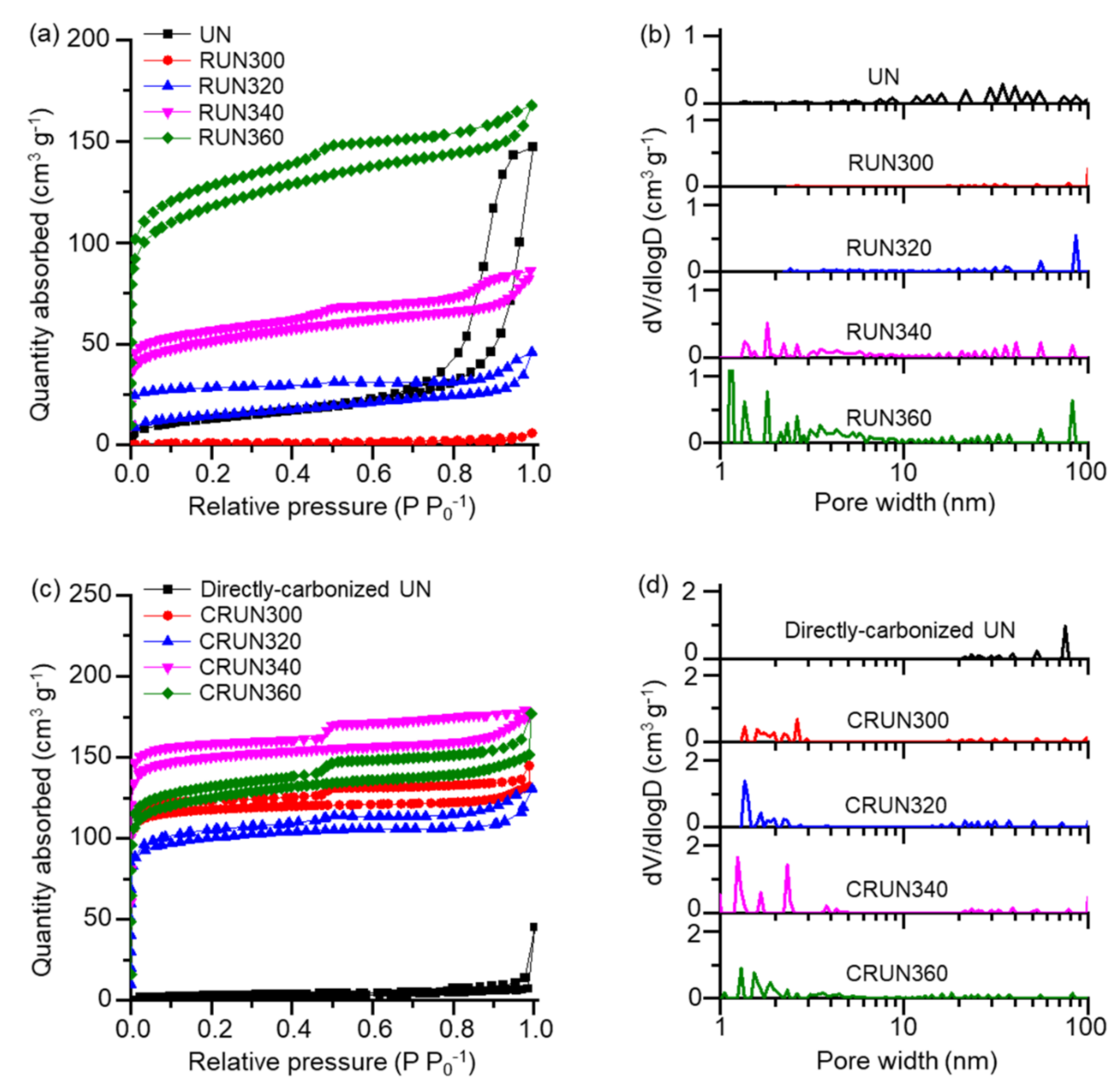{kind=link}
{kind=link}
{kind=link}
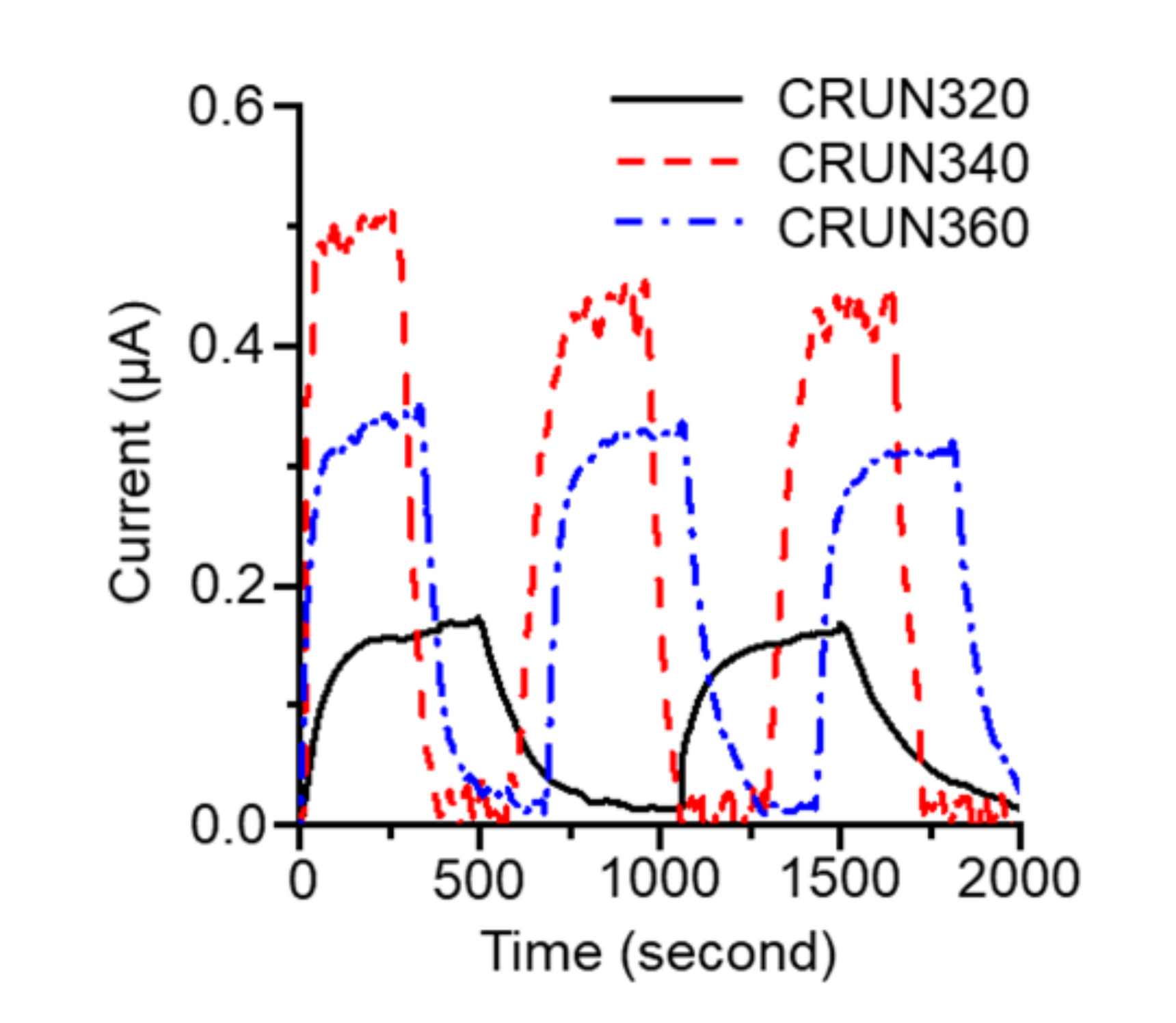{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of CRUNs
2.3. Characterizations
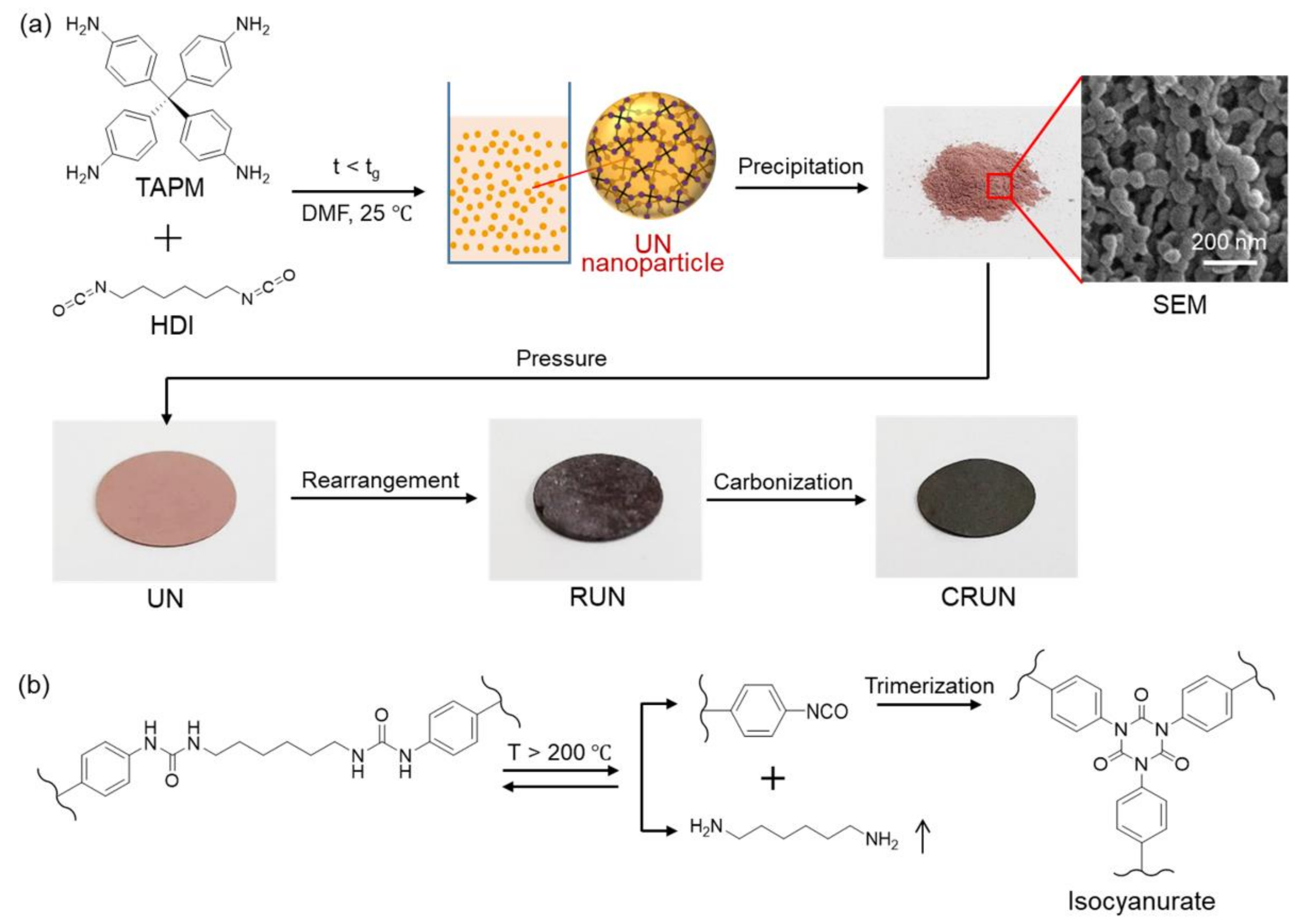
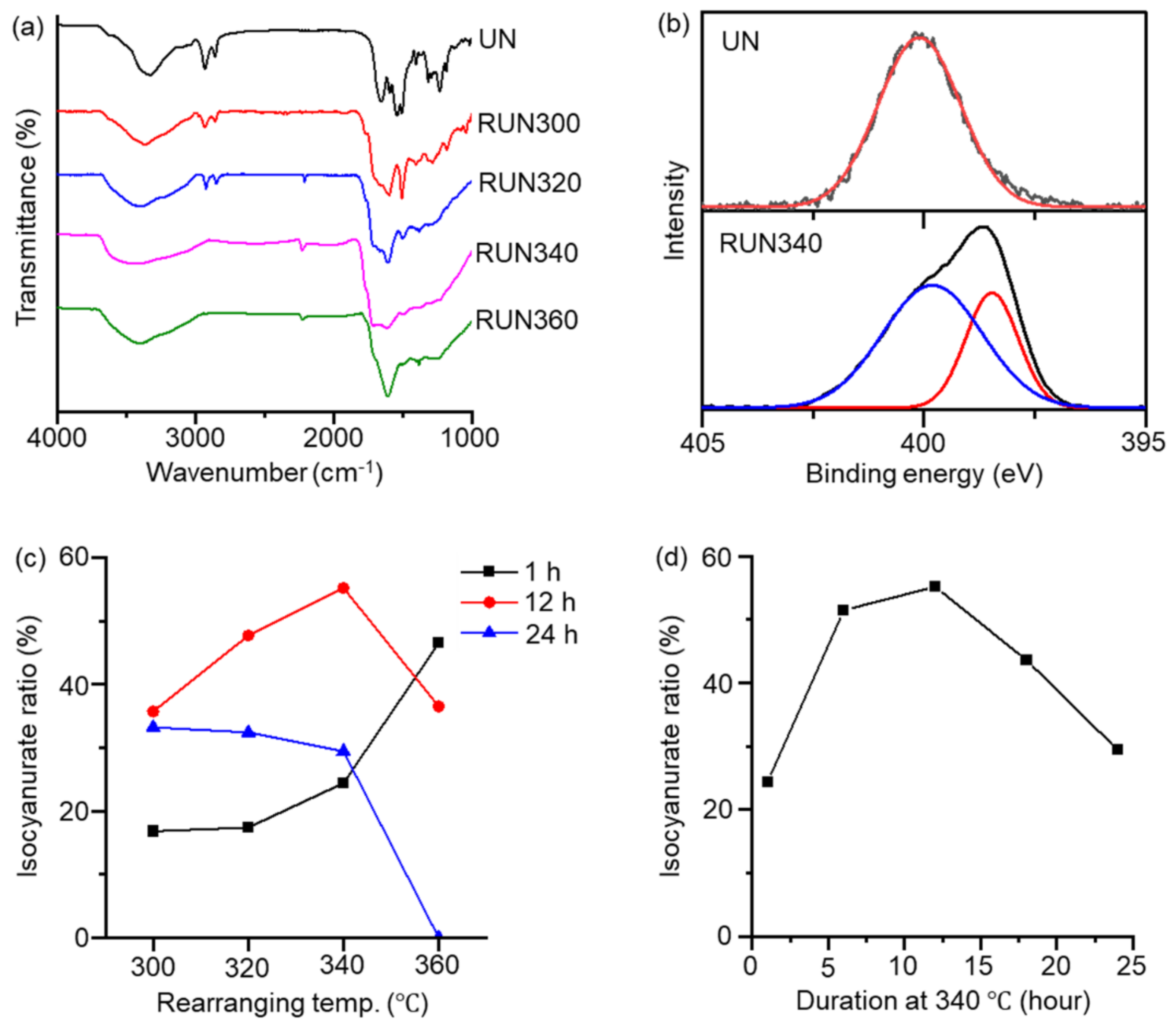
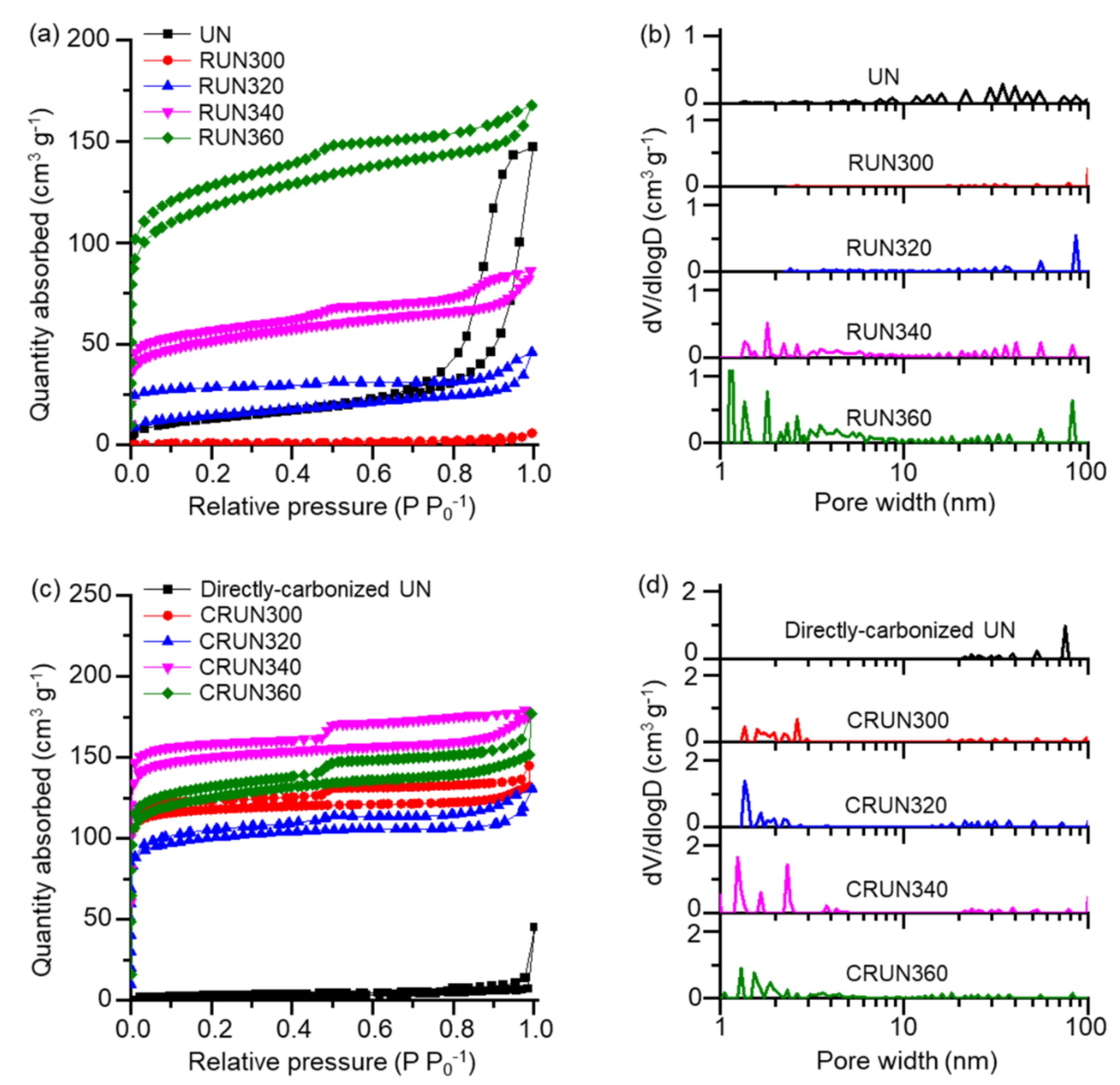
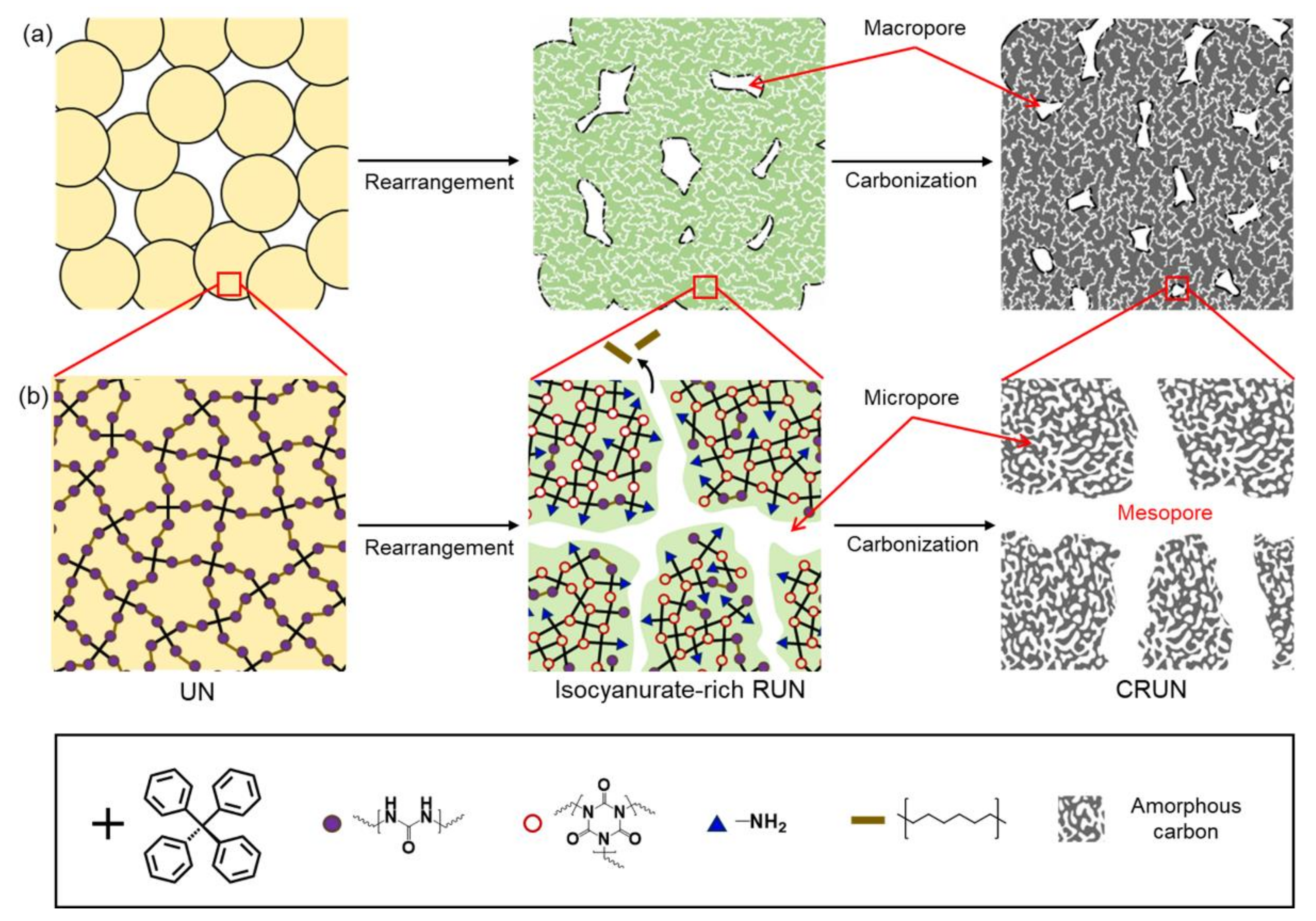
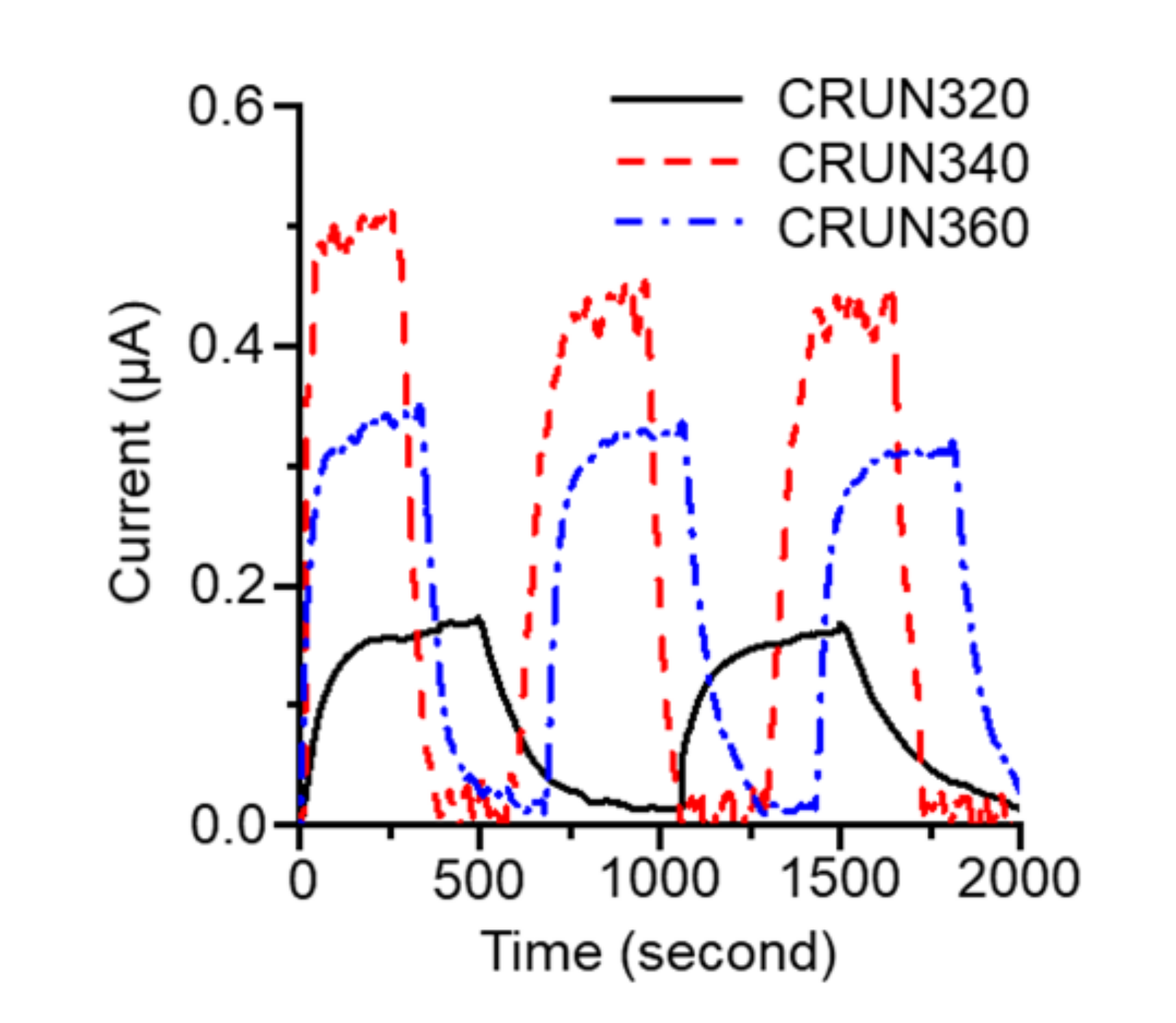
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jun, S.; Joo, S.H.; Ryoo, R.; Kruk, M.; Jaroniec, M.; Liu, Z.; Ohsuna, T.; Terasaki, O. Synthesis of New, Nanoporous Carbon with Hexagonally Ordered Mesostructure. J. Am. Chem. Soc. 2000, 122, 10712–10713. [Google Scholar] [CrossRef]
- Meng, Y.; Gu, D.; Zhang, F.; Shi, Y.; Yang, H.; Li, Z.; Yu, C.; Tu, B.; Zhao, D. Ordered Mesoporous Polymers and Homologous Carbon Frameworks: Amphiphilic Surfactant Templating and Direct Transformation. Angew. Chem. Int. Ed. 2005, 44, 7053–7059. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.D.; Li, X.; Zhang, H. Porous carbon spheres and monoliths: Morphology control, pore size tuning and their applications as Li-ion battery anode materials. Chem. Soc. Rev. 2014, 43, 4341–4356. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Bhaumik, A.; Wu, K.C.W. Hierarchically porous carbon derived from polymers and biomass: Effect of interconnected pores on energy applications. Energy Environ. Sci. 2014, 7, 3574–3592. [Google Scholar] [CrossRef]
