
The Interplay between PARP Inhibitors and Immunotherapy in Ovarian Cancer: The Rationale behind a New Combination Therapy

 ,
, 
 , and
, and
Abstract
:1. Introduction
2. BRCA Mutations and PARPis in OC
2.1. The ‘Synthetic Lethality’ in OC
2.2. Clinical Applications of PARPis in OC
3. Immune Checkpoint Inhibitors in OC
3.1. ICIs Pathways and Clinical Applications in OC
3.2. Predictive Factors for ICIs in OC
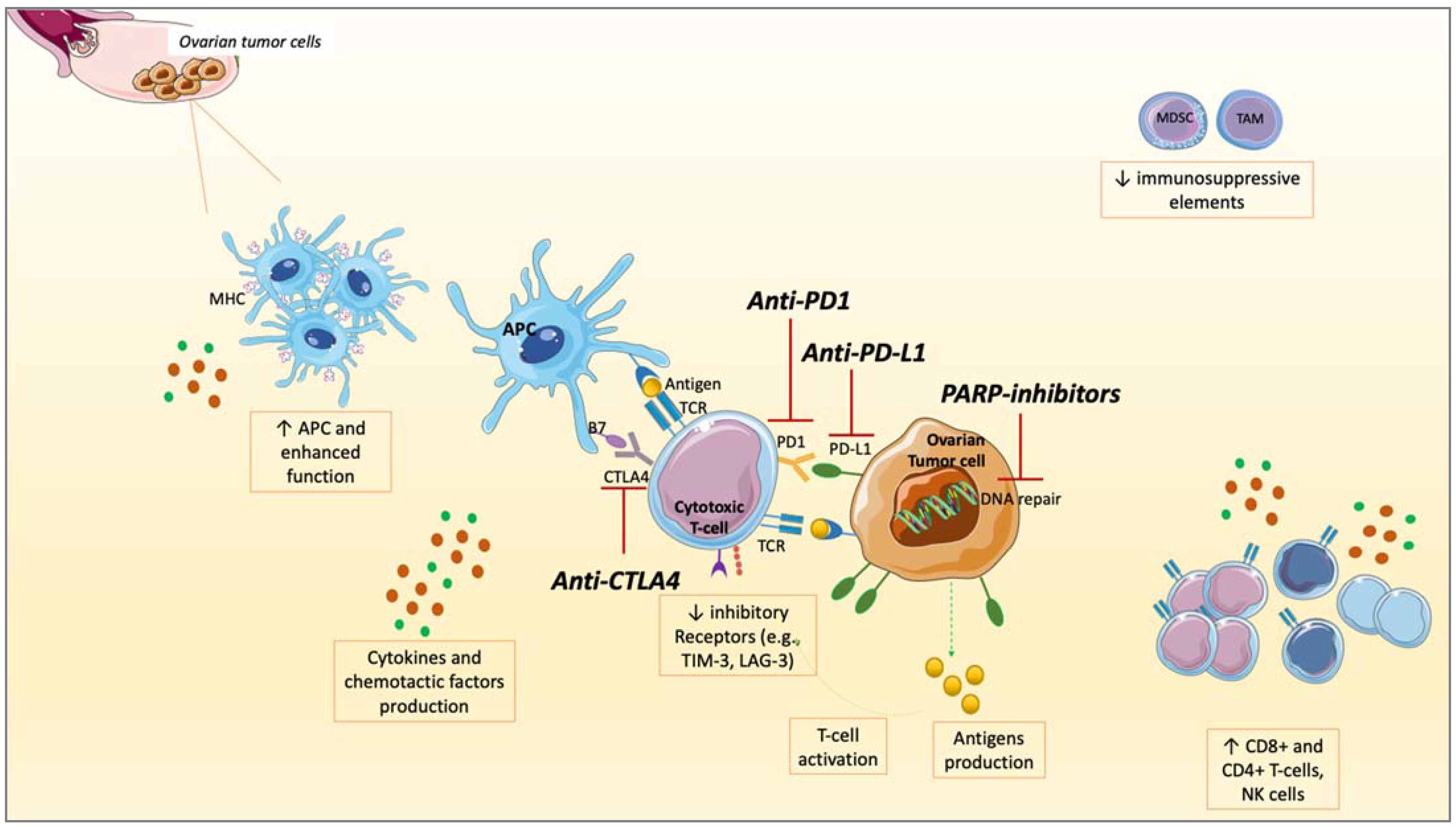
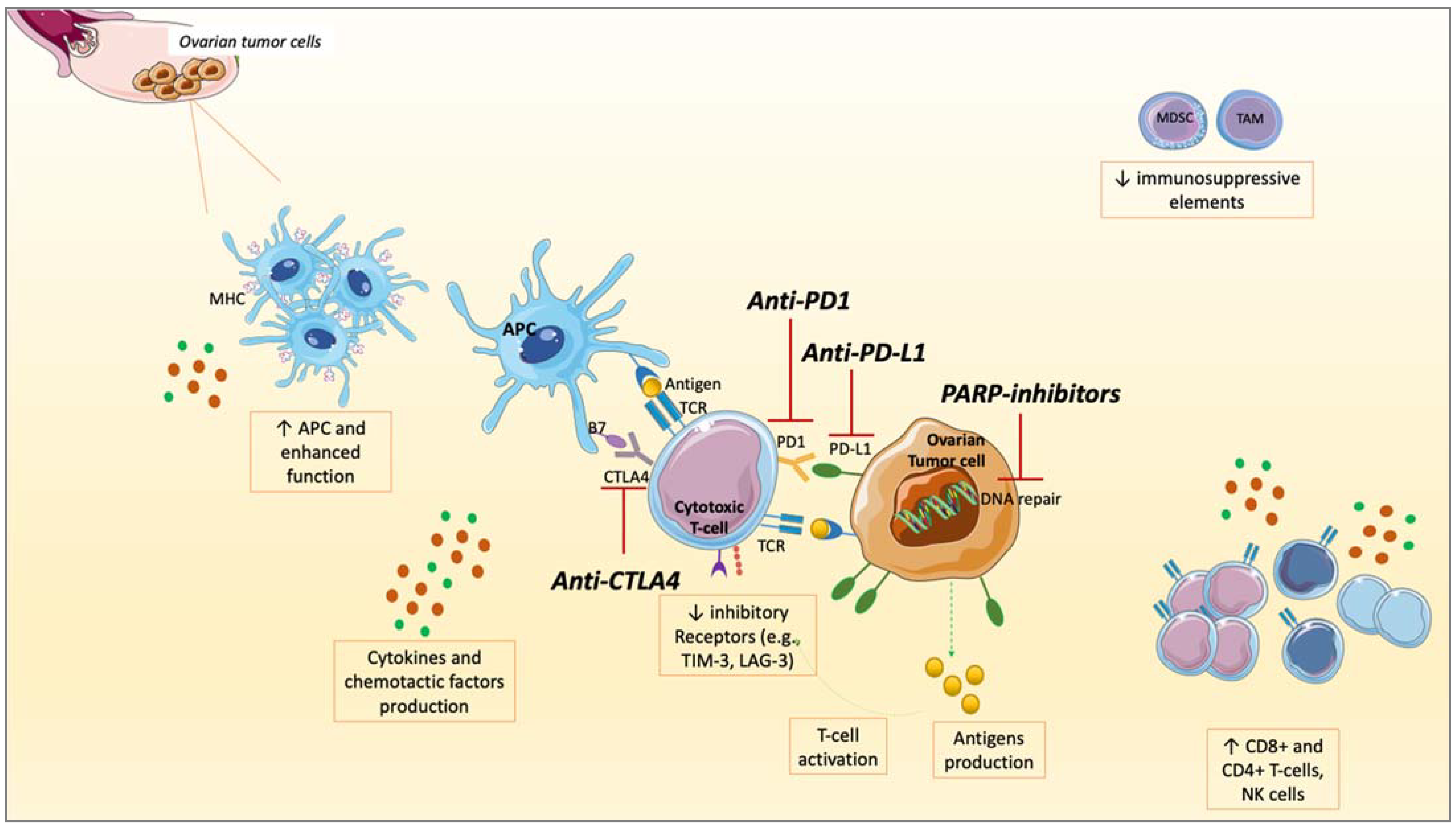
4. Rationale to Combine PARPis and ICIs in OC
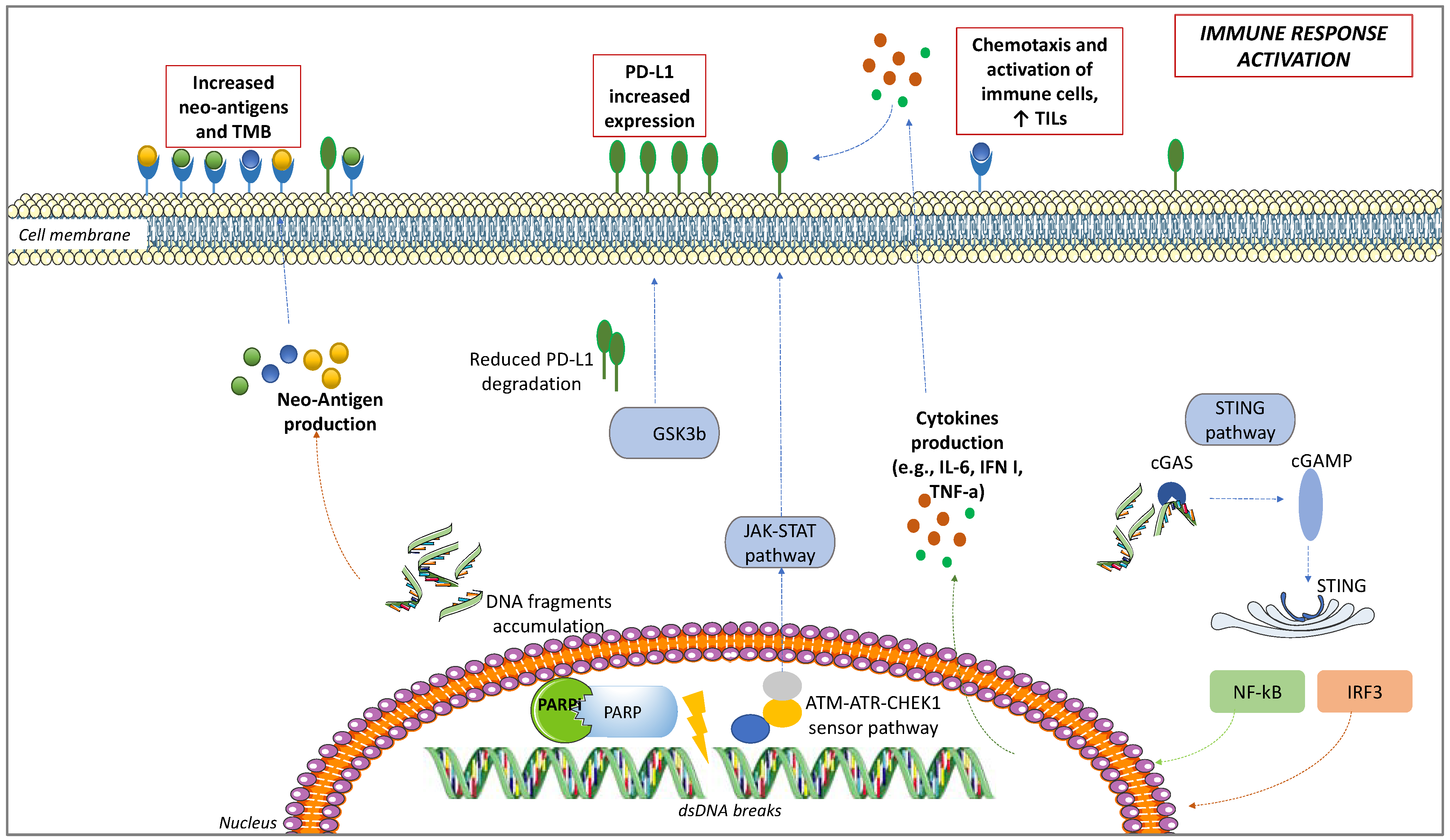
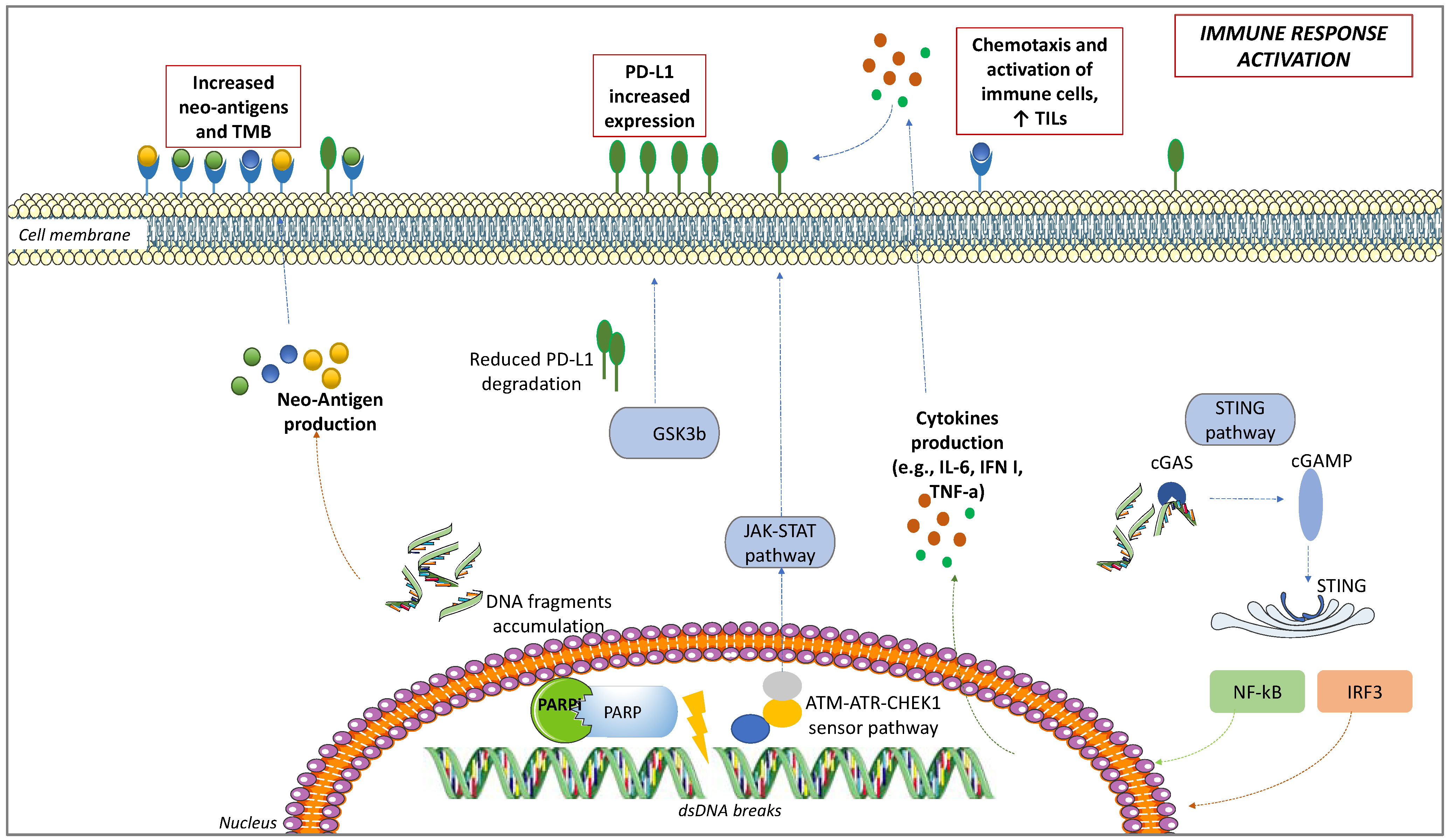
4.1. PD-L1 Upregulation
4.2. Interactions between PARPis and TME
4.3. TILs Increase
4.4. Neo-Antigens and TMB Increase
4.5. STING Pathway
5. Clinical Trials of PARP Inhibitors and ICIs Combination
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Globocan 2020. Ovary. Available online: https://gco.iarc.fr/today/data/factsheets/cancers/25-Ovary-fact-sheet.pdf (accessed on 15 June 2021).
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian Cancer Statistics, 2018. CA A Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Integrated Genomic Analyses of Ovarian Carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Piccart, M.J.; Bertelsen, K.; James, K.; Cassidy, J.; Mangioni, C.; Simonsen, E.; Stuart, G.; Kaye, S.; Vergote, I.; Blom, R.; et al. Randomized Intergroup Trial of Cisplatin-Paclitaxel Versus Cisplatin-Cyclophosphamide in Women With Advanced Epithelial Ovarian Cancer: Three-Year Results. J. Nat. Cancer Inst. 2000, 92, 699–708. [Google Scholar] [CrossRef] [Green Version]
- Ozols, R.F.; Bundy, B.N.; Greer, B.E.; Fowler, J.M.; Clarke-Pearson, D.; Burger, R.A.; Mannel, R.S.; DeGeest, K.; Hartenbach, E.M.; Baergen, R. Phase III Trial of Carboplatin and Paclitaxel Compared With Cisplatin and Paclitaxel in Patients With Optimally Resected Stage III Ovarian Cancer: A Gynecologic Oncology Group Study. JCO 2003, 21, 3194–3200. [Google Scholar] [CrossRef]
- Neijt, J.P.; Engelholm, S.A.; Tuxen, M.K.; Sørensen, P.G.; Hansen, M.; Sessa, C.; de Swart, C.A.M.; Hirsch, F.R.; Lund, B.; van Houwelingen, H.C. Exploratory Phase III Study of Paclitaxel and Cisplatin Versus Paclitaxel and Carboplatin in Advanced Ovarian Cancer. JCO 2000, 18, 3084–3092. [Google Scholar] [CrossRef] [PubMed]
- McGuire, W.P.; Hoskins, W.J.; Brady, M.F.; Kucera, P.R.; Partridge, E.E.; Look, K.Y.; Clarke-Pearson, D.L.; Davidson, M. Cyclophosphamide and Cisplatin Compared with Paclitaxel and Cisplatin in Patients with Stage III and Stage IV Ovarian Cancer. N. Engl. J. Med. 1996, 334, 1–6. [Google Scholar] [CrossRef]
- Bolton, K.L.; Chenevix-Trench, G.; Goh, C.; Sadetzki, S.; Ramus, S.J.; Karlan, B.Y.; Lambrechts, D.; Despierre, E.; Barrowdale, D.; McGuffog, L.; et al. Association Between BRCA1 and BRCA2 Mutations and Survival in Women With Invasive Epithelial Ovarian Cancer. JAMA 2012, 307, 382–389. [Google Scholar] [CrossRef] [Green Version]
- Konstantinopoulos, P.A.; Ceccaldi, R.; Shapiro, G.I.; D’Andrea, A.D. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov. 2015, 5, 1137–1154. [Google Scholar] [CrossRef] [Green Version]
- Ruscito, I.; Bellati, F.; Ray-Coquard, I.; Mirza, M.R.; du Bois, A.; Gasparri, M.L.; Costanzi, F.; De Marco, M.P.; Nuti, M.; Caserta, D.; et al. Incorporating Parp-inhibitors in Primary and Recurrent Ovarian Cancer: A Meta-analysis of 12 phase II/III randomized controlled trials. Cancer Treat. Rev. 2020, 87, 102040. [Google Scholar] [CrossRef]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef] [Green Version]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef] [Green Version]
