
Stress Granules and Acute Ischemic Stroke: Beyond mRNA Translation

 , , ,
, , ,  ,
,  , and
, and 
{kind=link}
{kind=link}
Abstract
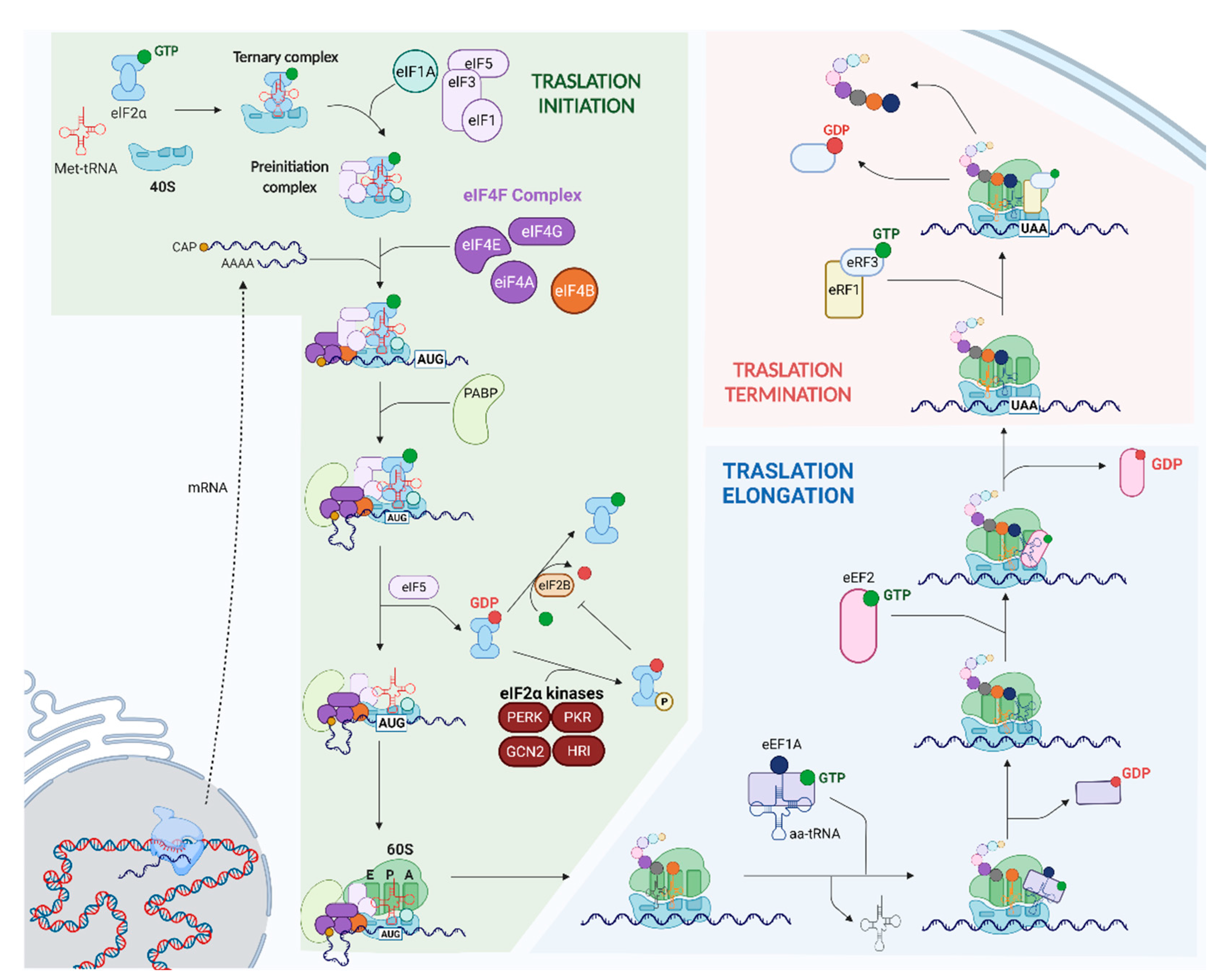
:1. Introduction
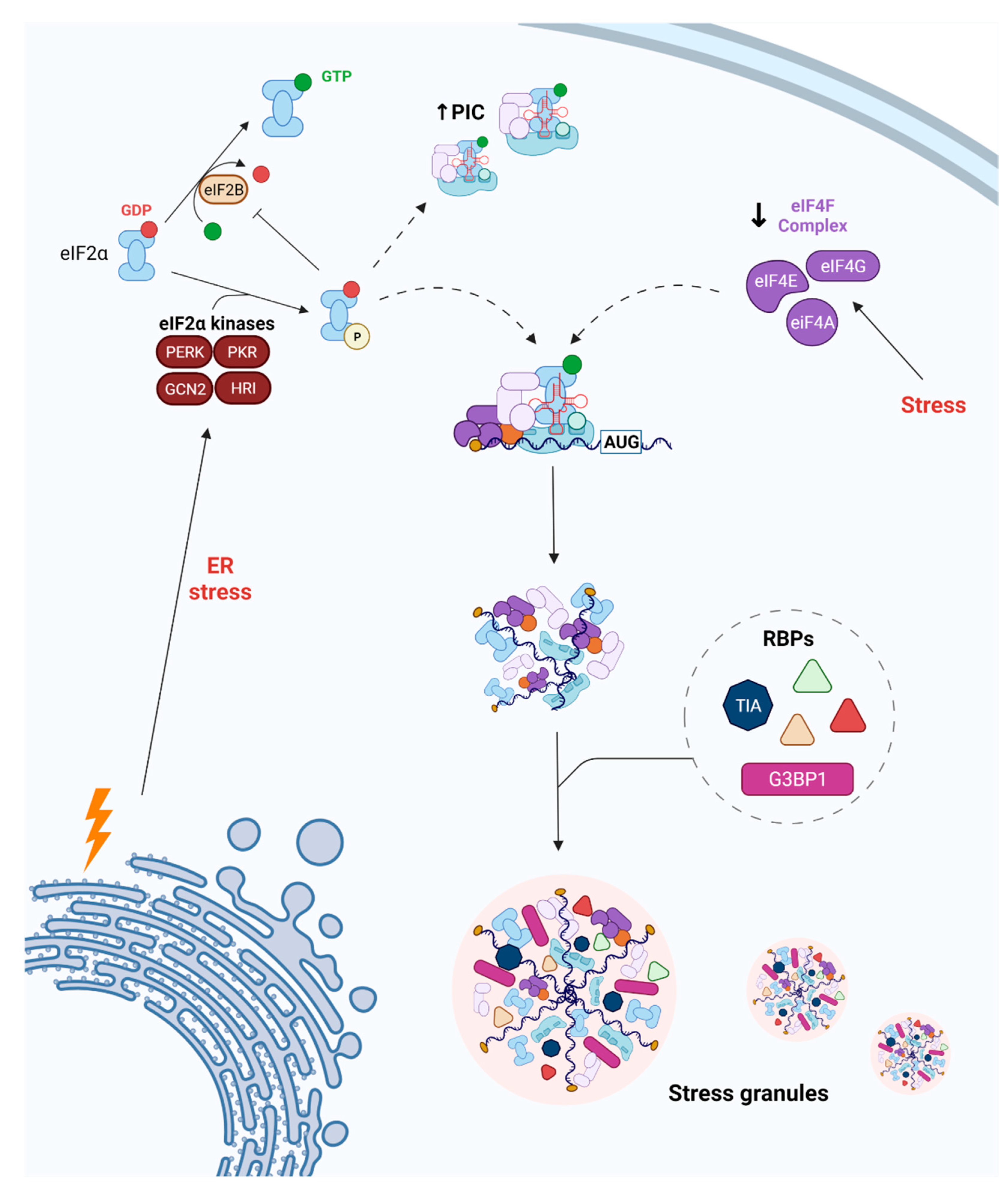
2. SG Dynamics following Cerebral Ischemia
2.1. Changes in RBP Expression
2.2. Modulation of RBP Function and Expression
2.3. Endothelial Cells and SG Dynamics
3. Two Different Ways SG Dynamics Act in Triggering Neuroprotection after Ischemia
4. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Feigin, V.L.; Vos, T.; Nichols, E.; Owolabi, M.O.; Carroll, W.M.; Dichgans, M.; Deuschl, G.; Parmar, P.; Brainin, M.; Murray, C. The global burden of neurological disorders: Translating evidence into policy. Lancet Neurol. 2020, 19, 255–265. [Google Scholar] [CrossRef]
- Ramos-Cabrer, P.; Campos, F.; Sobrino, T.; Castillo, J. Targeting the ischemic penumbra. Stroke 2011, 42, S7–S11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of Ischaemic Stroke: An Integrated View. Trends Neurosci. 1999, 22, 391–397. [Google Scholar] [CrossRef]
- Fredrick, K.; Ibba, M. PROTEIN SYNTHESIS: Errors Rectified in Retrospect. Nature 2009, 457, 157. [Google Scholar] [CrossRef] [Green Version]
- Kapur, M.; Monaghan, C.E.; Ackerman, S.L. Regulation of MRNA Translation in Neurons-A Matter of Life and Death. Neuron 2017, 96, 616–637. [Google Scholar] [CrossRef] [Green Version]
- Merrick, W.C.; Pavitt, G.D. Protein Synthesis Initiation in Eukaryotic Cells. Cold Spring Harb. Perspect. Biol. 2018, 10, a033092. [Google Scholar] [CrossRef]
- Saini, A.K.; Nanda, J.S.; Lorsch, J.R.; Hinnebusch, A.G. Regulatory Elements in EIF1A Control the Fidelity of Start Codon Selection by Modulating TRNAiMet Binding to the Ribosome. Genes Dev. 2010, 24, 97. [Google Scholar] [CrossRef] [Green Version]
- Andersen, G.R.; Nissen, P.; Nyborg, J. Elongation Factors in Protein Biosynthesis. Trends Biochem. Sci. 2003, 28, 434–441. [Google Scholar] [CrossRef]