- Zhao, X.; Li, W.; Chen, H.; Wang, S.; Kong, F.; Liu, S. Facile Control of the Porous Structure of Larch-Derived Mesoporous Carbons via Self-Assembly for Supercapacitors. Materials 2017, 10, 1330. [Google Scholar] [CrossRef] [Green Version]
- Uriburu-Gray, M.; Pinar-Serrano, A.; Cavus, G.; Knipping, E.; Aucher, C.; Conesa-Cabeza, A.; Satti, A.; Amantia, D.; Martínez-Crespiera, S. Mesoporous Carbons from Polysaccharides and Their Use in Li-O2 Batteries. Nanomaterials 2020, 10, 2036. [Google Scholar] [CrossRef]
- Schlumberger, C.; Thommes, M. Characterization of Hierarchically Ordered Porous Materials by Physisorption and Mercury Porosimetry—A Tutorial Review. Adv. Mater. Interfaces 2021, 8, 2002181. [Google Scholar] [CrossRef]
- Nakanishi, K.; Tanaka, N. Sol–Gel with Phase Separation. Hierarchically Porous Materials Optimized for High-Performance Liquid Chromatography Separations. Acc. Chem. Res. 2007, 40, 863–873. [Google Scholar] [CrossRef]
- Ko, Y.G.; Lee, H.J.; Kim, J.Y.; Choi, U.S. Hierarchically Porous Aminosilica Monolith as a CO2 Adsorbent. ACS Appl. Mater. Interfaces 2014, 6, 12988–12996. [Google Scholar] [CrossRef]
- Daud, W.M.A.W.; Ali, W.S.W.; Sulaiman, M.Z. The effects of carbonization temperature on pore development in palm-shell-based activated carbon. Carbon 2000, 38, 1925–1932. [Google Scholar] [CrossRef]