- Mirza, M.R.; Åvall Lundqvist, E.; Birrer, M.J.; dePont Christensen, R.; Nyvang, G.B.; Malander, S.; Anttila, M.; Werner, T.L.; Lund, B.; Lindahl, G.; et al. Niraparib plus bevacizumab versus niraparib alone for platinum-sensitive recurrent ovarian cancer (NSGO-AVANOVA2/ENGOT-ov24): A randomised, phase 2, superiority trial. Lancet Oncol. 2019, 20, 1409–1419. [Google Scholar] [CrossRef]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef] [PubMed]
- González-Martín, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. PAOLA-1 Investigators. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.L.; Fleming, G.F.; Brady, M.F.; Swisher, E.M.; Steffensen, K.D.; Friedlander, M.; Okamoto, A.; Moore, K.N.; Efrat Ben-Baruch, N.; Werner, T.L.; et al. Veliparib with First-Line Chemotherapy and as Maintenance Therapy in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2403–2415. [Google Scholar] [CrossRef] [PubMed]
- Maiorano, B.A.; Maiorano, M.F.P.; Lorusso, D.; Maiello, E. Ovarian Cancer in the Era of Immune Checkpoint Inhibitors: State of the Art and Future Perspectives. Cancers 2021, 13, 4438. [Google Scholar] [CrossRef]
- Moore, K.N.; Bookman, M.; Sehouli, J.; Miller, A.; Anderson, C.; Scambia, G.; Myers, T.; Taskiran, C.; Robison, K.; Mäenpää, J.; et al. Atezolizumab, Bevacizumab, and Chemotherapy for Newly Diagnosed Stage III or IV Ovarian Cancer: Placebo-Controlled Randomized Phase III Trial (IMagyn050/GOG 3015/ENGOT-OV39). JCO 2021, 39, 1842–1855. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Fujiwara, K.; Ledermann, J.A.; Oza, A.M.; Kristeleit, R.S.; Ray-Coquard, I.L.; Richardson, G.E.; Sessa, C.; Yonemori, K.; Banerjee, S.; et al. Avelumab Alone or in Combination with Pegylated Liposomal Doxorubicin versus Pegylated Liposomal Doxorubicin Alone in Platinum-Resistant or Refractory Epithelial Ovarian Cancer: Primary and Biomarker Analysis of the Phase III JAVELIN Ovarian 200 Trial. Gynecol. Oncol. 2019, 154, 21–22. [Google Scholar] [CrossRef]
- Omatsu, K.; Hamanishi, J.; Katsumata, N.; Nishio, S.; Sawada, K.; Takeuchi, S.; Aoki, D.; Fujiwara, K.; Sugiyama, T.; Konishi, I. 807O Nivolumab versus Gemcitabine or Pegylated Liposomal Doxorubicin for Patients with Platinum-Resistant (Advanced or Recurrent) Ovarian Cancer: Open-Label, Randomized Trial in Japan (NINJA Trial). Ann. Oncol. 2020, 31, S611. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Waggoner, S.; Vidal, G.A.; Mita, M.; Moroney, J.W.; Holloway, R.; Van Le, L.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination With Pembrolizumab in Patients With Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol. 2019, 5, 1141–1149. [Google Scholar] [CrossRef] [Green Version]
- Lampert, E.J.; Zimmer, A.; Padget, M.; Cimino-Mathews, A.; Nair, J.R.; Liu, Y.; Swisher, E.M.; Hodge, J.W.; Nixon, A.B.; Nichols, E.; et al. Combination of PARP Inhibitor Olaparib, and PD-L1 Inhibitor Durvalumab, in Recurrent Ovarian Cancer: A Proof-of-Concept Phase II Study. Clin. Cancer Res. 2020, 26, 4268–4279. [Google Scholar] [CrossRef] [PubMed]
- Drew, Y.; Kaufman, B.; Banerjee, S.; Lortholary, A.; Hong, S.H.; Park, Y.H.; Zimmermann, S.; Roxburgh, P.; Ferguson, M.; Alvarez, R.H.; et al. Phase II Study of Olapatib + Durvalumab (MEDIOLA): Updated Results in Germline BRCA-mutated Platinum-Sensitive Relapsed (PSR) Ovarian Cancer (OC). Ann. Oncol. 2019, 30, 485–486. [Google Scholar] [CrossRef]
