
Comparison between Nanoparticle Encapsulation and Surface Loading for Lysosomal Enzyme Replacement Therapy

 , ,
, ,
Abstract
1. Introduction
2. Results
2.1. Characterization and Optimization of PLGA NPs Encapsulating a Model Enzyme
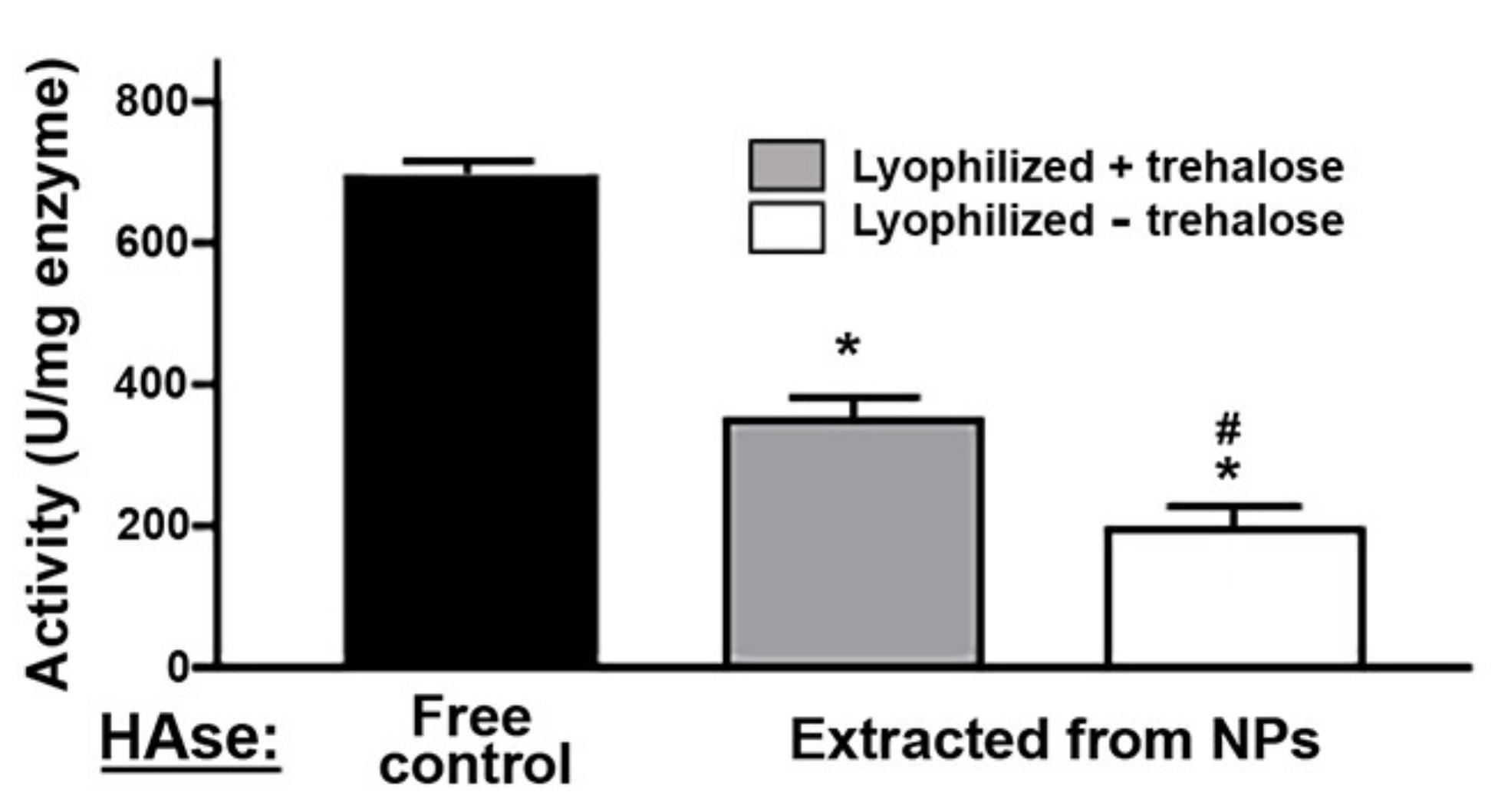
2.2. Activity, Antibody Coating, and Stability of the PLGA NPs Encapsulating a Model Enzyme
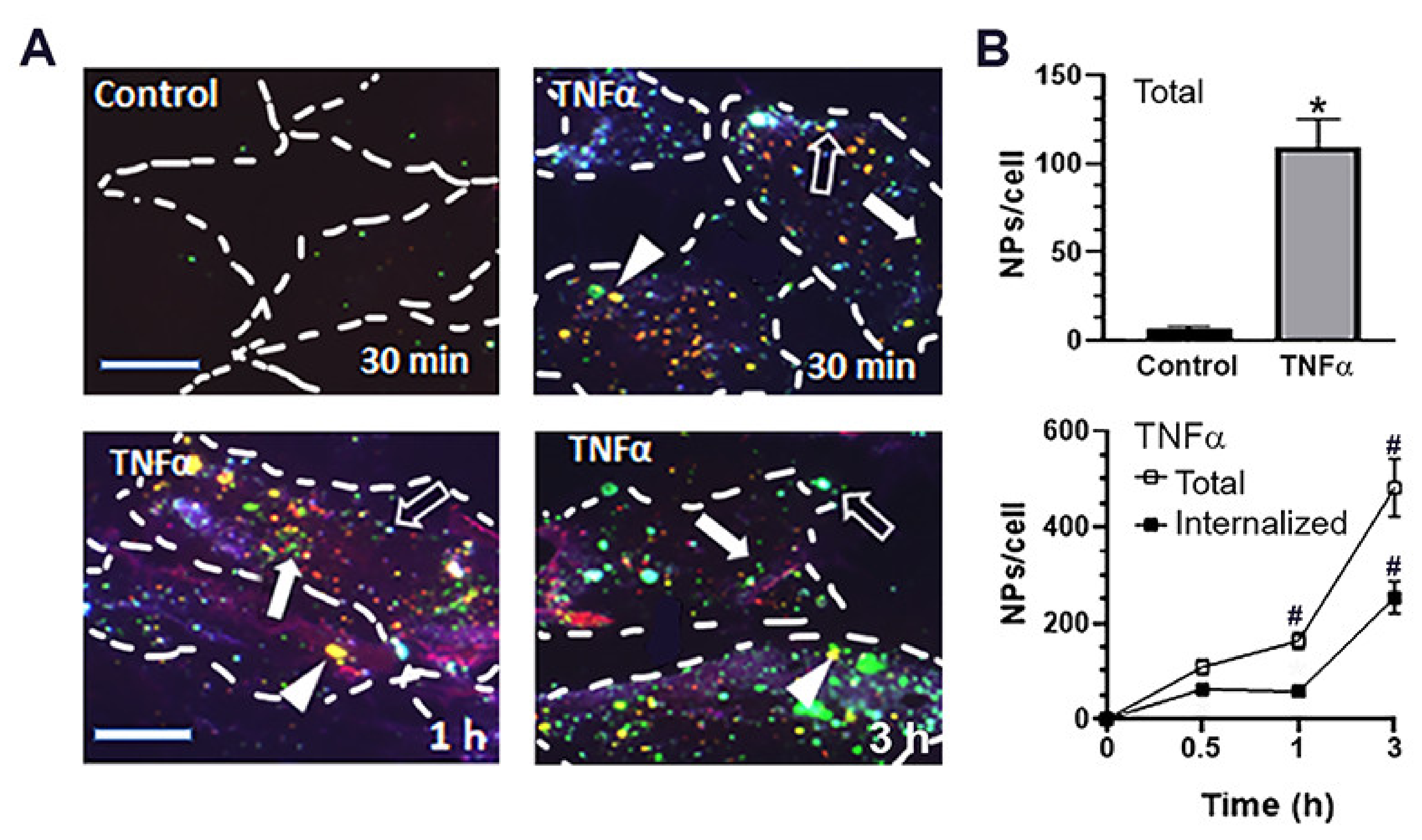
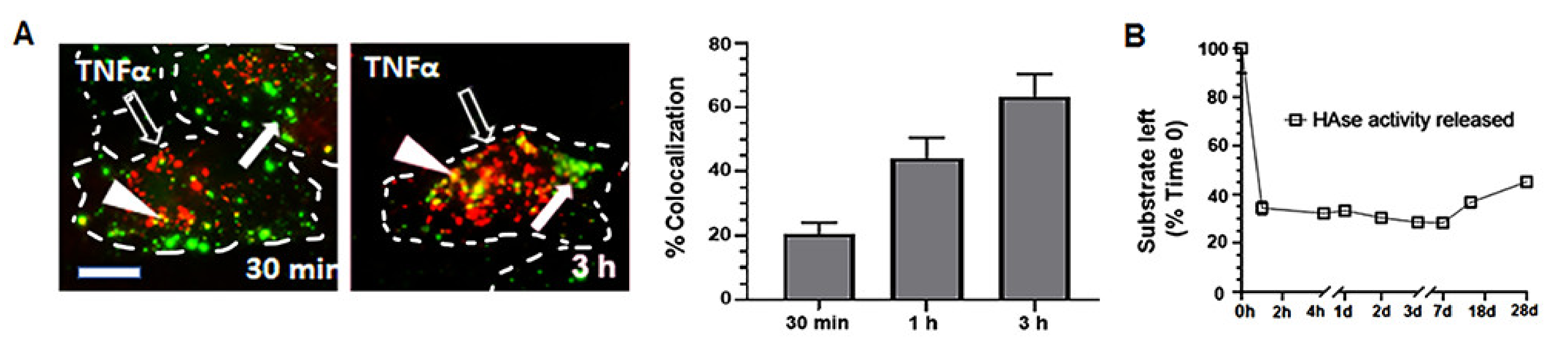
2.3. Cellular Interactions and Intracellular Enzyme Delivery by anti-ICAM PLGA NPs
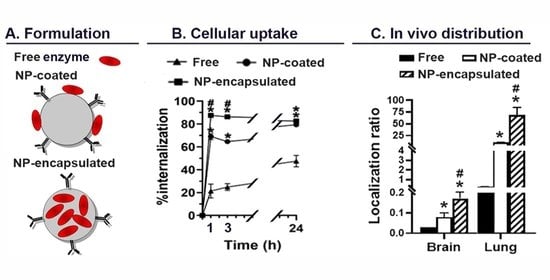
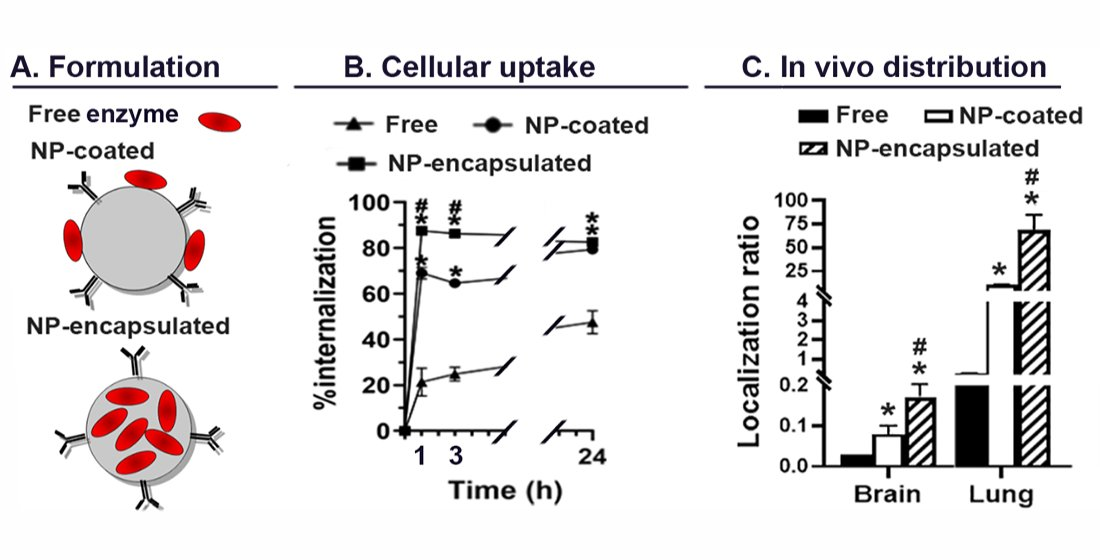
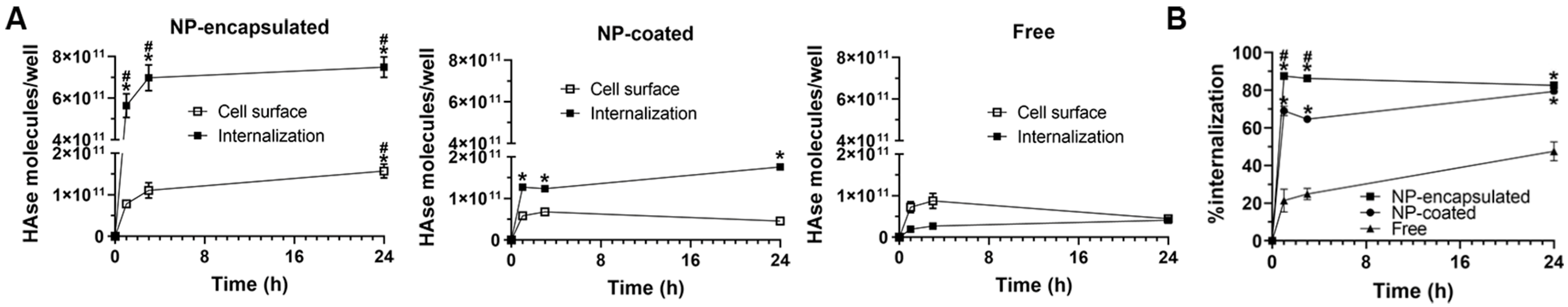
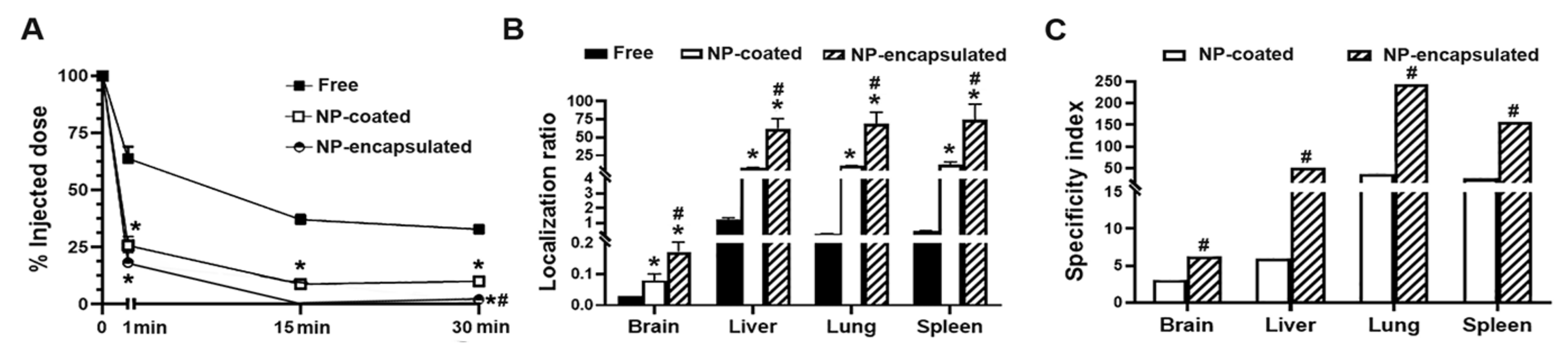
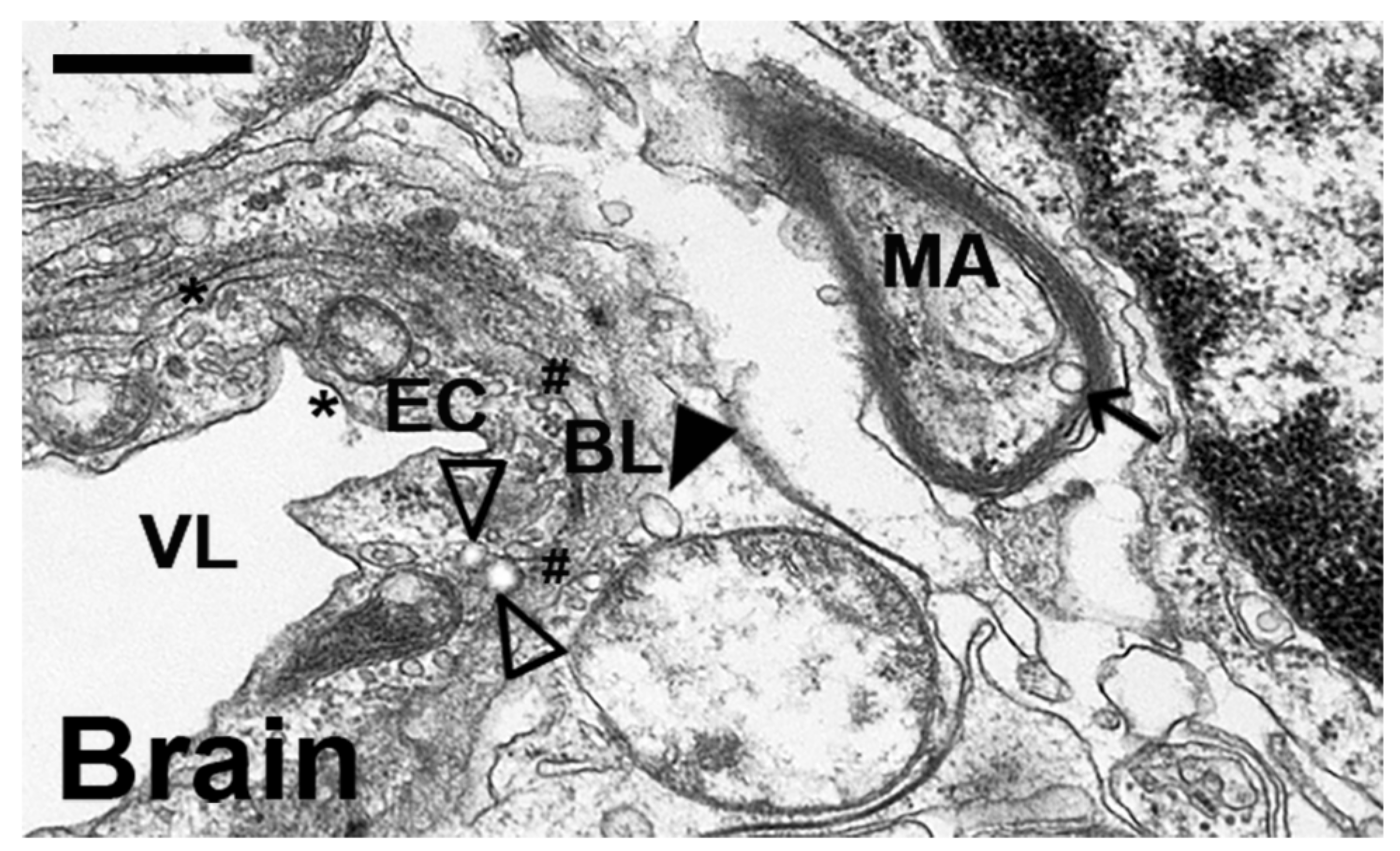
2.4. Comparative Characterization and Enzyme Delivery in Cells and Mice of Enzyme-Encapsulating vs. Enzyme-Coating Anti-ICAM PLGA NPs
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Labeling of Antibodies and Enzymes
4.3. Preparation of HAse NPs
4.4. NP Characterization
4.5. Encapsulation Efficiency and Enzyme Extraction
4.6. Lyophilization
4.7. Enzyme Activity
4.8. Coating with Targeting Antibodies or Enzyme + Antibodies
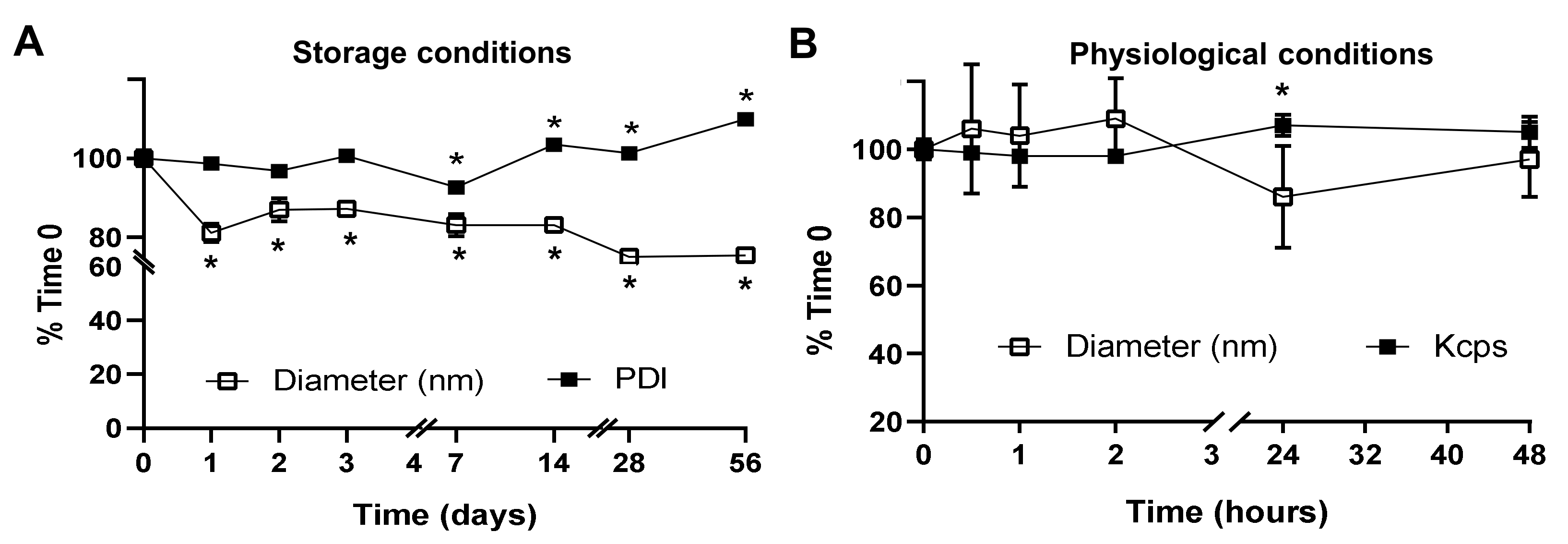
4.9. NP Stability under Storage and Physiological Conditions
4.10. Cell Cultures
4.11. Cellular Uptake
4.12. Lysosomal Trafficking
4.13. Enzyme Release under Lysosomal Conditions
4.14. In Vivo Biodistribution in Mice
4.15. Visualization of Mouse Tissues
4.16. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Antosova, Z.; Mackova, M.; Kral, V.; Macek, T. Therapeutic application of peptides and proteins: Parenteral forever? Trends. Biotechnol. 2009, 27, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Johnson-Léger, C.; Power, C.A.; Shomade, G.; Shaw, J.P.; Proudfoot, A.E. Protein therapeutics–lessons learned and a view of the future. Expert Opin. Biol. Ther. 2006, 6, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Leader, B.; Baca, Q.J.; Golan, D.E. Protein therapeutics: A summary and pharmacological classification. Nat. Rev. Drug Discov. 2008, 7, 21–39. [Google Scholar] [CrossRef] [PubMed]
- Usmani, S.S.; Bedi, G.; Samuel, J.S.; Singh, S.; Kalra, S.; Kumar, P.; Ahuja, A.A.; Sharma, M.; Gautam, A.; Raghava, G.P. THPdb: Database of FDA-approved peptide and protein therapeutics. PLoS ONE 2017, 12, e0181748. [Google Scholar] [CrossRef] [PubMed]
- Krejsa, C.; Rogge, M.; Sadee, W. Protein therapeutics: New applications for pharmacogenetics. Nat. Rev. Drug Discov. 2006, 5, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Singh, S.; Lillard, J.W. Past, present, and future technologies for oral delivery of therapeutic proteins. J. Pharm. Sci. 2008, 97, 2497–2523. [Google Scholar] [CrossRef]
- Amsden, B.; Goosen, M. Transdermal delivery of peptide and protein drugs: An overview. AIChE J. 1995, 41, 1972–1997. [Google Scholar] [CrossRef]