- Liang, D.; Xie, Q.; Liu, J.; Xie, F.; Liu, D.; Wan, C. Mechanism of the evolution of pore structure during the preparation of activated carbon from Zhundong high-alkali coal based on gas–solid diffusion and activation reactions. RSC Adv. 2020, 10, 33566–33575. [Google Scholar] [CrossRef]
- Kim, T.W.; Park, I.S.; Ryoo, R. A Synthetic Route to Ordered Mesoporous Carbon Materials with Graphitic Pore Walls. Angew. Chem. Int. Ed. 2003, 42, 4375–4379. [Google Scholar] [CrossRef] [PubMed]
- Altay, E.; Nykypanchuk, D.; Rzayev, J. Mesoporous Polymer Frameworks from End-Reactive Bottlebrush Copolymers. ACS Nano 2017, 11, 8207–8214. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.G.; Bein, T. Conducting Carbon Wires in Ordered, Nanometer-Sized Channels. Science 1994, 266, 1013–1015. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Yoon, S.B.; Yu, J.S. Template Synthesis of a New Mesostructured Silica from Highly Ordered Mesoporous Carbon Molecular Sieves. Chem. Mater. 2003, 15, 1932–1934. [Google Scholar] [CrossRef]
- Wei, H.; Lv, Y.; Han, L.; Tu, B.; Zhao, D. Facile Synthesis of Transparent Mesostructured Composites and Corresponding Crack-free Mesoporous Carbon/Silica Monoliths. Chem. Mater. 2011, 23, 2353–2360. [Google Scholar] [CrossRef]