- Han, Y.; Yuan, M.; Guo, Y.S.; Shen, X.Y.; Gao, Z.K.; Bi, X. Mechanism of Endoplasmic Reticulum Stress in Cerebral Ischemia. Front. Cell. Neurosci. 2021, 15, 294. [Google Scholar] [CrossRef]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated Translation Initiation Controls Stress-Induced Gene Expression in Mammalian Cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Wang, Y.C.; Li, X.; Shen, Y.; Lyu, J.; Sheng, H.; Paschen, W.; Yang, W. PERK (Protein Kinase RNA-Like ER Kinase) Branch of the Unfolded Protein Response Confers Neuroprotection in Ischemic Stroke by Suppressing Protein Synthesis. Stroke 2020, 51, 1570–1577. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, N.; Gorman, A.M.; Gupta, S.; Samali, A. The EIF2α Kinases: Their Structures and Functions. Cell. Mol. Life Sci. CMLS 2013, 70, 3493–3511. [Google Scholar] [CrossRef] [PubMed]
- Kwong, M.; Kan, Y.W.; Chan, J.Y. The CNC Basic Leucine Zipper Factor, Nrf1, Is Essential for Cell Survival in Response to Oxidative Stress-Inducing Agents. Role for Nrf1 in Gamma-Gcs(l) and Gss Expression in Mouse Fibroblasts. J. Biol. Chem. 1999, 274, 37491–37498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lange, P.S.; Chavez, J.C.; Pinto, J.T.; Coppola, G.; Sun, C.W.; Townes, T.M.; Geschwind, D.H.; Ratan, R.R. ATF4 Is an Oxidative Stress-Inducible, Prodeath Transcription Factor in Neurons in Vitro and in Vivo. J. Exp. Med. 2008, 205, 1227–1242. [Google Scholar] [CrossRef] [Green Version]
- Vattem, K.M.; Wek, R.C. Reinitiation Involving Upstream ORFs Regulates ATF4 MRNA Translation in Mammalian Cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11269–11274. [Google Scholar] [CrossRef] [Green Version]
- Wolozin, B.; Ivanov, P. Stress Granules and Neurodegeneration. Nat. Rev. Neurosci. 2019, 20, 649–666. [Google Scholar] [CrossRef]
- Shin, Y.; Brangwynne, C.P. Liquid Phase Condensation in Cell Physiology and Disease. Science 2017, 357, eaaf4382. [Google Scholar] [CrossRef] [Green Version]
- Protter, D.S.W.; Parker, R. Principles and Properties of Stress Granules. Trends Cell Biol. 2016, 26, 668–679. [Google Scholar] [CrossRef] [Green Version]
- Si, W.; Li, Y.; Ye, S.; Li, Z.; Liu, Y.; Kuang, W.; Chen, D.; Zhu, M. Methyltransferase 3 Mediated MiRNA M6A Methylation Promotes Stress Granule Formation in the Early Stage of Acute Ischemic Stroke. Front. Mol. Neurosci. 2020, 13, 103. [Google Scholar] [CrossRef]