- Adams, S.F.; Rixe, O.; Lee, J.-H.; McCance, D.J.; Westgate, S.; Eberhardt, S.C.; Rutledge, T.; Muller, C. Phase I Study Combining Olaparib and Tremelimumab for the Treatment of Women with BRCA-Deficient Recurrent Ovarian Cancer. JCO 2017, 35, e17052. [Google Scholar] [CrossRef]
- Huertas, P. DNA Resection in Eukaryotes: Deciding How to Fix the Break. Nat. Struct. Mol. Biol. 2010, 17, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Lieber, M.R. The Mechanism of Human Nonhomologous DNA End Joining. J. Biol. Chem. 2008, 283, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Moynahan, M.E.; Jasin, M. Mitotic Homologous Recombination Maintains Genomic Stability and Suppresses Tumorigenesis. Nat. Rev. Mol. Cell Biol. 2010, 11, 196–207. [Google Scholar] [CrossRef] [Green Version]
- Gudmundsdottir, K.; Ashworth, A. The Roles of BRCA1 and BRCA2 and Associated Proteins in the Maintenance of Genomic Stability. Oncogene 2006, 25, 5864–5874. [Google Scholar] [CrossRef] [Green Version]
- Yun, M.H.; Hiom, K. CtIP-BRCA1 Modulates the Choice of DNA Double-Strand-Break Repair Pathway throughout the Cell Cycle. Nature 2009, 459, 460–463. [Google Scholar] [CrossRef]
- Dong, Y.; Hakimi, M.-A.; Chen, X.; Kumaraswamy, E.; Cooch, N.S.; Godwin, A.K.; Shiekhattar, R. Regulation of BRCC, a Holoenzyme Complex Containing BRCA1 and BRCA2, by a Signalosome-like Subunit and Its Role in DNA Repair. Mol. Cell 2003, 12, 1087–1099. [Google Scholar] [CrossRef]
- Yang, D.; Khan, S.; Sun, Y.; Hess, K.; Shmulevich, I.; Sood, A.K.; Zhang, W. Association of BRCA1 and BRCA2 Mutations With Survival, Chemotherapy Sensitivity, and Gene Mutator Phenotype in Patients With Ovarian Cancer. JAMA 2011, 306, 1557–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lord, C.J.; Ashworth, A. BRCAness Revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. PARP Inhibitors: Synthetic Lethality in the Clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Bai, P. Biology of Poly(ADP-Ribose) Polymerases: The Factotums of Cell Maintenance. Mol. Cell 2015, 58, 947–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helleday, T. The Underlying Mechanism for the PARP and BRCA Synthetic Lethality: Clearing up the Misunderstandings. Mol. Oncol. 2011, 5, 387–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Yu, X. Function of BRCA1 in the DNA Damage Response Is Mediated by ADP-Ribosylation. Cancer Cell 2013, 23, 693–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA Repair Defect in BRCA Mutant Cells as a Therapeutic Strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, T.A.; Shi, Y.; Rodriguez, L.E.; Solomon, L.R.; Donawho, C.K.; DiGiammarino, E.L.; Panchal, S.C.; Wilsbacher, J.L.; Gao, W.; Olson, A.M.; et al. Mechanistic Dissection of PARP1 Trapping and the Impact on In Vivo Tolerability and Efficacy of PARP Inhibitors. Mol. Cancer Res. 2015, 13, 1465–1477. [Google Scholar] [CrossRef] [Green Version]
- Murai, J.; Huang, S.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.G.; Sarkaria, J.N.; Kaufmann, S.H. Nonhomologous End Joining Drives Poly(ADP-Ribose) Polymerase (PARP) Inhibitor Lethality in Homologous Recombination-Deficient Cells. Proc. Natl. Acad. Sci. USA 2011, 108, 3406–3411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peyraud, F.; Italiano, A. Combined PARP Inhibition and Immune Checkpoint Therapy in Solid Tumors. Cancers 2020, 12, 1502. [Google Scholar] [CrossRef] [PubMed]
