
PDGF/PDGFR: A Possible Molecular Target in Scleroderma Fibrosis
 ,
,  , , and
, , and
Abstract
1. Introduction
2. Platelet-Derived Growth Factor (PDGF)
3. PDGF Receptor (PDGFR)
4. PDGF/PDGFR in Physiology and in Organ Fibrosis
5. PDGF/PDGFR in Preclinical Models of Systemic Sclerosis
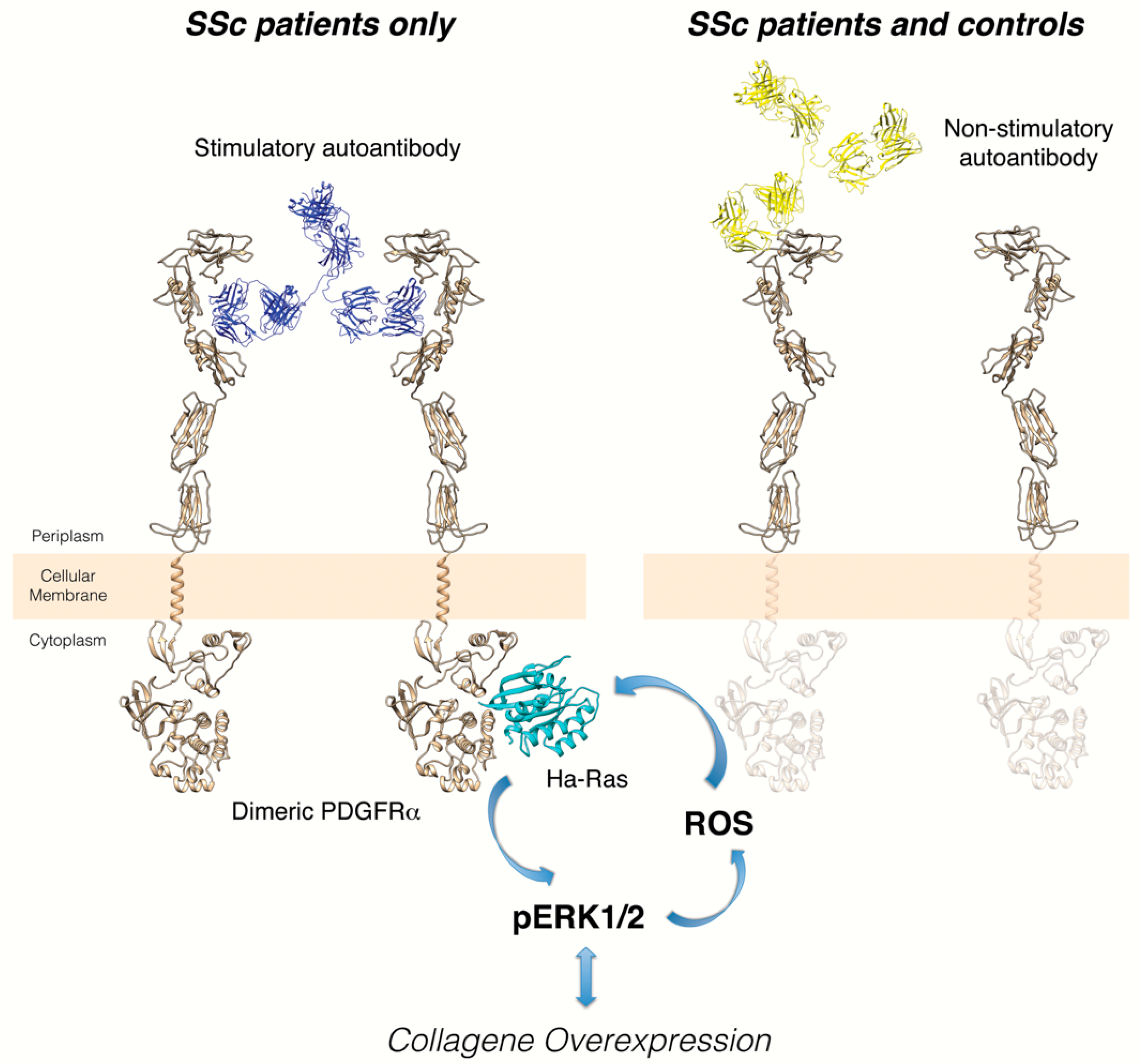
6. Anti-PDGFRα Autoantibodies in Systemic Sclerosis
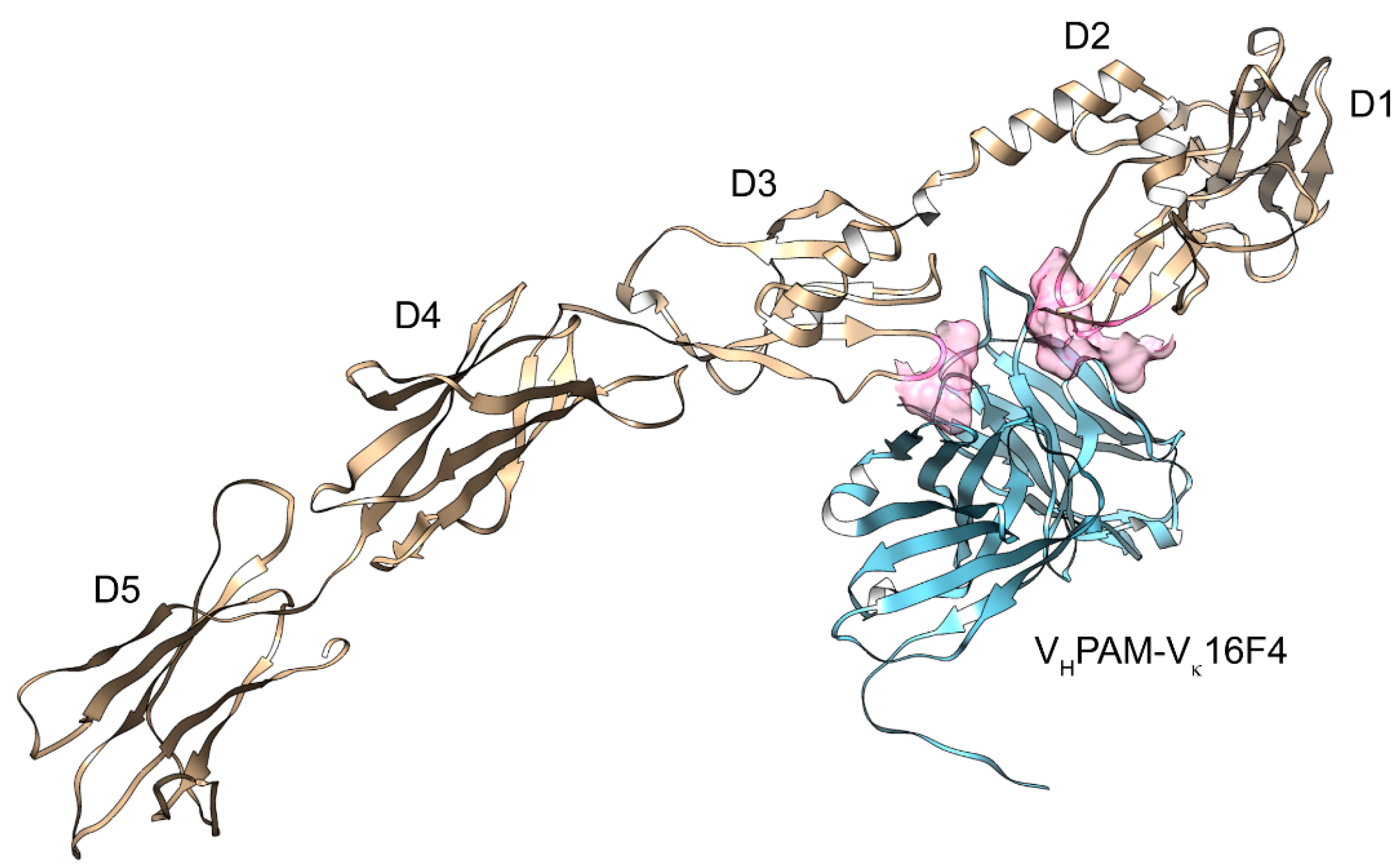
7. Structural Analysis of PDGFRα and its Ligands
8. PDGF/PDGFR Targeting Approaches in Systemic Sclerosis
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Calderon, L.M.; Pope, J.E. Scleroderma epidemiology update. Curr. Opin. Rheumatol. 2021, 33, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Varga, J.; Trojanowska, M.; Kuwana, M. Pathogenesis of systemic sclerosis: Recent insights of molecular and cellular mechanisms and therapeutic opportunities. J. Scleroderma Relat. Disord. 2017, 2, 137–152. [Google Scholar] [CrossRef]
- Hoffmann-Vold, A.-M.; Fretheim, H.; Meier, C.; Maurer, B. Circulating biomarkers of systemic sclerosis—Interstitial lung disease. J. Scleroderma Relat. Disord. 2020, 5 (Suppl. S2), 41–47. [Google Scholar] [CrossRef]
- Nihtyanova, S.I.; Denton, C.P. Pathogenesis of systemic sclerosis associated interstitial lung disease. J. Scleroderma Relat. Disord. 2020, 5 (Suppl. S2), 6–16. [Google Scholar] [CrossRef]
- Chairta, P.; Nicolaou, P.; Christodoulou, K. Genomic and genetic studies of systemic sclerosis: A systematic review. Hum. Immunol. 2017, 78, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Dai, B.; Ding, L.; Zhao, L.; Zhu, H.; Luo, H. Contributions of Immune Cells and Stromal Cells to the Pathogenesis of Systemic Sclerosis: Recent Insights. Front. Pharmacol. 2022, 13, 826839. [Google Scholar] [CrossRef] [PubMed]
- Kuret, T.; Sodin-Semrl, S.; Leskosek, B.; Ferk, P. Single Cell RNA Sequencing in Autoimmune Inflammatory Rheumatic Diseases: Current Applications, Challenges and a Step Toward Precision Medicine. Front. Med. 2021, 8, 822804. [Google Scholar] [CrossRef]
- Szabo, I.; Muntean, L.; Crisan, T.; Rednic, V.; Sirbe, C.; Rednic, S. Novel Concepts in Systemic Sclerosis Pathogenesis: Role for miRNAs. Biomedicines 2021, 9, 1471. [Google Scholar] [CrossRef]
- Blagojevic, J.; Abignano, G.; Avouac, J.; Cometi, L.; Frerix, M.; Bellando-Randone, S.; Guiducci, S.; Bruni, C.; Huscher, D.; Jaeger, V.K.; et al. Use of vasoactive/vasodilating drugs for systemic sclerosis (SSc)-related digital ulcers (DUs) in expert tertiary centres: Results from the analysis of the observational real-life DeSScipher study. Clin. Rheumatol. 2020, 39, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Kowal-Bielecka, O.; Fransen, J.; Avouac, J.; Becker, M.; Kulak, A.; Allanore, Y.; Distler, O.; Clements, P.; Cutolo, M.; Czirjak, L.; et al. Update of EULAR recommendations for the treatment of systemic sclerosis. Ann. Rheum. Dis. 2017, 76, 1327–1339. [Google Scholar] [CrossRef] [PubMed]