- Feng, D.; Lv, Y.; Wu, Z.; Dou, Y.; Han, L.; Sun, Z.; Xia, Y.; Zheng, G.; Zhao, D. Free-Standing Mesoporous Carbon Thin Films with Highly Ordered Pore Architectures for Nanodevices. J. Am. Chem. Soc. 2011, 133, 15148–15156. [Google Scholar] [CrossRef]
- An, Z.; Kong, S.; Zhang, W.; Yuan, M.; An, Z.; Chen, D. Synthesis and Adsorption Performance of a Hierarchical Micro-Mesoporous Carbon for Toluene Removal under Ambient Conditions. Materials 2020, 13, 716. [Google Scholar] [CrossRef] [Green Version]
- Park, H.B.; Jung, C.H.; Lee, Y.M.; Hill, A.J.; Pas, S.J.; Mudie, S.T.; Van Wagner, E.; Freeman, B.D.; Cookson, D.J. Polymers with Cavities Tuned for Fast Selective Transport of Small Molecules and Ions. Science 2007, 318, 254–258. [Google Scholar] [CrossRef]
- Liaw, D.J.; Wang, K.L.; Huang, Y.C.; Lee, K.R.; Lai, J.Y.; Ha, C.S. Advanced polyimide materials: Syntheses, physical properties and applications. Prog. Polym. Sci. 2012, 37, 907–974. [Google Scholar] [CrossRef]
- Jung, Y.; Byun, S.; Park, S.; Lee, H. Polyimide–Organosilicate Hybrids with Improved Thermal and Optical Properties. ACS Appl. Mater. Interfaces 2014, 6, 6054–6061. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.Y.; Bae, J.S.; Jeon, E.; Park, J.W. Organic Sol–Gel Synthesis: Solution-Processable Microporous Organic Networks. Angew. Chem. Int. Ed. 2010, 49, 9504–9508. [Google Scholar] [CrossRef]