- Scheuch, G.; Siekmeier, R. Novel approaches to enhance pulmonary delivery of proteins and peptides. J. Physiol. Pharmacol. 2007, 58, 615–625. [Google Scholar]
- Brasnjevic, I.; Steinbusch, H.W.; Schmitz, C.; Martinez-Martinez, P.; Initiative, E.N.R. Delivery of peptide and protein drugs over the blood–brain barrier. Prog. Neurobiol. 2009, 87, 212–251. [Google Scholar] [CrossRef]
- Schuster, J.; Koulov, A.; Mahler, H.-C.; Detampel, P.; Huwyler, J.; Singh, S.; Mathaes, R. In vivo stability of therapeutic proteins. Pharm. Res. 2020, 37, 23. [Google Scholar] [CrossRef]
- Schellekens, H. Immunogenicity of therapeutic proteins: Clinical implications and future prospects. Clin. Ther. 2002, 24, 1720–1740. [Google Scholar] [CrossRef]
- Rehman, K.; Sajid Hamid Akash, M.; Akhtar, B.; Tariq, M.; Mahmood, A.; Ibrahim, M. Delivery of therapeutic proteins: Challenges and strategies. Curr. Drug Targets 2016, 17, 1172–1188. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V. Intracellular delivery of protein and peptide therapeutics. Drug Discov. Today Technol. 2008, 5, e95–e103. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Lin, Z.Y.; Yildirimer, L.; Dhinakar, A.; Zhao, X.; Wu, J. Polymer-based nanoparticles for protein delivery: Design, strategies and applications. J. Mater. Chem. B 2016, 4, 4060–4071. [Google Scholar] [CrossRef]
- Xie, J.; Lee, S.; Chen, X. Nanoparticle-based theranostic agents. Adv. Drug Deliv. Rev. 2010, 62, 1064–1079. [Google Scholar] [CrossRef]
- Johnston, A.P.; Kamphuis, M.M.; Such, G.K.; Scott, A.M.; Nice, E.C.; Heath, J.K.; Caruso, F. Targeting cancer cells: Controlling the binding and internalization of antibody-functionalized capsules. ACS Nano 2012, 6, 6667–6674. [Google Scholar] [CrossRef]
- Bertrand, N.; Wu, J.; Xu, X.; Kamaly, N.; Farokhzad, O.C. Cancer nanotechnology: The impact of passive and active targeting in the era of modern cancer biology. Adv. Drug Deliv. Rev. 2014, 66, 2–25. [Google Scholar] [CrossRef]
- Muro, S. Challenges in design and characterization of ligand-targeted drug delivery systems. J. Control. Release 2012, 164, 125–137. [Google Scholar] [CrossRef]
- Pisal, D.S.; Kosloski, M.P.; Balu-Iyer, S.V. Delivery of therapeutic proteins. J. Pharm. Sci. 2010, 99, 2557–2575. [Google Scholar] [CrossRef]
- Saito, N.; Usui, Y.; Aoki, K.; Narita, N.; Shimizu, M.; Hara, K.; Ogiwara, N.; Nakamura, K.; Ishigaki, N.; Kato, H. Carbon nanotubes: Biomaterial applications. Chem. Soc. Rev. 2009, 38, 1897–1903. [Google Scholar] [CrossRef]
- Atanase, L.I. Micellar drug delivery systems based on natural biopolymers. Polymers 2021, 13, 477. [Google Scholar] [CrossRef]
- Hamid Akash, M.S.; Rehman, K.; Chen, S. Natural and synthetic polymers as drug carriers for delivery of therapeutic proteins. Polym. Rev. 2015, 55, 371–406. [Google Scholar] [CrossRef]