- Distler, O.; Gahlemann, M.; Maher, T.M. Nintedanib for Systemic Sclerosis-Associated Interstitial Lung Disease. Reply. N. Engl. J. Med. 2019, 381, 1596–1597. [Google Scholar] [PubMed]
- Andrae, J.; Gallini, R.; Betsholtz, C. Role of platelet-derived growth factors in physiology and medicine. Genes. Dev. 2008, 22, 1276–1312. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Westermark, B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol. Rev. 1999, 79, 1283–1316. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Hamashima, T.; Ishii, Y.; Yamamoto, S.; Okuno, N.; Yoshida, N.; Yamada, M.; Huang, T.T.; Shioda, N.; Tomihara, K.; et al. Different PDGF Receptor Dimers Drive Distinct Migration Modes of the Mouse Skin Fibroblast. Cell Physiol. Biochem. 2018, 51, 1461–1479. [Google Scholar] [CrossRef]
- Kazlauskas, A. PDGFs and their receptors. Gene 2017, 614, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Tallquist, M.; Kazlauskas, A. PDGF signaling in cells and mice. Cytokine Growth Factor Rev. 2004, 15, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Klinkhammer, B.M.; Floege, J.; Boor, P. PDGF in organ fibrosis. Mol. Aspects Med. 2018, 62, 44–62. [Google Scholar] [CrossRef]
- Zhou, J.; Shao, L.; Yu, J.; Huang, J.; Feng, Q. PDGF-BB promotes vascular smooth muscle cell migration by enhancing Pim-1 expression via inhibiting miR-214. Ann. Transl. Med. 2021, 9, 1728. [Google Scholar] [CrossRef]
- Alvarez, R.H.; Kantarjian, H.M.; Cortes, J.E. Biology of platelet-derived growth factor and its involvement in disease. In Mayo Clinic Proceedings; Elsevier: Amsterdam, The Netherlands, 2006; Volume 81, pp. 1241–1257. [Google Scholar]
- Rogers, M.A.; Fantauzzo, K.A. The emerging complexity of PDGFRs: Activation, internalization and signal attenuation. Biochem. Soc. Trans. 2020, 48, 1167–1176. [Google Scholar] [CrossRef] [PubMed]
- Valius, M.; Kazlauskas, A. Phospholipase C-gamma 1 and phosphatidylinositol 3 kinase are the downstream mediators of the PDGF receptor’s mitogenic signal. Cell 1993, 73, 321–334. [Google Scholar] [CrossRef]
- Chiariello, M.; Marinissen, M.J.; Gutkind, J.S. Regulation of c-myc expression by PDGF through Rho GTPases. Nat. Cell Biol. 2001, 3, 580–586. [Google Scholar] [CrossRef]
- Chen, P.H.; Chen, X.; He, X. Platelet-derived growth factors and their receptors: Structural and functional perspectives. Biochim. Biophys. Acta 2013, 1834, 2176–2186. [Google Scholar] [CrossRef] [PubMed]
- Olson, L.E.; Soriano, P. Increased PDGFRalpha activation disrupts connective tissue development and drives systemic fibrosis. Dev. Cell 2009, 16, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Rheaume, M.A.; Kazlauskas, A. Recent developments in our understanding of how platelet-derived growth factor (PDGF) and its receptors contribute to proliferative vitreoretinopathy. Exp. Eye Res. 2010, 90, 376–381. [Google Scholar] [CrossRef]