- Oh, W.; Bae, J.S.; Park, J.W. The Interplay between Phase Separation and Gelation Controlling the Morphologies of the Reactive Covalent Network/Polymer Blends. Macromolecules 2021, 54, 1192–1202. [Google Scholar] [CrossRef]
- Zhang, Y.; Riduan, S.N.; Ying, J.Y. Microporous Polyisocyanurate and Its Application in Heterogeneous Catalysis. Chem. Eur. J. 2009, 15, 1077–1081. [Google Scholar] [CrossRef]
- Moon, S.Y.; Jeon, E.; Bae, J.S.; Park, M.K.; Kim, C.; Noh, D.Y.; Lee, E.; Park, J.W. Thermo-processable covalent scaffolds with reticular hierarchical porosity and their high efficiency capture of carbon dioxide. J. Mater. Chem. A 2015, 3, 14871–14875. [Google Scholar] [CrossRef]
- Nam, J.; Jeon, E.; Moon, S.Y.; Park, J.W. Rearranged Copolyurea Networks for Selective Carbon Dioxide Adsorption at Room Temperature. Polymers 2021, 13, 4004. [Google Scholar] [CrossRef] [PubMed]
- Heidarinejad, Z.; Dehghani, M.H.; Heidari, M.; Javedan, G.; Ali, I.; Sillanpää, M. Methods for preparation and activation of activated carbon: A review. Environ. Chem. Lett. 2020, 18, 393–415. [Google Scholar] [CrossRef]
- Patawat, C.; Silakate, K.; Chuan-Udom, S.; Supanchaiyamat, N.; Hunt, A.J.; Ngernyen, Y. Preparation of activated carbon from Dipterocarpus alatus fruit and its application for methylene blue adsorption. RSC Adxv. 2020, 10, 21082–21091. [Google Scholar] [CrossRef]