- Gogola, E.; Duarte, A.A.; de Ruiter, J.R.; Wiegant, W.W.; Schmid, J.A.; de Bruijn, R.; James, D.I.; Llobet, S.G.; Vis, D.J.; Annunziato, S.; et al. Selective Loss of PARG Restores PARylation and Counteracts PARP Inhibitor-Mediated Synthetic Lethality. Cancer Cell 2018, 33, 1078–1093.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray Chaudhuri, A.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.-E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication Fork Stability Confers Chemoresistance in BRCA-Deficient Cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Rondinelli, B.; Gogola, E.; Yücel, H.; Duarte, A.A.; van de Ven, M.; van der Sluijs, R.; Konstantinopoulos, P.A.; Jonkers, J.; Ceccaldi, R.; Rottenberg, S.; et al. EZH2 Promotes Degradation of Stalled Replication Forks by Recruiting MUS81 through Histone H3 Trimethylation. Nat. Cell Biol. 2017, 19, 1371–1378. [Google Scholar] [CrossRef]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.-K.; Hsu, J.-M.; Hsu, J.L.; Yu, W.-H.; Du, Y.; Lee, H.-H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yélamos, J.; Moreno-Lama, L.; Jimeno, J.; Ali, S.O. Immunomodulatory Roles of PARP-1 and PARP-2: Impact on PARP-Centered Cancer Therapies. Cancers 2020, 12, 392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strickland, K.C.; Howitt, B.E.; Shukla, S.A.; Rodig, S.; Ritterhouse, L.L.; Liu, J.F.; Garber, J.E.; Chowdhury, D.; Wu, C.J.; D’Andrea, A.D.; et al. Association and Prognostic Significance of BRCA1/2-Mutation Status with Neoantigen Load, Number of Tumor-Infiltrating Lymphocytes and Expression of PD-1/PD-L1 in High Grade Serous Ovarian Cancer. Oncotarget 2016, 7, 13587–13598. [Google Scholar] [CrossRef] [Green Version]
- Turinetto, M.; Scotto, G.; Tuninetti, V.; Giannone, G.; Valabrega, G. The Role of PARP Inhibitors in the Ovarian Cancer Microenvironment: Moving Forward From Synthetic Lethality. Front. Oncol. 2021, 11, 689829. [Google Scholar] [CrossRef]
- Ding, L.; Kim, H.-J.; Wang, Q.; Kearns, M.; Jiang, T.; Ohlson, C.E.; Li, B.B.; Xie, S.; Liu, J.F.; Stover, E.H.; et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in BRCA1-Deficient Ovarian Cancer. Cell Rep. 2018, 25, 2972–2980.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.; Zhao, W.; Ju, Z.; Wang, L.; Peng, Y.; Labrie, M.; Yap, T.A.; Mills, G.B.; Peng, G. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res. 2019, 79, 311–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Flies, D.B. Molecular Mechanisms of T Cell Co-Stimulation and Co-Inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Chambers, C.A.; Kuhns, M.S.; Egen, J.G.; Allison, J.P. CTLA-4-Mediated Inhibition in Regulation of T Cell Responses: Mechanisms and Manipulation in Tumor Immunotherapy. Annu. Rev. Immunol. 2001, 19, 565–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinartz, S.; Schumann, T.; Finkernagel, F.; Wortmann, A.; Jansen, J.M.; Meissner, W.; Krause, M.; Schwörer, A.; Wagner, U.; Müller-Brüsselbach, S.; et al. Mixed-polarization Phenotype of Ascites-associated Macrophages in Human Ovarian Carcinoma: Correlation of CD163 Expression, Cytokine Levels and Early Relapse. Int. J. Cancer 2014, 134, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; He, Y.; Sun, X.; Li, Q.; Wang, W.; Zhao, A.; Di, W. A High M1/M2 Ratio of Tumor-Associated Macrophages Is Associated with Extended Survival in Ovarian Cancer Patients. J. Ovarian Res. 2014, 7, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, S. Alternative Activation of Macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Fan, X.; Houghton, J. Tumor Microenvironment: The Role of the Tumor Stroma in Cancer. J. Cell. Biochem. 2007, 101, 805–815. [Google Scholar] [CrossRef]