- Martinez, A.P.; Qamar, B.; Fuerst, T.R.; Muro, S.; Andrianov, A.K. Biodegradable “smart” polyphosphazenes with intrinsic multifunctionality as intracellular protein delivery vehicles. Biomacromolecules 2017, 18, 2000–2011. [Google Scholar] [CrossRef] [PubMed]
- Qamar, B.; Solomon, M.; Marin, A.; Fuerst, T.R.; Andrianov, A.K.; Muro, S. Intracellular delivery of active proteins by polyphosphazene polymers. Pharmaceutics 2021, 13, 249. [Google Scholar] [CrossRef]
- Nitta, S.K.; Numata, K. Biopolymer-based nanoparticles for drug/gene delivery and tissue engineering. Int. J. Mol. Sci. 2013, 14, 1629–1654. [Google Scholar] [CrossRef] [PubMed]
- Sun, A. Lysosomal storage disease overview. Ann Transl. Med. 2018, 6, 476. [Google Scholar] [CrossRef] [PubMed]
- Solomon, M.; Muro, S. Lysosomal enzyme replacement therapies: Historical development, clinical outcomes, and future perspectives. Adv. Drug Deliv. Rev. 2017, 118, 109–134. [Google Scholar] [CrossRef]
- Muro, S. Strategies for delivery of therapeutics into the central nervous system for treatment of lysosomal storage disorders. Drug Deliv. Transl. Res. 2012, 2, 169–186. [Google Scholar] [CrossRef]
- Muro, S.; Schuchman, E.H.; Muzykantov, V.R. Lysosomal enzyme delivery by ICAM-1-targeted nanocarriers bypassing glycosylation-and clathrin-dependent endocytosis. Mol. Ther. 2006, 13, 135–141. [Google Scholar] [CrossRef]
- Garnacho, C.; Dhami, R.; Simone, E.; Dziubla, T.; Leferovich, J.; Schuchman, E.H.; Muzykantov, V.; Muro, S. Delivery of acid sphingomyelinase in normal and Niemann-pick disease mice using intercellular adhesion molecule-1-targeted polymer nanocarriers. J. Pharmacol. Exp. Ther. 2008, 325, 400–408. [Google Scholar] [CrossRef]
- Hsu, J.; Serrano, D.; Bhowmick, T.; Kumar, K.; Shen, Y.; Kuo, Y.C.; Garnacho, C.; Muro, S. Enhanced endothelial delivery and biochemical effects of α-galactosidase by ICAM-1-targeted nanocarriers for Fabry disease. J. Control. Release 2011, 149, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.; Northrup, L.; Bhowmick, T.; Muro, S. Enhanced delivery of α-glucosidase for Pompe disease by ICAM-1-targeted nanocarriers: Comparative performance of a strategy for three distinct lysosomal storage disorders. Nanomedicine 2012, 8, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Garnacho, C.; Muro, S. ICAM-1 targeting, intracellular trafficking, and functional activity of polymer nanocarriers coated with a fibrinogen-derived peptide for lysosomal enzyme replacement. J. Drug Target. 2017, 25, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Garnacho, C.; Dhami, R.; Solomon, M.; Schuchman, E.H.; Muro, S. Enhanced delivery and effects of acid sphingomyelinase by ICAM-1-targeted nanocarriers in type B Niemann-Pick disease mice. Mol. Ther. 2017, 25, 1686–1696. [Google Scholar] [CrossRef]