- Ganesan, P.; Yang, X.; Loos, J.; Savenije, T.J.; Abellon, R.D.; Zuilhof, H.; Sudhölter, E.J.R. Tetrahedral n-Type Materials: Efficient Quenching of the Excitation of p-Type Polymers in Amorphous Films. J. Am. Chem. Soc. 2005, 127, 14530–14531. [Google Scholar] [CrossRef]
- Moon, S.Y.; Mo, H.R.; Ahn, M.K.; Bae, J.S.; Jeon, E.; Park, J.W. Organic sol–gel synthesis of microporous molecular networks containing spirobifluorene and tetraphenylmethane nodes. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 1758–1766. [Google Scholar] [CrossRef]
- Moon, S.Y.; Jeon, E.; Bae, J.S.; Byeon, M.; Park, J.W. Polyurea networks via organic sol–gel crosslinking polymerization of tetrafunctional amines and diisocyanates and their selective adsorption and filtration of carbon dioxide. Polym. Chem. 2014, 5, 1124–1131. [Google Scholar] [CrossRef]
- Oh, W.; Park, J.W. Facile Synthesis of Robust and Pore-Size-Tunable Nanoporous Covalent Framework Membrane by Simultaneous Gelation and Phase Separation of Covalent Network/Poly(methyl methacrylate) Mixture. ACS Appl. Mater. Interfaces 2019, 11, 32398–32407. [Google Scholar] [CrossRef]
- Schaber, P.M.; Colson, J.; Higgins, S.; Thielen, D.; Anspach, B.; Brauer, J. Thermal decomposition (pyrolysis) of urea in an open reaction vessel. Thermochim. Acta 2004, 424, 131–142. [Google Scholar] [CrossRef]