- Nasta, F.; Laudisi, F.; Sambucci, M.; Rosado, M.M.; Pioli, C. Increased Foxp3+ Regulatory T Cells in Poly(ADP-Ribose) Polymerase-1 Deficiency. J. Immunol. 2010, 184, 3470–3477. [Google Scholar] [CrossRef]
- Labidi-Galy, S.I.; Sisirak, V.; Meeus, P.; Gobert, M.; Treilleux, I.; Bajard, A.; Combes, J.-D.; Faget, J.; Mithieux, F.; Cassignol, A.; et al. Quantitative and Functional Alterations of Plasmacytoid Dendritic Cells Contribute to Immune Tolerance in Ovarian Cancer. Cancer Res. 2011, 71, 5423–5434. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch Repair Deficiency Predicts Response of Solid Tumors to PD-1 Blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Havel, J.J.; Chowell, D.; Chan, T.A. The Evolving Landscape of Biomarkers for Checkpoint Inhibitor Immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-W.; Lim, S.-O.; Xia, W.; Lee, H.-H.; Chan, L.-C.; Kuo, C.-W.; Khoo, K.-H.; Chang, S.-S.; Cha, J.-H.; Kim, T.; et al. Glycosylation and Stabilization of Programmed Death Ligand-1 Suppresses T-Cell Activity. Nat. Commun. 2016, 7, 12632. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Yang, J.; Cai, Y.; Fu, S.; Zhang, N.; Fu, X.; Li, L. IFN-γ-Mediated Inhibition of Lung Cancer Correlates with PD-L1 Expression and Is Regulated by PI3K-AKT Signaling: IFN-γ in Lung Adenocarcinoma. Int. J. Cancer 2018, 143, 931–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA Double-Strand Break Repair Pathway Regulates PD-L1 Expression in Cancer Cells. Nat. Commun. 2017, 8, 1751. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y.; Ziv, Y. The ATM Protein Kinase: Regulating the Cellular Response to Genotoxic Stress, and More. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Shiotani, B.; Zou, L. Single-Stranded DNA Orchestrates an ATM-to-ATR Switch at DNA Breaks. Mol. Cell 2009, 33, 547–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yélamos, J.; Monreal, Y.; Saenz, L.; Aguado, E.; Schreiber, V.; Mota, R.; Fuente, T.; Minguela, A.; Parrilla, P.; de Murcia, G.; et al. PARP-2 Deficiency Affects the Survival of CD4+CD8+ Double-Positive Thymocytes. EMBO J. 2006, 25, 4350–4360. [Google Scholar] [CrossRef] [PubMed]
- Navarro, J.; Gozalbo-López, B.; Méndez, A.C.; Dantzer, F.; Schreiber, V.; Martínez, C.; Arana, D.M.; Farrés, J.; Revilla-Nuin, B.; Bueno, M.F.; et al. PARP-1/PARP-2 Double Deficiency in Mouse T Cells Results in Faulty Immune Responses and T Lymphomas. Sci. Rep. 2017, 7, 41962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sambucci, M.; Laudisi, F.; Novelli, F.; Bennici, E.; Rosado, M.M.; Pioli, C. Effects of PARP-1 Deficiency on Th1 and Th2 Cell Differentiation. Sci. World J. 2013, 2013, 375024. [Google Scholar] [CrossRef] [PubMed]
- Samstein, R.M.; Krishna, C.; Ma, X.; Pei, X.; Lee, K.-W.; Makarov, V.; Kuo, F.; Chung, J.; Srivastava, R.M.; Purohit, T.A.; et al. Mutations in BRCA1 and BRCA2 Differentially Affect the Tumor Microenvironment and Response to Checkpoint Blockade Immunotherapy. Nat. Cancer 2020, 1, 1188–1203. [Google Scholar] [CrossRef]
- Lee, E.K.; Konstantinopoulos, P.A. PARP Inhibition and Immune Modulation: Scientific Rationale and Perspectives for the Treatment of Gynecologic Cancers. Ther. Adv. Med. Oncol. 2020, 12, 175883592094411. [Google Scholar] [CrossRef] [PubMed]