- Serrano, D.; Muro, S. Endothelial cell adhesion molecules and drug delivery applications. In Mechanobiology of the Endothelium, 1st ed.; Aranda-Espinoza, H., Ed.; CRC Press: Boca Raton, FL, USA, 2015. [Google Scholar]
- Hsu, J.; Hoenicka, J.; Muro, S. Targeting, endocytosis, and lysosomal delivery of active enzymes to model human neurons by ICAM-1-targeted nanocarriers. Pharm. Res. 2015, 32, 1264–1278. [Google Scholar] [CrossRef][Green Version]
- Hsu, J.; Rappaport, J.; Muro, S. Specific binding, uptake, and transport of ICAM-1-targeted nanocarriers across endothelial and subendothelial cell components of the blood–brain barrier. Pharm. Res. 2014, 31, 1855–1866. [Google Scholar] [CrossRef]
- Manthe, R.L.; Loeck, M.; Bhowmick, T.; Solomon, M.; Muro, S. Intertwined mechanisms define transport of anti-ICAM nanocarriers across the endothelium and brain delivery of a therapeutic enzyme. J. Control. Release 2020, 324, 181–193. [Google Scholar] [CrossRef]
- Mane, V.; Muro, S. Biodistribution and endocytosis of ICAM-1-targeting antibodies versus nanocarriers in the gastrointestinal tract in mice. Int. J. Nanomed. 2012, 7, 4223. [Google Scholar]
- Ghaffarian, R.; Pérez-Herrero, E.; Oh, H.; Raghavan, S.R.; Muro, S. Chitosan–Alginate Microcapsules Provide Gastric Protection and Intestinal Release of ICAM-1-Targeting Nanocarriers, Enabling GI Targeting In Vivo. Adv. Funct. Mater. 2016, 26, 3382–3393. [Google Scholar] [CrossRef]
- Lee, H.J.; Park, H.H.; Sohn, Y.; Ryu, J.; Park, J.H.; Rhee, W.J.; Park, T.H. α-Galactosidase delivery using 30Kc19-human serum albumin nanoparticles for effective treatment of Fabry disease. Appl. Microbiol. Biotechnol. 2016, 100, 10395–10402. [Google Scholar] [CrossRef]
- Giannotti, M.I.; Esteban, O.; Oliva, M.; García-Parajo, M.F.; Sanz, F. pH-responsive polysaccharide-based polyelectrolyte complexes as nanocarriers for lysosomal delivery of therapeutic proteins. Biomacromolecules 2011, 12, 2524–2533. [Google Scholar] [CrossRef] [PubMed]
- Sarrazin, S.; Wilson, B.; Sly, W.S.; Tor, Y.; Esko, J.D. Guanidinylated neomycin mediates heparan sulfate–dependent transport of active enzymes to lysosomes. Mol. Ther. 2010, 18, 1268–1274. [Google Scholar] [CrossRef] [PubMed]
- Mühlstein, A.; Gelperina, S.; Kreuter, J. Development of nanoparticle-bound arylsulfatase B for enzyme replacement therapy of mucopolysaccharidosis VI. Die Pharm.-Int. J. Pharm. Sci 2013, 68, 549–554. [Google Scholar] [PubMed]
- Muro, S.; Dziubla, T.; Qiu, W.; Leferovich, J.; Cui, X.; Berk, E.; Muzykantov, V.R. Endothelial targeting of high-affinity multivalent polymer nanocarriers directed to intercellular adhesion molecule 1. J. Pharmacol. Exp. Ther. 2006, 317, 1161–1169. [Google Scholar] [CrossRef]