- Kahl, A.; Blanco, I.; Jackman, K.; Baskar, J.; Milaganur Mohan, H.; Rodney-Sandy, R.; Zhang, S.; Iadecola, C.; Hochrainer, K. Cerebral Ischemia Induces the Aggregation of Proteins Linked to Neurodegenerative Diseases. Sci. Rep. 2018, 8, 2701. [Google Scholar] [CrossRef]
- Liu, X.; Yamashita, T.; Shi, X.; Bian, Y.; Bian, Z.; Omote, Y.; Takemoto, M.; Hishikawa, N.; Ohta, Y.; Abe, K. Dynamic Changes and Mislocalizations of Neurodegenerative Disease-Related Proteins in Mice Stroke Model. Brain Res. 2020, 1742, 146862. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Zuo, Y.; Ai-Zakwani, K.; Luo, J.; Zhu, H.; Yan, X.X.; Liu, F. Subarachnoid Hemorrhage Enhances the Expression of TDP-43 in the Brain of Experimental Rats and Human Subjects. Exp. Ther. Med. 2018, 16, 3363–3368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombrita, C.; Zennaro, E.; Fallini, C.; Weber, M.; Sommacal, A.; Buratti, E.; Silani, V.; Ratti, A. TDP-43 Is Recruited to Stress Granules in Conditions of Oxidative Insult. J. Neurochem. 2009, 111, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Ayuso, M.I.; Martínez-Alonso, E.; Regidor, I.; Alcázar, A. Stress Granule Induction after Brain Ischemia is Independent of Eukaryotic Translation Initiation Factor (EIF) 2α Phosphorylation and Is Correlated with a Decrease in EIF4B and EIF4E Proteins. J. Biol. Chem. 2016, 291, 27252–27264. [Google Scholar] [CrossRef] [Green Version]
- DeGracia, D.J.; Rudolph, J.; Roberts, G.G.; Rafols, J.A.; Wang, J. Convergence of Stress Granules and Protein Aggregates in Hippocampal Cornu Ammonis 1 at Later Reperfusion Following Global Brain Ischemia. Neuroscience 2007, 146, 562–572. [Google Scholar] [CrossRef] [Green Version]
- Kayali, F.; Montie, H.L.; Rafols, J.A.; DeGracia, D.J. Prolonged Translation Arrest in Reperfused Hippocampal Cornu Ammonis 1 Is Mediated by Stress Granules. Neuroscience 2005, 134, 1223–1245. [Google Scholar] [CrossRef]
- Thammisetty, S.S.; Pedragosa, J.; Weng, Y.C.; Calon, F.; Planas, A.; Kriz, J. Age-Related Deregulation of TDP-43 after Stroke Enhances NF-ΚB-Mediated Inflammation and Neuronal Damage. J. Neuroinflammation 2018, 15, 312. [Google Scholar] [CrossRef]