- Pantelidou, C.; Sonzogni, O.; De Oliveria Taveira, M.; Mehta, A.K.; Kothari, A.; Wang, D.; Visal, T.; Li, M.K.; Pinto, J.; Castrillon, J.A.; et al. PARP Inhibitor Efficacy Depends on CD8+ T-Cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov. 2019, 9, 722–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouw, K.W.; Goldberg, M.S.; Konstantinopoulos, P.A.; D’Andrea, A.D. DNA Damage and Repair Biomarkers of Immunotherapy Response. Cancer Discov. 2017, 7, 675–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Li, L.; Chen, L.; Yuan, W.; Dong, L.; Zhang, Y.; Wu, H.; Wang, C. PARP-1 Mediates LPS-Induced HMGB1 Release by Macrophages through Regulation of HMGB1 Acetylation. J. Immunol. 2014, 193, 6114–6123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crusz, S.M.; Balkwill, F.R. Inflammation and Cancer: Advances and New Agents. Nat. Rev. Clin. Oncol. 2015, 12, 584–596. [Google Scholar] [CrossRef]
- Pietrzak, J.; Gronkowska, K.; Robaszkiewicz, A. PARP Traps Rescue the Pro-Inflammatory Response of Human Macrophages in the In Vitro Model of LPS-Induced Tolerance. Pharmaceuticals 2021, 14, 170. [Google Scholar] [CrossRef]
- Kunze, F.A.; Bauer, M.; Komuczki, J.; Lanzinger, M.; Gunasekera, K.; Hopp, A.-K.; Lehmann, M.; Becher, B.; Müller, A.; Hottiger, M.O. ARTD1 in Myeloid Cells Controls the IL-12/18–IFN-γ Axis in a Model of Sterile Sepsis, Chronic Bacterial Infection, and Cancer. J. Immunol. 2019, 202, 1406–1416. [Google Scholar] [CrossRef]
- Tang, M.L.F.; Khan, M.K.N.; Croxford, J.L.; Tan, K.W.; Angeli, V.; Gasser, S. The DNA Damage Response Induces Antigen Presenting Cell-like Functions in Fibroblasts: Immunomodulation. Eur. J. Immunol. 2014, 44, 1108–1118. [Google Scholar] [CrossRef]
- Huang, J.; Wang, L.; Cong, Z.; Amoozgar, Z.; Kiner, E.; Xing, D.; Orsulic, S.; Matulonis, U.; Goldberg, M.S. The PARP1 Inhibitor BMN 673 Exhibits Immunoregulatory Effects in a BRCA1 −/− Murine Model of Ovarian Cancer. Biochem. Biophys. Res. Commun. 2015, 463, 551–556. [Google Scholar] [CrossRef]
- Morse, C.B.; Toukatly, M.N.; Kilgore, M.R.; Agnew, K.J.; Bernards, S.S.; Norquist, B.M.; Pennington, K.P.; Garcia, R.L.; Liao, J.B.; Swisher, E.M. Tumor Infiltrating Lymphocytes and Homologous Recombination Deficiency Are Independently Associated with Improved Survival in Ovarian Carcinoma. Gynecol. Oncol. 2019, 153, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific Recruitment of Regulatory T Cells in Ovarian Carcinoma Fosters Immune Privilege and Predicts Reduced Survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef]
- Hamanishi, J.; Mandai, M.; Iwasaki, M.; Okazaki, T.; Tanaka, Y.; Yamaguchi, K.; Higuchi, T.; Yagi, H.; Takakura, K.; Minato, N.; et al. Programmed Cell Death 1 Ligand 1 and Tumor-Infiltrating CD8+ T Lymphocytes Are Prognostic Factors of Human Ovarian Cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 3360–3365. [Google Scholar] [CrossRef] [Green Version]
- Chan, N.; Koritzinsky, M.; Zhao, H.; Bindra, R.; Glazer, P.M.; Powell, S.; Belmaaza, A.; Wouters, B.; Bristow, R.G. Chronic Hypoxia Decreases Synthesis of Homologous Recombination Proteins to Offset Chemoresistance and Radioresistance. Cancer Res. 2008, 68, 605–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Furness, A.J.S.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal Neoantigens Elicit T Cell Immunoreactivity and Sensitivity to Immune Checkpoint Blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germano, G.; Lamba, S.; Rospo, G.; Barault, L.; Magrì, A.; Maione, F.; Russo, M.; Crisafulli, G.; Bartolini, A.; Lerda, G.; et al. Inactivation of DNA Repair Triggers Neoantigen Generation and Impairs Tumour Growth. Nature 2017, 552, 116–120. [Google Scholar] [CrossRef]