- Salvalaio, M.; Rigon, L.; Belletti, D.; D’Avanzo, F.; Pederzoli, F.; Ruozi, B.; Marin, O.; Vandelli, M.A.; Forni, F.; Scarpa, M. Targeted polymeric nanoparticles for brain delivery of high molecular weight molecules in lysosomal storage disorders. PLoS ONE 2016, 11, e0156452. [Google Scholar] [CrossRef]
- Del Grosso, A.; Galliani, M.; Angella, L.; Santi, M.; Tonazzini, I.; Parlanti, G.; Signore, G.; Cecchini, M. Brain-targeted enzyme-loaded nanoparticles: A breach through the blood-brain barrier for enzyme replacement therapy in Krabbe disease. Sci. Adv. 2019, 5, eaax7462. [Google Scholar] [CrossRef]
- Galliani, M.; Santi, M.; Del Grosso, A.; Cecchettini, A.; Santorelli, F.M.; Hofmann, S.L.; Lu, J.-Y.; Angella, L.; Cecchini, M.; Signore, G. Cross-Linked enzyme aggregates as versatile tool for enzyme delivery: Application to polymeric nanoparticles. Bioconjug. Chem. 2018, 29, 2225–2231. [Google Scholar] [CrossRef]
- Bourdenx, M.; Daniel, J.; Genin, E.; Soria, F.N.; Blanchard-Desce, M.; Bezard, E.; Dehay, B. Nanoparticles restore lysosomal acidification defects: Implications for Parkinson and other lysosomal-related diseases. Autophagy 2016, 12, 472–483. [Google Scholar] [CrossRef]
- Martin, D.C.; Atmuri, V.; Hemming, R.J.; Farley, J.; Mort, J.S.; Byers, S.; Hombach-Klonisch, S.; Csoka, A.B.; Stern, R.; Triggs-Raine, B.L. A mouse model of human mucopolysaccharidosis IX exhibits osteoarthritis. Hum. Mol. Genet. 2008, 17, 1904–1915, Erratum in: Hum. Mol. Genet. 2008, 17, 2919. [Google Scholar] [CrossRef]
- Stern, R.; Jedrzejas, M.J. Hyaluronidases: Their genomics, structures, and mechanisms of action. Chem. Rev. 2006, 106, 818–839. [Google Scholar] [CrossRef]
- Fleischer, C.C.; Payne, C.K. Nanoparticle–cell interactions: Molecular structure of the protein corona and cellular outcomes. Acc. Chem. Res. 2014, 47, 2651–2659. [Google Scholar] [CrossRef] [PubMed]
- Spada, A.; Emami, J.; Tuszynski, J.A.; Lavasanifar, A. The Uniqueness of Albumin as a Carrier in Nanodrug Delivery. Mol. Pharm. 2021, 18, 1862–1894. [Google Scholar] [CrossRef] [PubMed]
- Muro, S.; Cui, X.; Gajewski, C.; Murciano, J.-C.; Muzykantov, V.R.; Koval, M. Slow intracellular trafficking of catalase nanoparticles targeted to ICAM-1 protects endothelial cells from oxidative stress. Am. J. Physiol. Cell Physiol. 2003, 285, C1339–C1347. [Google Scholar] [CrossRef]
- Schuchman, E. The pathogenesis and treatment of acid sphingomyelinase-deficient Niemann–Pick disease. J. Inherit. Metab. Dis. 2007, 30, 654–663. [Google Scholar] [CrossRef] [PubMed]
- Horinouchi, K.; Erlich, S.; Perl, D.P.; Ferlinz, K.; Bisgaier, C.L.; Sandhoff, K.; Desnick, R.J.; Stewart, C.L.; Schuchman, E.H. Acid sphingomyelinase deficient mice: A model of types A and B Niemann-Pick disease. Nat. Genet. 1995, 10, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Panigrahi, D.; Sahu, P.K.; Swain, S.; Verma, R.K. Quality by design prospects of pharmaceuticals application of double emulsion method for PLGA loaded nanoparticles. SN Appl. Sci. 2021, 3, 638. [Google Scholar] [CrossRef]