- Guerit, E.; Arts, F.; Dachy, G.; Boulouadnine, B.; Demoulin, J.B. PDGF receptor mutations in human diseases. Cell Mol. Life Sci. 2021, 78, 3867–3881. [Google Scholar] [CrossRef]
- Ishii, Y.; Hamashima, T.; Yamamoto, S.; Sasahara, M. Pathogenetic significance and possibility as a therapeutic target of platelet derived growth factor. Pathol. Int. 2017, 67, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Medamana, J.; Clark, R.A.; Butler, J. Platelet-Derived Growth Factor in Heart Failure. Handb. Exp. Pharmacol. 2017, 243, 355–369. [Google Scholar] [PubMed]
- Rosenbloom, J.; Macarak, E.; Piera-Velazquez, S.; Jimenez, S.A. Human Fibrotic Diseases: Current Challenges in Fibrosis Research. Methods Mol. Biol. 2017, 1627, 1–23. [Google Scholar] [PubMed]
- Distler, J.H.W.; Gyorfi, A.H.; Ramanujam, M.; Whitfield, M.L.; Konigshoff, M.; Lafyatis, R. Shared and distinct mechanisms of fibrosis. Nat. Rev. Rheumatol. 2019, 15, 705–730. [Google Scholar] [CrossRef] [PubMed]
- Roehlen, N.; Crouchet, E.; Baumert, T.F. Liver Fibrosis: Mechanistic Concepts and Therapeutic Perspectives. Cells 2020, 9, 875. [Google Scholar] [CrossRef] [PubMed]
- White, S.J.; Chong, J.J.H. Growth factor therapy for cardiac repair: An overview of recent advances and future directions. Biophys. Rev. 2020, 12, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Gallini, R.; Lindblom, P.; Bondjers, C.; Betsholtz, C.; Andrae, J. PDGF-A and PDGF-B induces cardiac fibrosis in transgenic mice. Exp. Cell Res. 2016, 349, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Mueller, A.A.; van Velthoven, C.T.; Fukumoto, K.D.; Cheung, T.H.; Rando, T.A. Intronic polyadenylation of PDGFRalpha in resident stem cells attenuates muscle fibrosis. Nature 2016, 540, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Takamura, N.; Renaud, L.; da Silveira, W.A.; Feghali-Bostwick, C. PDGF Promotes Dermal Fibroblast Activation via a Novel Mechanism Mediated by Signaling Through MCHR1. Front. Immunol. 2021, 12, 745308. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.E.; Lopez, H.; Abdi, B.A.; Guerra, S.G.; Shiwen, X.; Khan, K.; Etomi, O.; Martin, G.R.; Abraham, D.J.; Denton, C.P.; et al. Multiplex cytokine analysis of dermal interstitial blister fluid defines local disease mechanisms in systemic sclerosis. Arthritis Res. Ther. 2015, 17, 73. [Google Scholar] [CrossRef]
- Yamakage, A.; Kikuchi, K.; Smith, E.A.; LeRoy, E.C.; Trojanowska, M. Selective upregulation of platelet-derived growth factor alpha receptors by transforming growth factor beta in scleroderma fibroblasts. J. Exp. Med. 1992, 175, 1227–1234. [Google Scholar] [CrossRef]