- Jeon, E.; Moon, S.Y.; Bae, J.S.; Park, J.W. In situ Generation of Reticulate Micropores through Covalent Network/Polymer Nanocomposite Membranes for Reverse-Selective Separation of Carbon Dioxide. Angew. Chem. Int. Ed. 2016, 55, 1318–1323. [Google Scholar] [CrossRef] [PubMed]
- Rolph, M.S.; Markowska, A.L.J.; Warriner, C.N.; O’Reilly, R.K. Blocked isocyanates: From analytical and experimental considerations to non-polyurethane applications. Polym. Chem. 2016, 7, 7351–7364. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Cui, Y.; Chen, P.; Liu, S.; Zhou, N.; Ding, K.; Fan, L.; Peng, P.; Min, M.; Cheng, Y.; et al. Chapter 14—Gasification Technologies and Their Energy Potentials. In Sustainable Resource Recovery and Zero Waste Approaches; Taherzadeh, M.J., Bolton, K., Wong, J., Pandey, A., Eds.; Elsevier: Berlin/Heidelberg, Germany, 2019; pp. 193–206. [Google Scholar]
- Caruso, S.; Foti, S.; Maravigna, P.; Montaudo, G. Mass spectral characterization of polymers. Primary thermal fragmentation processes in polyureas. J. Polym. Sci. Polym. Chem. Ed. 1982, 20, 1685–1696. [Google Scholar] [CrossRef]
- Delebecq, E.; Pascault, J.P.; Boutevin, B.; Ganachaud, F. On the Versatility of Urethane/Urea Bonds: Reversibility, Blocked Isocyanate, and Non-isocyanate Polyurethane. Chem. Rev. 2013, 113, 80–118. [Google Scholar] [CrossRef]