- Jahan, S.T.; Haddadi, A. Investigation and optimization of formulation parameters on preparation of targeted anti-CD205 tailored PLGA nanoparticles. Int. J. Nanomed. 2015, 10, 7371. [Google Scholar]
- Blanco, D.; Alonso, M.A.J. Protein encapsulation and release from poly (lactide-co-glycolide) microspheres: Effect of the protein and polymer properties and of the co-encapsulation of surfactants. Eur. J. Pharm. Biopharm. 1998, 45, 285–294. [Google Scholar] [CrossRef]
- Ravi, S.; Peh, K.; Darwis, Y.; Murthy, B.K.; Singh, T.R.R.; Mallikarjun, C. Development and characterization of polymeric microspheres for controlled release protein loaded drug delivery system. Indian J. Pharm. Sci. 2008, 70, 303. [Google Scholar] [CrossRef]
- Nafee, N.; Taetz, S.; Schneider, M.; Schaefer, U.F.; Lehr, C.-M. Chitosan-coated PLGA nanoparticles for DNA/RNA delivery: Effect of the formulation parameters on complexation and transfection of antisense oligonucleotides. Nanomedicine 2007, 3, 173–183. [Google Scholar] [CrossRef]
- Sahana, D.; Mittal, G.; Bhardwaj, V.; Kumar, M.R. PLGA nanoparticles for oral delivery of hydrophobic drugs: Influence of organic solvent on nanoparticle formation and release behavior in vitro and in vivo using estradiol as a model drug. J. Pharm. Sci. 2008, 97, 1530–1542. [Google Scholar] [CrossRef] [PubMed]
- Foged, C.; Brodin, B.; Frokjaer, S.; Sundblad, A. Particle size and surface charge affect particle uptake by human dendritic cells in an in vitro model. Int. J. Pharm. 2005, 298, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Amini, Y.; Amel Jamehdar, S.; Sadri, K.; Zare, S.; Musavi, D.; Tafaghodi, M. Different methods to determine the encapsulation efficiency of protein in PLGA nanoparticles. Biomed. Mater. Eng. 2017, 28, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.K.; Labhasetwar, V. Nanoparticle-mediated delivery of superoxide dismutase to the brain: An effective strategy to reduce ischemia-reperfusion injury. FASEB J. 2009, 23, 1384–1395. [Google Scholar] [CrossRef]
- Davda, J.; Labhasetwar, V. Sustained proangiogenic activity of vascular endothelial growth factor following encapsulation in nanoparticles. J. Biomed. Nanotechnol. 2005, 1, 74–82. [Google Scholar] [CrossRef]
- Xiong, Z.-J.; Huang, J.; Poda, G.; Pomès, R.; Privé, G.G. Structure of human acid sphingomyelinase reveals the role of the saposin domain in activating substrate hydrolysis. J. Mol. Biol. 2016, 428, 3026–3042. [Google Scholar] [CrossRef]
- Hai-Chau, C.; Lih-Chung, W. A simple proof of Thue’s theorem on circle packing. arXiv 2010, arXiv:1009.4322. [Google Scholar]