- Kanazawa, M.; Kakita, A.; Igarashi, H.; Takahashi, T.; Kawamura, K.; Takahashi, H.; Nakada, T.; Nishizawa, M.; Shimohata, T. Biochemical and Histopathological Alterations in TAR DNA-Binding Protein-43 after Acute Ischemic Stroke in Rats. J. Neurochem. 2011, 116, 957–965. [Google Scholar] [CrossRef]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a Risk Factor for Neurodegenerative Disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Liu, Y.; Gao, Y.; Wu, Y.; Wu, Y.; Wang, H.; Zhang, C. Histochemical Mapping of HnRNP A2/B1 in Rat Brain after Ischemia-Reperfusion Insults. J. Histochem. Cytochem. Off. J. Histochem. Soc. 2010, 58, 695–705. [Google Scholar] [CrossRef] [Green Version]
- Ávila-Gómez, P.; Vieites-Prado, A.; Dopico-López, A.; Bashir, S.; Fernández-Susavila, H.; Gubern, C.; Pérez-Mato, M.; Correa-Paz, C.; Iglesias-Rey, R.; Sobrino, T.; et al. Cold Stress Protein RBM3 Responds to Hypothermia and Is Associated with Good Stroke Outcome. Brain Commun. 2020, 2, fcaa078. [Google Scholar] [CrossRef] [PubMed]
- Ávila-Gómez, P.; Pérez-Mato, M.; Hervella, P.; Dopico-López, A.; da Silva-Candal, A.; Bugallo-Casal, A.; López-Amoedo, S.; Candamo-Lourido, M.; Sobrino, T.; Iglesias-Rey, R.; et al. Clinical Medicine Associations between RNA-Binding Motif Protein 3, Fibroblast Growth Factor 21, and Clinical Outcome in Patients with Stroke. J. Clin. Med 2022, 2022, 949. [Google Scholar] [CrossRef] [PubMed]
- Si, W.; Li, Z.; Huang, Z.; Ye, S.; Li, X.; Li, Y.; Kuang, W.; Chen, D.; Zhu, M. RNA Binding Protein Motif 3 Inhibits Oxygen-Glucose Deprivation/Reoxygenation-Induced Apoptosis Through Promoting Stress Granules Formation in PC12 Cells and Rat Primary Cortical Neurons. Front. Cell. Neurosci. 2020, 14, 287. [Google Scholar] [CrossRef] [PubMed]
- Voelz, C.; Habib, P.; Köberlein, S.; Beyer, C.; Slowik, A. Alteration of MiRNA Biogenesis Regulating Proteins in the Human Microglial Cell Line HMC-3 After Ischemic Stress. Mol. Neurobiol. 2021, 58, 1535–1549. [Google Scholar] [CrossRef]
- Si, W.; Ye, S.; Ren, Z.; Liu, X.; Wu, Z.; Li, Y.; Zhou, J.; Zhang, S.; Li, Y.; Deng, R.; et al. MiR 335 Promotes Stress Granule Formation to Inhibit Apoptosis by Targeting ROCK2 in Acute Ischemic Stroke. Int. J. Mol. Med. 2019, 43, 1452–1466. [Google Scholar] [CrossRef]