- Ludwicka, A.; Ohba, T.; Trojanowska, M.; Yamakage, A.; Strange, C.; Smith, E.A.; Leroy, E.C.; Sutherland, S.; Silver, R.M. Elevated levels of platelet derived growth factor and transforming growth factor-beta 1 in bronchoalveolar lavage fluid from patients with scleroderma. J. Rheumatol. 1995, 22, 1876–1883. [Google Scholar]
- Liu, T.; Zhang, J.; Zhang, J.; Mu, X.; Su, H.; Hu, X.; Liu, W.; Zhao, E.; Li, W. RNA interference against platelet-derived growth factor receptor alpha mRNA inhibits fibroblast transdifferentiation in skin lesions of patients with systemic sclerosis. PLoS ONE 2013, 8, e60414. [Google Scholar]
- Tanaka, S.; Suto, A.; Ikeda, K.; Sanayama, Y.; Nakagomi, D.; Iwamoto, T.; Suzuki, K.; Kambe, N.; Matsue, H.; Matsumura, R.; et al. Alteration of circulating miRNAs in SSc: miR-30b regulates the expression of PDGF receptor beta. Rheumatology 2013, 52, 1963–1972. [Google Scholar] [CrossRef]
- Orlando, F.; Paolini, C.; Agarbati, S.; Tonnini, C.; Grieco, A.; Capelli, C.; Introna, M.; Provinciali, M.; Gabrielli, A.; Moroncini, G. Induction of Mouse Lung Injury by Endotracheal Injection of Bleomycin. J. Vis. Exp. 2019, 30, e58922. [Google Scholar] [CrossRef]
- Baroni, S.S.; Santillo, M.; Bevilacqua, F.; Luchetti, M.; Spadoni, T.; Mancini, M.; Fraticelli, P.; Sambo, P.; Funaro, A.; Kazlauskas, A.; et al. Stimulatory autoantibodies to the PDGF receptor in systemic sclerosis. N. Engl. J. Med. 2006, 354, 2667–2676. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, Y. Autoantibody profiles in systemic sclerosis: Predictive value for clinical evaluation and prognosis. J. Dermatol. 2010, 37, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Gabrielli, A.; Svegliati, S.; Moroncini, G.; Avvedimento, E.V. Pathogenic autoantibodies in systemic sclerosis. Curr. Opin. Immunol. 2007, 19, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Moroncini, G.; Mori, S.; Tonnini, C.; Gabrielli, A. Role of viral infections in the etiopathogenesis of systemic sclerosis. Clin. Exp. Rheumatol. 2013, 31, 3–7. [Google Scholar]
- Moroncini, G.; Grieco, A.; Nacci, G.; Paolini, C.; Tonnini, C.; Pozniak, K.N.; Cuccioloni, M.; Mozzicafreddo, M.; Svegliati, S.; Angeletti, M.; et al. Epitope Specificity Determines Pathogenicity and Detectability of Anti-Platelet-Derived Growth Factor Receptor alpha Autoantibodies in Systemic Sclerosis. Arthritis Rheumatol. 2015, 67, 1891–1903. [Google Scholar] [CrossRef] [PubMed]
- Moroncini, G.; Cuccioloni, M.; Mozzicafreddo, M.; Pozniak, K.N.; Grieco, A.; Paolini, C.; Tonnini, C.; Spadoni, T.; Svegliati, S.; Funaro, A.; et al. Characterization of binding and quantification of human autoantibodies to PDGFRalpha using a biosensor-based approach. Anal. Biochem. 2017, 528, 26–33. [Google Scholar] [CrossRef][Green Version]