- Kim, J.; Sinha, S.; Solomon, M.; Perez-Herrero, E.; Hsu, J.; Tsinas, Z.; Muro, S. Co-coating of receptor-targeted drug nanocarriers with anti-phagocytic moieties enhances specific tissue uptake versus non-specific phagocytic clearance. Biomaterials 2017, 147, 14–25. [Google Scholar] [CrossRef]
- Fonte, P.; Soares, S.; Costa, A.; Andrade, J.C.; Seabra, V.; Reis, S.; Sarmento, B. Effect of cryoprotectants on the porosity and stability of insulin-loaded PLGA nanoparticles after freeze-drying. Biomatter 2012, 2, 329–339. [Google Scholar] [CrossRef]
- Ken-ichi, I.; Sumie, Y.; Yasushi, T. The effects of additives on the stability of freeze-dried β-galactosidase stored at elevated temperature. Int. J. Pharm. 1991, 71, 137–146. [Google Scholar] [CrossRef]
- Allison, S.D.; Chang, B.; Randolph, T.W.; Carpenter, J.F. Hydrogen bonding between sugar and protein is responsible for inhibition of dehydration-induced protein unfolding. Arch. Biochem. Biophys. 1999, 365, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Jung, H. Hyaluronidase: An overview of its properties, applications, and side effects. Arch. Plast. Surg. 2020, 47, 297. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, M.E.; Frank, C.W. Antibody adsorption and orientation on hydrophobic surfaces. Langmuir 2012, 28, 1765–1774. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Copolymer | Surfactant | Diameter (nm) | PDI | ζ (mV) | EE (%) |
|---|---|---|---|---|---|
| Resomer | PVA | 178.1 ± 3.1 | 0.5 ± 0.01 * | 1.6 ± 0.1 | 67.3 ± 3.0 |
| Lactel I | PVA | 158.3 ± 5.8 * | 0.4 ± 0.03 * | 1.9 ± 0.03 | 43.0 ± 3.6 * |
| Lactel II | PVA | 189.1 ± 2.1 # | 0.3 ± 0.01 *,# | 0.03 ± 0.01 *,# | 59.6 ± 2.5 |
| Lactel II | DMAB | 99.7 ± 1.1 # | 0.2 ± 0.01 | 63.7 ± 0.6 # | 26.0 ± 2.0 # |
| Lactel II | Pluronic F68 | 211.0 ± 11.0 # | 0.2 ± 0.01 ns | −18.7 ± 0.2 # | 49.2 ± 2.3 |
| Protein Input (µg/mg Copolymer) | EE (%) (Indirect) | EE (%) (Direct) | HAse (molec/NP) | Diameter (nm) | PDI | ζ-Potential (mV) | ||
|---|---|---|---|---|---|---|---|---|
| HAse | BSA | Total | ||||||
| 177.8 e | 88.9 | 266.7 | 52.1 ± 0.4 | 2.5 ± 0.2 $ | 719.0 ± 114.2 | 211.0 ± 11.0 | 0.1 ± 0.01 | −30.0 ± 1.3 |
| 22.2 f | 11.1 | 33.3 | 45.8 ± 0.9 | 11.5 ± 1.0 $ | 885.2 ± 270.8 | 189.3 ± 1.8 | 0.2 ± 0.02 | ND |
| 17.8 | - | 17.8 | 58.7 ± 1.5 | 14.2 ± 2.2 | 284.3 ± 46.7 | 148.0 ± 3.5 | 0.1 ± 0.01 | −34.03 ± 1.1 |
| 8.9 a | 4.4 | 13.3 | 92.1 ± 0.1 | 29.4 ± 2.4 $ | 538.0 ± 68.0 | 180.6 ± 7.9 | 0.1 ± 0.01 | −36.57 ± 0.5 |
| 8.9 b | - | 8.9 | 94.5 ± 0.1 | 20.7 ± 2.0 * | 697.0 ± 363.0 | 194.1 ± 11.2 | 0.1 ± 0.02 | −27.98 ± 0.2 |
| 7.1 | 3.6 | 10.7 | 86.2 ± 3.1 | 8.1 ± 0.8 | 208.5 ± 25.4 | 174.3 ± 2.1 | 0.1 ± 0.01 | −35.78 ± 0.6 |
| 5.9 c | 3 | 8.9 | 89.1 ± 0.2 | 12.0 ± 0.8 # | 313.0 ± 165.0 | 172.8 ± 8.8 | 0.1 ± 0.01 | −35.77 ± 0.3 |
| 5.9 d | 4.7 | 10.7 | 88.0 ± 4.5 | 9.9 ± 2.4 ns | 228.0 ± 44.0 | 177.3 ± 3.2 | 0.1 ± 0.01 | −35.07 ± 0.4 |
| 3.6 a′ | 1.8 | 5.3 | 95.0 ± 0.0 | 14.3 ± 1.0 | 120.5 ± 10.5 | 226.8 ± 26.7 | 0.1 ± 0.02 | −36.15 ± 0.2 |