- Liu, Q.; Hu, Y.; Zhang, M.; Yan, Y.; Yu, H.; Ge, L. MicroRNA-451 Protects Neurons against Ischemia/Reperfusion Injury-Induced Cell Death by Targeting CELF2. Neuropsychiatr. Dis. Treat. 2018, 14, 2773–2782. [Google Scholar] [CrossRef] [Green Version]
- Kadry, H.; Noorani, B.; Cucullo, L. A Blood-Brain Barrier Overview on Structure, Function, Impairment, and Biomarkers of Integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef]
- Bernardo-Castro, S.; Sousa, J.A.; Brás, A.; Cecília, C.; Rodrigues, B.; Almendra, L.; Machado, C.; Santo, G.; Silva, F.; Ferreira, L.; et al. Pathophysiology of Blood–Brain Barrier Permeability Throughout the Different Stages of Ischemic Stroke and Its Implication on Hemorrhagic Transformation and Recovery. Front. Neurol. 2020, 11, 1605. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, C.; Jiang, J.; Xin, M.; Hao, J. The TDP43 CFTS Affect Brain Endothelial Cell Functions by Regulating YAP and Tight Junction Proteins in Cerebral Ischemic Injury. Res. Sq. 2021. [Google Scholar] [CrossRef]
- Thilmann, R.; Xie, Y.; Kleihues, P.; Kiessling, M. Persistent Inhibition of Protein Synthesis Precedes Delayed Neuronal Death in Postischemic Gerbil Hippocampus. Acta Neuropathol. 1986, 71, 88–93. [Google Scholar] [CrossRef]
- Dienel, G.A.; Pulsinelli, W.A.; Duffy, T.E. Regional Protein Synthesis in Rat Brain Following Acute Hemispheric Ischemia. J. Neurochem. 1980, 35, 1216–1226. [Google Scholar] [CrossRef]
- Bodsch, W.; Takahashi, K.; Barbier, A.; Ophoff, B.G.; Hossmann, K.A. Cerebral Protein Synthesis and Ischemia. Prog. Brain Res. 1985, 63, 197–210. [Google Scholar] [CrossRef]
- Arimoto-Matsuzaki, K.; Saito, H.; Takekawa, M. TIA1 Oxidation Inhibits Stress Granule Assembly and Sensitizes Cells to Stress-Induced Apoptosis. Nat. Commun. 2016, 7, 10252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, G.G.; di Loreto, M.J.; Marshall, M.; Wang, J.; DeGracia, D.J. Hippocampal Cellular Stress Responses after Global Brain Ischemia and Reperfusion. Antioxid. Redox Signal. 