
Epigenetics of Cutaneous T-Cell Lymphomas

Abstract
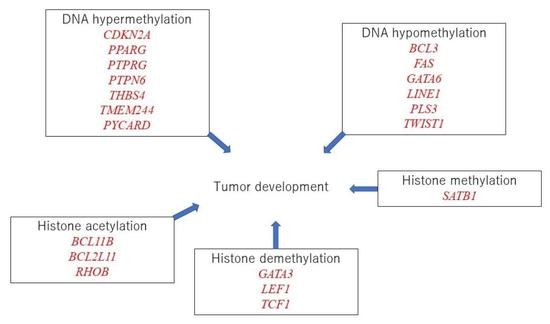
:
1. Introduction
1.1. DNA Methylation
1.2. Histone Methylation
1.3. Histone Acetylation
1.4. MicroRNA (miRNA)
2. Cutaneous T-Cell Lymphomas
3. Epigenetics Modification in CTCL
3.1. DNA Hypermethylation in Cyclin Dependent Kinase Inhibitor 2A (CDKN2A)
3.2. DNA Hypermethylation in PYD and CARD Domain Containing (PYCARD)
3.3. DNA Hypermethylation in Peroxisome Proliferator Activated Receptor Gamma (PPARG)
3.4. DNA Hypomethylation in Fas Cell Surface Death Receptor (FAS)
3.5. DNA Hypomethylation in Twist Family bHLH Transcription Factor 1 (TWIST1)
3.6. Histone Acetylation in BCL2 like 11 (BCL2L11)
3.7. The Enhancer of Zeste Homolog 2 (EZH2)-Mediated Histone Methylation
3.8. Histone Demethylation in GATA Binding Protein 3 (GATA3)
3.9. Histone Demethylation in Lymphoid Enhancer-Binding Factor 1 (LEF1)
3.10. miRNA in CTCL
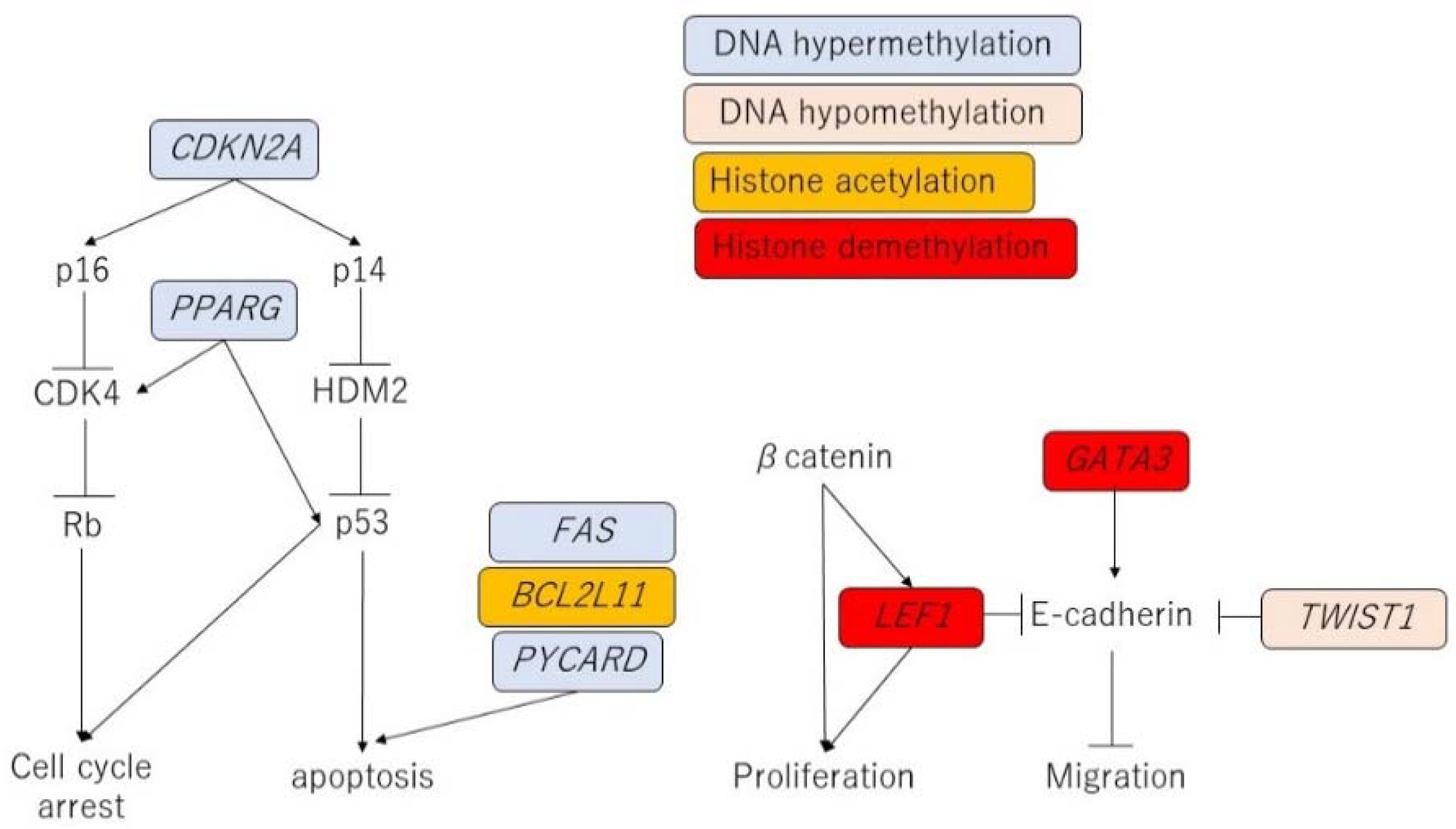
3.11. Possible Linkage between Epigenetic Modifications
3.12. The Therapeutic Target for Epigenetics in Cutaneous Lymphomas
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sawada, Y.; Gallo, R.L. Role of Epigenetics in the Regulation of Immune Functions of the Skin. J. Investig. Derm. 2021, 141, 1157–1166. [Google Scholar]
- Sawada, Y.; Nakatsuji, T.; Dokoshi, T.; Kulkarni, N.N.; Liggins, M.C.; Sen, G.; Gallo, R.L. Cutaneous innate immune tolerance is mediated by epigenetic control of MAP2K3 by HDAC8/9. Sci. Immunol. 2021, 6, eabe1935. [Google Scholar] [PubMed]
- Dor, Y.; Cedar, H. Principles of DNA methylation and their implications for biology and medicine. Lancet 2018, 392, 777–786. [Google Scholar] [PubMed]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [PubMed] [Green Version]
- Jeltsch, A. Beyond Watson and Crick: DNA methylation and molecular enzymology of DNA methyltransferases. ChemBioChem Eur. J. Chem. Biol. 2002, 3, 274–293. [Google Scholar]
- Frank, D.; Keshet, I.; Shani, M.; Levine, A.; Razin, A.; Cedar, H. Demethylation of CpG islands in embryonic cells. Nature 1991, 351, 239–241. [Google Scholar]
- Nanamori, H.; Sawada, Y. Epigenetic Modification of PD-1/PD-L1-Mediated Cancer Immunotherapy against Melanoma. Int. J. Mol. Sci. 2022, 23, 1119. [Google Scholar]
- Rice, J.C.; Allis, C.D. Histone methylation versus histone acetylation: New insights into epigenetic regulation. Curr. Opin. Cell Biol. 2001, 13, 263–273. [Google Scholar]