- Härtlova, A.; Erttmann, S.F.; Raffi, F.A.M.; Schmalz, A.M.; Resch, U.; Anugula, S.; Lienenklaus, S.; Nilsson, L.M.; Kröger, A.; Nilsson, J.A.; et al. DNA Damage Primes the Type I Interferon System via the Cytosolic DNA Sensor STING to Promote Anti-Microbial Innate Immunity. Immunity 2015, 42, 332–343. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Chen, Z.J. The CGAS–CGAMP–STING Pathway Connects DNA Damage to Inflammation, Senescence, and Cancer. J. Exp. Med. 2018, 215, 1287–1299. [Google Scholar] [CrossRef]
- Corrales, L.; McWhirter, S.M.; Dubensky, T.W.; Gajewski, T.F. The Host STING Pathway at the Interface of Cancer and Immunity. J. Clin. Investig. 2016, 126, 2404–2411. [Google Scholar] [CrossRef] [Green Version]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I Interferons in Anticancer Immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Reisländer, T.; Lombardi, E.P.; Groelly, F.J.; Miar, A.; Porru, M.; Di Vito, S.; Wright, B.; Lockstone, H.; Biroccio, A.; Harris, A.; et al. BRCA2 Abrogation Triggers Innate Immune Responses Potentiated by Treatment with PARP Inhibitors. Nat. Commun. 2019, 10, 3143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Färkkilä, A.; Gulhan, D.C.; Casado, J.; Jacobson, C.A.; Nguyen, H.; Kochupurakkal, B.; Maliga, Z.; Yapp, C.; Chen, Y.-A.; Schapiro, D.; et al. Immunogenomic Profiling Determines Responses to Combined PARP and PD-1 Inhibition in Ovarian Cancer. Nat. Commun. 2020, 11, 1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aysal, A.; Karnezis, A.; Medhi, I.; Grenert, J.P.; Zaloudek, C.J.; Rabban, J.T. Ovarian Endometrioid Adenocarcinoma: Incidence and Clinical Significance of the Morphologic and Immunohistochemical Markers of Mismatch Repair Protein Defects and Tumor Microsatellite Instability. Am. J. Surg. Pathol. 2012, 36, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Marcus, L.; Lemery, S.J.; Keegan, P.; Pazdur, R. FDA Approval Summary: Pembrolizumab for the Treatment of Microsatellite Instability-High Solid Tumors. Clin. Cancer Res. 2019, 25, 3753–3758. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Cortez, D.; Yazdi, P.; Neff, N.; Elledge, S.J.; Qin, J. BASC, a Super Complex of BRCA1-Associated Proteins Involved in the Recognition and Repair of Aberrant DNA Structures. Genes Dev. 2000, 14, 927–939. [Google Scholar] [CrossRef]
- Tentori, L.; Lacal, P.M.; Muzi, A.; Dorio, A.S.; Leonetti, C.; Scarsella, M.; Ruffini, F.; Xu, W.; Min, W.; Stoppacciaro, A.; et al. Poly(ADP-Ribose) Polymerase (PARP) Inhibition or PARP-1 Gene Deletion Reduces Angiogenesis. Eur. J. Cancer 2007, 43, 2124–2133. [Google Scholar] [CrossRef] [PubMed]
- Rahma, O.E.; Hodi, F.S. The Intersection between Tumor Angiogenesis and Immune Suppression. Clin. Cancer Res. 2019, 25, 5449–5457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perren, T.J.; Swart, A.M.; Pfisterer, J.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; Kurzeder, C.; et al. A Phase 3 Trial of Bevacizumab in Ovarian Cancer. N. Engl. J. Med. 2011, 365, 2484–2496. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Study Name/NCT Identifier | Phase | Target Population (n) | ICI (Target) | PARPi | Results |
|---|---|---|---|---|---|
| TOPACIO/Keynote-162/NCT02657889 [23] | I-II | PR-ROC (n = 62), not selected for BRCA/HR status | Pembrolizumab 200 mg q3w (anti-PD1) | Niraparib 200 mg OD | ORR 25%, DCR 68% BRCAm (21%): ORR 45%, DCR 73% 8 CR (5 BRCAwt patients) |