- Moroncini, G.; Svegliati Baroni, S.; Gabrielli, A. Agonistic antibodies in systemic sclerosis. Immunol. Lett. 2018, 195, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Svegliati, S.; Amico, D.; Spadoni, T.; Fischetti, C.; Finke, D.; Moroncini, G.; Paolini, C.; Tonnini, C.; Grieco, A.; Rovinelli, M.; et al. Agonistic Anti-PDGF Receptor Autoantibodies from Patients with Systemic Sclerosis Impact Human Pulmonary Artery Smooth Muscle Cells Function In Vitro. Front. Immunol. 2017, 8, 75. [Google Scholar] [PubMed]
- Luchetti, M.M.; Moroncini, G.; Jose Escamez, M.; Svegliati Baroni, S.; Spadoni, T.; Grieco, A.; Paolini, C.; Funaro, A.; Avvedimento, E.V.; Larcher, F.; et al. Induction of Scleroderma Fibrosis in Skin-Humanized Mice by Administration of Anti-Platelet-Derived Growth Factor Receptor Agonistic Autoantibodies. Arthritis Rheumatol. 2016, 68, 2263–2273. [Google Scholar] [CrossRef] [PubMed]
- Kschonsak, M.; Rouge, L.; Arthur, C.P.; Hoangdung, H.; Patel, N.; Kim, I.; Johnson, M.C.; Kraft, E.; Rohou, A.L.; Gill, A.; et al. Structures of HCMV Trimer reveal the basis for receptor recognition and cell entry. Cell 2021, 184, 1232–1244.e16. [Google Scholar] [CrossRef]
- Shim, A.H.; Liu, H.; Focia, P.J.; Chen, X.; Lin, P.C.; He, X. Structures of a platelet-derived growth factor/propeptide complex and a platelet-derived growth factor/receptor complex. Proc. Natl. Acad. Sci. USA 2010, 107, 11307–11312. [Google Scholar] [CrossRef] [PubMed]
- Yuzawa, S.; Opatowsky, Y.; Zhang, Z.; Mandiyan, V.; Lax, I.; Schlessinger, J. Structural basis for activation of the receptor tyrosine kinase KIT by stem cell factor. Cell 2007, 130, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Markovic-Mueller, S.; Stuttfeld, E.; Asthana, M.; Weinert, T.; Bliven, S.; Goldie, K.N.; Kisko, K.; Capitani, G.; Ballmer-Hofer, K. Structure of the Full-length VEGFR-1 Extracellular Domain in Complex with VEGF-A. Structure 2017, 25, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Oefner, C.; D’Arcy, A.; Winkler, F.K.; Eggimann, B.; Hosang, M. Crystal structure of human platelet-derived growth factor BB. EMBO J. 1992, 11, 3921–3926. [Google Scholar] [CrossRef]
- Liang, L.; Yan, X.E.; Yin, Y.; Yun, C.H. Structural and biochemical studies of the PDGFRA kinase domain. Biochem. Biophys. Res. Commun. 2016, 477, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, N.; Lennartsson, J. The PDGF/PDGFR pathway as a drug target. Mol. Aspects Med. 2018, 62, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Kuai, J.; Mosyak, L.; Brooks, J.; Cain, M.; Carven, G.J.; Ogawa, S.; Ishino, T.; Tam, M.; Lavallie, E.R.; Yang, Z.; et al. Characterization of binding mode of action of a blocking anti-platelet-derived growth factor (PDGF)-B monoclonal antibody, MOR8457, reveals conformational flexibility and avidity needed for PDGF-BB to bind PDGF receptor-beta. Biochemistry 2015, 54, 1918–1929. [Google Scholar] [CrossRef] [PubMed]