2007, 9, 2265–2275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottschald, O.R.; Malec, V.; Krasteva, G.; Hasan, D.; Kamlah, F.; Herold, S.; Rose, F.; Seeger, W.; Hänze, J. TIAR and TIA-1 MRNA-Binding Proteins Co-Aggregate under Conditions of Rapid Oxygen Decline and Extreme Hypoxia and Suppress the HIF-1α Pathway. J. Mol. Cell Biol. 2010, 2, 345–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, K.; Li, W.; Nagayama, T.; He, X.; Sinor, A.D.; Chang, J.; Mao, X.; Graham, S.H.; Simon, R.P.; Greenberg, D.A. Expression of the RNA-Binding Protein TIAR is Increased in Neurons after Ischemic Cerebral Injury. J. Neurosci. Res. 2000, 59, 767–774. [Google Scholar] [CrossRef]
- Gorospe, M.; Tominaga, K.; Wu, X.; Fähling, M.; Ivan, M. Post-Transcriptional Control of the Hypoxic Response by RNA-Binding Proteins and MicroRNAs. Front. Mol. Neurosci. 2011, 4, 7. [Google Scholar] [CrossRef] [Green Version]
- Helton, R.; Cui, J.; Scheel, J.R.; Ellison, J.A.; Ames, C.; Gibson, C.; Blouw, B.; Ouyang, L.; Dragatsis, I.; Zeitlin, S.; et al. Brain-Specific Knock-out of Hypoxia-Inducible Factor-1alpha Reduces Rather than Increases Hypoxic-Ischemic Damage. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 4099–4107. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aramburu-Núñez, M.; Custodia, A.; Pérez-Mato, M.; Iglesias-Rey, R.; Campos, F.; Castillo, J.; Ouro, A.; Romaus-Sanjurjo, D.; Sobrino, T. Stress Granules and Acute Ischemic Stroke: Beyond mRNA Translation. Int. J. Mol. Sci. 2022, 23, 3747. https://doi.org/10.3390/ijms23073747
Aramburu-Núñez M, Custodia A, Pérez-Mato M, Iglesias-Rey R, Campos F, Castillo J, Ouro A, Romaus-Sanjurjo D, Sobrino T. Stress Granules and Acute Ischemic Stroke: Beyond mRNA Translation. International Journal of Molecular Sciences. 2022; 23(7):3747. https://doi.org/10.3390/ijms23073747
Chicago/Turabian StyleAramburu-Núñez, Marta, Antía Custodia, María Pérez-Mato, Ramón Iglesias-Rey, Francisco Campos, José Castillo, Alberto Ouro, Daniel Romaus-Sanjurjo, and Tomás Sobrino. 2022. "Stress Granules and Acute Ischemic Stroke: Beyond mRNA Translation" International Journal of Molecular Sciences 23, no. 7: 3747. https://doi.org/10.3390/ijms23073747
APA StyleAramburu-Núñez, M., Custodia, A., Pérez-Mato, M., Iglesias-Rey, R., Campos, F., Castillo, J., Ouro, A., Romaus-Sanjurjo, D., & Sobrino, T. (2022). Stress Granules and Acute Ischemic Stroke: Beyond mRNA Translation. International Journal of Molecular Sciences, 23(7), 3747. https://doi.org/10.3390/ijms23073747