| NCT02484404 [24] | II | ROC (n = 35: 30 PR-ROC + 5 PS-ROC): BRCAwt (n = 27) gBRCAmut (n = 6) sBRCAmut (n = 2) | Durvalumab 1500 mg q4w (anti-PD-L1) | Olaparib 300 mg BID | ORR 14%, DCR 71% 5 PRs (2 gBRCAmut, 2 BRCAwt, 1 sBRCAmut) mPFS 3.9 mos |
| MEDIOLA/NCT02734004 [25] | II | gBRCAmut PS-ROC (n = 32) | Durvalumab 1500 mg q4w | Olaparib 300 mg BID | ORR 71.9% mPFS 11.1 mos 12-wks DCR 81% 7/32 CR (21.8%) |
| PS-ROC BRCAwt (n = 32) | Durvalumab | Olaparib | ORR 31.3%, 24-wks DCR of 28.1% mPFS 5.5 mos | ||
| PS-ROC BRCAwt (n = 31) | Durvalumab | Olaparib + Bevacizumab 10 mg/kg q2w | ORR 77.4%, 24-wks DCR 77.4% mPFS 14.7 mos | ||
| NCT02571725 [26] | Ib/II | gBRCAmut ROC (n = 3) | Tremelimumab 10 mg/kg q4w (anti-CTLA4) | Olaparib 300 mg BID | ORR 100% 3 PRs |
| Clinicaltrials. Gov Registration/NAME | Phase | Setting | Combination |
|---|---|---|---|
| NCT03602859/FIRST | III | First-line, Stage III/IV EOC | Niraparib + Dostarlimab |
| NCT03522246/ATHENA | III | Maintenance after first-line platinum-based CT | Rucaparib + Nivolumab |
| NCT04679064/NItCHE-MITO33 | III | Platinum-ineligible ROC | Niraparib + Dostarlimab |
| NCT04361370/DUO-O | III | Maintenance after first-line platinum-based CT | Olaparib + Durvalumab + Bevacizumab |
| NCT03740165/KEYLYNK-001 | III | Maintenance after first-line platinum-based CT | Olaparib + Pembrolizumab |
| NCT02873962 | II | ROC | Rucaparib + Nivolumab + Bevacizumab |
| NCT03955471/MOONSTONE | II | PS-ROC | Niraparib + Dostarlimab |
| NCT03695380 (Cohort 2) | I/II | PS-ROC | Niraparib + Cobimetinib + Atezolizumab |
| NCT04673448 | I | BRCAmut-ROC | Niraparib + Dostarlimab |
| NCT03651206/ROCSAN | II | Ovarian Carcinosarcoma | Niraparib + Dostarlimab |
| NCT04361370/OPEB-01 | II | Maintenance in PS-ROC BRCAwt | Olaparib + Pembrolizumab + Bevacizumab |
| NCT04417192/OLAPem | II | Neoadjuvant OC | Olaparib + Pembrolizumab |
| NCT02953457 | II | BRCAmut-ROC | Olaparib + Durvalumab + Tremelimumab |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maiorano, B.A.; Lorusso, D.; Maiorano, M.F.P.; Ciardiello, D.; Parrella, P.; Petracca, A.; Cormio, G.; Maiello, E. The Interplay between PARP Inhibitors and Immunotherapy in Ovarian Cancer: The Rationale behind a New Combination Therapy. Int. J. Mol. Sci. 2022, 23, 3871. https://doi.org/10.3390/ijms23073871
Maiorano BA, Lorusso D, Maiorano MFP, Ciardiello D, Parrella P, Petracca A, Cormio G, Maiello E. The Interplay between PARP Inhibitors and Immunotherapy in Ovarian Cancer: The Rationale behind a New Combination Therapy. International Journal of Molecular Sciences. 2022; 23(7):3871. https://doi.org/10.3390/ijms23073871
Chicago/Turabian StyleMaiorano, Brigida Anna, Domenica Lorusso, Mauro Francesco Pio Maiorano, Davide Ciardiello, Paola Parrella, Antonio Petracca, Gennaro Cormio, and Evaristo Maiello. 2022. "The Interplay between PARP Inhibitors and Immunotherapy in Ovarian Cancer: The Rationale behind a New Combination Therapy" International Journal of Molecular Sciences 23, no. 7: 3871. https://doi.org/10.3390/ijms23073871
APA StyleMaiorano, B. A., Lorusso, D., Maiorano, M. F. P., Ciardiello, D., Parrella, P., Petracca, A., Cormio, G., & Maiello, E. (2022). The Interplay between PARP Inhibitors and Immunotherapy in Ovarian Cancer: The Rationale behind a New Combination Therapy. International Journal of Molecular Sciences, 23(7), 3871. https://doi.org/10.3390/ijms23073871