- Musa, H.; Kaur, K.; O’Connell, R.; Klos, M.; Guerrero-Serna, G.; Avula, U.M.; Herron, T.J.; Kalifa, J.; Anumonwo, J.M.; Jalife, J. Inhibition of platelet-derived growth factor-AB signaling prevents electromechanical remodeling of adult atrial myocytes that contact myofibroblasts. Heart Rhythm. 2013, 10, 1044–1051. [Google Scholar] [CrossRef]
- Donovan, J.; Shiwen, X.; Norman, J.; Abraham, D. Platelet-derived growth factor alpha and beta receptors have overlapping functional activities towards fibroblasts. Fibrogenesis Tissue Repair 2013, 6, 10. [Google Scholar] [CrossRef]
- Gerber, D.E.; Gupta, P.; Dellinger, M.T.; Toombs, J.E.; Peyton, M.; Duignan, I.; Malaby, J.; Bailey, T.; Burns, C.; Brekken, R.A.; et al. Stromal platelet-derived growth factor receptor alpha (PDGFRalpha) provides a therapeutic target independent of tumor cell PDGFRalpha expression in lung cancer xenografts. Mol. Cancer Ther. 2012, 11, 2473–2482. [Google Scholar] [CrossRef] [PubMed]
- Moroncini, G.; Maccaroni, E.; Fiordoliva, I.; Pellei, C.; Gabrielli, A.; Berardi, R. Developments in the management of advanced soft-tissue sarcoma—Olaratumab in context. Onco Targets Ther. 2018, 11, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Tap, W.D.; Wagner, A.J.; Schoffski, P.; Martin-Broto, J.; Krarup-Hansen, A.; Ganjoo, K.N.; Yen, C.C.; Abdul Razak, A.R.; Spira, A.; Kawai, A.; et al. Effect of Doxorubicin Plus Olaratumab vs Doxorubicin Plus Placebo on Survival in Patients With Advanced Soft Tissue Sarcomas: The ANNOUNCE Randomized Clinical Trial. JAMA 2020, 323, 1266–1276. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, N.; Iqbal, N. Imatinib: A breakthrough of targeted therapy in cancer. Chemother. Res. Pract. 2014, 2014, 357027. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, F.A.; Piera-Velazquez, S.; Jimenez, S.A. Tyrosine kinases in the pathogenesis of tissue fibrosis in systemic sclerosis and potential therapeutic role of their inhibition. Transl. Res. 2021, 231, 139–158. [Google Scholar] [CrossRef] [PubMed]
- Daniels, C.E.; Wilkes, M.C.; Edens, M.; Kottom, T.J.; Murphy, S.J.; Limper, A.H.; Leof, E.B. Imatinib mesylate inhibits the profibrogenic activity of TGF-beta and prevents bleomycin-mediated lung fibrosis. J. Clin. Investig. 2004, 114, 1308–1316. [Google Scholar] [CrossRef] [PubMed]
- Distler, J.H.; Jungel, A.; Huber, L.C.; Schulze-Horsel, U.; Zwerina, J.; Gay, R.E.; Michel, B.A.; Hauser, T.; Schett, G.; Gay, S.; et al. Imatinib mesylate reduces production of extracellular matrix and prevents development of experimental dermal fibrosis. Arthritis Rheum. 2007, 56, 311–322. [Google Scholar] [CrossRef]