| 3.6 b′ | - | 3.6 | 89.4 ± 1.1 | 11.5 ± 0.1 * | 130.5 ± 19.5 | 179.0 ± 12.9 | 0.5 ± 0.04 | −35.38 ± 0.4 |
| Nanoparticles | Ab | Original Coating | After Mechanical Stress | After Serum | |||
|---|---|---|---|---|---|---|---|
| Ab/NP | Ab/µm2 | Ab/NP | Ab/µm2 | Ab/NP | Ab/µm2 | ||
| Lactel II-PVA | R65 | 100.3 ± 4.0 * | 675.6 ± 27.0 * | 23.2 ± 2.4 * | 156.4 ± 16.0 * | 9.5 ± 0.1 * | 63.9 ± 0.9 * |
| Lactel II-F68 | R65 | 1229.6 ± 29.2 | 7974.4 ± 189.1 | 679.8 ± 21.1 | 4408.4 ± 136.5 | 121.9 ± 0.0 | 2530.0 ± 1739.7 |
| Lactel II-PVA | YN1 | 96.9 ± 7.9 # | 652.9 ± 53.3 # | 28.3 ± 2.4 # | 190.7 ± 16.3 # | 10.5 ± 0.1 # | 71.0 ± 1.0 # |
| Lactel II-F68 | YN1 | 880.0 ± 8.6 | 5706.9 ± 78.6 | 443.6 ± 67.5 | 2876.6 ± 437.7 | 252.9 ± 39.5 | 1640.1 ± 256.3 |
| PLGA Formulation | Diameter (nm) | PDI | ζ (mV) | HAse (molec./NP) |
|---|---|---|---|---|
| Pristine | 111.3 ± 3.7 | 0.16 ± 0.06 | −33.7 ± 1.9 | N/A |
| HAse and anti-ICAM coat | 140.7 ± 10.9 | 0.15 ± 0.02 | −25.6 ± 5.9 | 291.6 ± 54.1 |
| Encapsulated HAse | 180.6 ± 7.9 | 0.15 ± 0.01 | −36.5 ± 0.5 | 538.0 ± 68.0 |
| Encapsulated HAse and anti-ICAM coat | 236.1 ± 30.2 | 0.21 ± 0.02 | −29.8 ± 6.7 | (as above) |
| Specificity Index Tissues | Wildtype Mice | ASMKO Mice |
|---|---|---|
| Brain | 3.05 ± 0.62 | 2.97 ± 0.65 |
| Heart | 1.89 ± 0.34 | 2.18 ± 0.56 |
| Kidney | 3.01 ± 0.47 | 3.22 ± 0.45 |
| Liver | 5.97 ± 1.0 | 2.89 ± 1.06 |
| Lung | 35.64 ± 4.02 | 33.63 ± 6.46 |
| Spleen | 26.25 ± 7.16 | 4.64 ± 4.43 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muntimadugu, E.; Silva-Abreu, M.; Vives, G.; Loeck, M.; Pham, V.; del Moral, M.; Solomon, M.; Muro, S. Comparison between Nanoparticle Encapsulation and Surface Loading for Lysosomal Enzyme Replacement Therapy. Int. J. Mol. Sci. 2022, 23, 4034. https://doi.org/10.3390/ijms23074034
Muntimadugu E, Silva-Abreu M, Vives G, Loeck M, Pham V, del Moral M, Solomon M, Muro S. Comparison between Nanoparticle Encapsulation and Surface Loading for Lysosomal Enzyme Replacement Therapy. International Journal of Molecular Sciences. 2022; 23(7):4034. https://doi.org/10.3390/ijms23074034
Chicago/Turabian StyleMuntimadugu, Eameema, Marcelle Silva-Abreu, Guillem Vives, Maximilian Loeck, Vy Pham, Maria del Moral, Melani Solomon, and Silvia Muro. 2022. "Comparison between Nanoparticle Encapsulation and Surface Loading for Lysosomal Enzyme Replacement Therapy" International Journal of Molecular Sciences 23, no. 7: 4034. https://doi.org/10.3390/ijms23074034
APA StyleMuntimadugu, E., Silva-Abreu, M., Vives, G., Loeck, M., Pham, V., del Moral, M., Solomon, M., & Muro, S. (2022). Comparison between Nanoparticle Encapsulation and Surface Loading for Lysosomal Enzyme Replacement Therapy. International Journal of Molecular Sciences, 23(7), 4034. https://doi.org/10.3390/ijms23074034