- Star, A.; Han, T.R.; Joshi, V.; Gabriel, J.C.P.; Grüner, G. Nanoelectronic Carbon Dioxide Sensors. Adv. Mater. 2004, 16, 2049–2052. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nam, J.; Pak, Y.; Jung, G.Y.; Park, J.-W. Template-Free Preparation of a Mesopore-Rich Hierarchically Porous Carbon Monolith from a Thermally Rearrangeable Polyurea Network. Int. J. Mol. Sci. 2022, 23, 4271. https://doi.org/10.3390/ijms23084271
Nam J, Pak Y, Jung GY, Park J-W. Template-Free Preparation of a Mesopore-Rich Hierarchically Porous Carbon Monolith from a Thermally Rearrangeable Polyurea Network. International Journal of Molecular Sciences. 2022; 23(8):4271. https://doi.org/10.3390/ijms23084271
Chicago/Turabian StyleNam, Junsik, Yusin Pak, Gun Young Jung, and Ji-Woong Park. 2022. "Template-Free Preparation of a Mesopore-Rich Hierarchically Porous Carbon Monolith from a Thermally Rearrangeable Polyurea Network" International Journal of Molecular Sciences 23, no. 8: 4271. https://doi.org/10.3390/ijms23084271
APA StyleNam, J., Pak, Y., Jung, G. Y., & Park, J.-W. (2022). Template-Free Preparation of a Mesopore-Rich Hierarchically Porous Carbon Monolith from a Thermally Rearrangeable Polyurea Network. International Journal of Molecular Sciences, 23(8), 4271. https://doi.org/10.3390/ijms23084271