- Bournia, V.K.; Evangelou, K.; Sfikakis, P.P. Therapeutic inhibition of tyrosine kinases in systemic sclerosis: A review of published experience on the first 108 patients treated with imatinib. In Seminars in Arthritis and Rheumatism; WB Saunders: Philadelphia, PA, USA, 2013; Volume 42, pp. 377–390. [Google Scholar]
- Fraticelli, P.; Gabrielli, B.; Pomponio, G.; Valentini, G.; Bosello, S.; Riboldi, P.; Gerosa, M.; Faggioli, P.; Giacomelli, R.; Del Papa, N.; et al. Low-dose oral imatinib in the treatment of systemic sclerosis interstitial lung disease unresponsive to cyclophosphamide: A phase II pilot study. Arthritis Res. Ther. 2014, 16, R144. [Google Scholar] [CrossRef]
- Harrach, S.; Barz, V.; Pap, T.; Pavenstadt, H.; Schlatter, E.; Edemir, B.; Distler, J.; Ciarimboli, G.; Bertrand, J. Notch Signaling Activity Determines Uptake and Biological Effect of Imatinib in Systemic Sclerosis Dermal Fibroblasts. J. Investig. Dermatol. 2019, 139, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Makino, K.; Makino, T.; Stawski, L.; Mantero, J.C.; Lafyatis, R.; Simms, R.; Trojanowska, M. Blockade of PDGF Receptors by Crenolanib Has Therapeutic Effect in Patient Fibroblasts and in Preclinical Models of Systemic Sclerosis. J. Investig. Dermatol. 2017, 137, 1671–1681. [Google Scholar] [CrossRef] [PubMed]
- van der Kroef, M.; Carvalheiro, T.; Rossato, M.; de Wit, F.; Cossu, M.; Chouri, E.; Wichers, C.G.K.; Bekker, C.P.J.; Beretta, L.; Vazirpanah, N.; et al. CXCL4 triggers monocytes and macrophages to produce PDGF-BB, culminating in fibroblast activation: Implications for systemic sclerosis. J. Autoimmun. 2020, 111, 102444. [Google Scholar] [CrossRef] [PubMed]
- Martyanov, V.; Kim, G.J.; Hayes, W.; Du, S.; Ganguly, B.J.; Sy, O.; Lee, S.K.; Bogatkevich, G.S.; Schieven, G.L.; Schiopu, E.; et al. Novel lung imaging biomarkers and skin gene expression subsetting in dasatinib treatment of systemic sclerosis-associated interstitial lung disease. PLoS ONE 2017, 12, e0187580. [Google Scholar] [CrossRef]
- Gordon, J.K.; Martyanov, V.; Magro, C.; Wildman, H.F.; Wood, T.A.; Huang, W.T.; Crow, M.K.; Whitfield, M.L.; Spiera, R.F. Nilotinib (Tasigna) in the treatment of early diffuse systemic sclerosis: An open-label, pilot clinical trial. Arthritis Res. Ther. 2015, 17, 213. [Google Scholar] [CrossRef] [PubMed]
- Hilberg, F.; Roth, G.J.; Krssak, M.; Kautschitsch, S.; Sommergruber, W.; Tontsch-Grunt, U.; Garin-Chesa, P.; Bader, G.; Zoephel, A.; Quant, J.; et al. BIBF 1120: Triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. 2008, 68, 4774–4782. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Beyer, C.; Palumbo-Zerr, K.; Zhang, Y.; Ramming, A.; Distler, A.; Gelse, K.; Distler, O.; Schett, G.; Wollin, L.; et al. Nintedanib inhibits fibroblast activation and ameliorates fibrosis in preclinical models of systemic sclerosis. Ann. Rheum. Dis. 2016, 75, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Benfaremo, D.; Svegliati, S.; Paolini, C.; Agarbati, S.; Moroncini, G. Systemic Sclerosis: From Pathophysiology to Novel Therapeutic Approaches. Biomedicines 2022, 10, 163. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.R.; Wells, A.U.; Cottin, V.; Devaraj, A.; Walsh, S.L.F.; Inoue, Y.; Richeldi, L.; Kolb, M.; Tetzlaff, K.; Stowasser, S.; et al. Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. N. Engl. J. Med. 2019, 381, 1718–1727. [Google Scholar] [CrossRef] [PubMed]
- Wells, A.U.; Flaherty, K.R.; Brown, K.K.; Inoue, Y.; Devaraj, A.; Richeldi, L.; Moua, T.; Crestani, B.; Wuyts, W.A.; Stowasser, S.; et al. Nintedanib in patients with progressive fibrosing interstitial lung diseases-subgroup analyses by interstitial lung disease diagnosis in the INBUILD trial: A randomised, double-blind, placebo-controlled, parallel-group trial. Lancet Respir. Med. 2020, 8, 453–460. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Drug Name | Mechanism of Action | Current Clinical Indications |
|---|---|---|
| MOR8457 | PDGF-B blocking antibody | Pre-clinical use only |
| Olaratumab | Recombinant human IgG1 anti-PDGFR moAb | Withdrawn from market |
| Imatinib | Tyrosine kinase inhibitor | Chronic myeloid leukaemia (CML) Acute lymphoblastic leukaemia (ALL) Myelodysplastic/myeloproliferative diseases Hypereosinophilic syndrome (HES) Gastrointestinal stromal tumours (GIST) Dermatofibrosarcoma protuberans (DFSP) |
| Crenolanib besylate | Tyrosine kinase inhibitor | Orphan designation for acute myeloid leukaemia |
| Dasatinib | Tyrosine kinase inhibitor | Chronic myeloid leukaemia (CML) Acute lymphoblastic leukaemia (ALL) |
| Nilotinib | Tyrosine kinase inhibitor | Chronic myeloid leukaemia (CML) |
| Nintedanib | Tyrosine kinase inhibitor | IPF SSc-ILD PF-ILD |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paolini, C.; Agarbati, S.; Benfaremo, D.; Mozzicafreddo, M.; Svegliati, S.; Moroncini, G. PDGF/PDGFR: A Possible Molecular Target in Scleroderma Fibrosis. Int. J. Mol. Sci. 2022, 23, 3904. https://doi.org/10.3390/ijms23073904
Paolini C, Agarbati S, Benfaremo D, Mozzicafreddo M, Svegliati S, Moroncini G. PDGF/PDGFR: A Possible Molecular Target in Scleroderma Fibrosis. International Journal of Molecular Sciences. 2022; 23(7):3904. https://doi.org/10.3390/ijms23073904
Chicago/Turabian StylePaolini, Chiara, Silvia Agarbati, Devis Benfaremo, Matteo Mozzicafreddo, Silvia Svegliati, and Gianluca Moroncini. 2022. "PDGF/PDGFR: A Possible Molecular Target in Scleroderma Fibrosis" International Journal of Molecular Sciences 23, no. 7: 3904. https://doi.org/10.3390/ijms23073904
APA StylePaolini, C., Agarbati, S., Benfaremo, D., Mozzicafreddo, M., Svegliati, S., & Moroncini, G. (2022). PDGF/PDGFR: A Possible Molecular Target in Scleroderma Fibrosis. International Journal of Molecular Sciences, 23(7), 3904. https://doi.org/10.3390/ijms23073904