- Morgan, M.A.J.; Shilatifard, A. Reevaluating the roles of histone-modifying enzymes and their associated chromatin modifications in transcriptional regulation. Nat. Genet. 2020, 52, 1271–1281. [Google Scholar]
- Sun, W.; Lv, S.; Li, H.; Cui, W.; Wang, L. Enhancing the Anticancer Efficacy of Immunotherapy through Combination with Histone Modification Inhibitors. Genes 2018, 9, 633. [Google Scholar]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [PubMed] [Green Version]
- Lee, D.Y.; Hayes, J.J.; Pruss, D.; Wolffe, A.P. A positive role for histone acetylation in transcription factor access to nucleosomal DNA. Cell 1993, 72, 73–84. [Google Scholar] [PubMed]
- Gräff, J.; Tsai, L.H. Histone acetylation: Molecular mnemonics on the chromatin. Nat. Rev. Neurosci. 2013, 14, 97–111. [Google Scholar]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar]
- Yao, Q.; Chen, Y.; Zhou, X. The roles of microRNAs in epigenetic regulation. Curr. Opin. Chem. Biol. 2019, 51, 11–17. [Google Scholar]
- Epstein, E.H., Jr. Mycosis fungoides: Clinical course and cellular abnormalities. J. Investig. Derm. 1980, 75, 103–106. [Google Scholar]
- Hwang, S.T.; Janik, J.E.; Jaffe, E.S.; Wilson, W.H. Mycosis fungoides and Sézary syndrome. Lancet 2008, 371, 945–957. [Google Scholar]
- Sawada, Y.; Sugita, K.; Kabashima, R.; Hino, R.; Nakamura, M.; Koga, C.; Tokura, Y. CD8+ CD56+ mycosis fungoides with an indolent clinical behaviour: Case report and literature review. Acta Derm.-Venereol. 2010, 90, 525–526. [Google Scholar]
- Agar, N.S.; Wedgeworth, E.; Crichton, S.; Mitchell, T.J.; Cox, M.; Ferreira, S.; Robson, A.; Calonje, E.; Stefanato, C.M.; Wain, E.M.; et al. Survival outcomes and prognostic factors in mycosis fungoides/Sézary syndrome: Validation of the revised International Society for Cutaneous Lymphomas/European Organisation for Research and Treatment of Cancer staging proposal. J. Clin. Oncol. 2010, 28, 4730–4739. [Google Scholar]
- Jawed, S.I.; Myskowski, P.L.; Horwitz, S.; Moskowitz, A.; Querfeld, C. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): Part II. Prognosis, management, and future directions. J. Am. Acad. Derm. 2014, 70, 223.e1–223.e.17; quiz 240–242. [Google Scholar] [PubMed]
- Lopez, A.T.; Bates, S.; Geskin, L. Current Status of HDAC Inhibitors in Cutaneous T-cell Lymphoma. Am. J. Clin. Dermatol. 2018, 19, 805–819. [Google Scholar] [PubMed]
- Bigler, R.D.; Crilley, P.; Micaily, B.; Brady, L.W.; Topolsky, D.; Bulova, S.; Vonderheid, E.C.; Brodsky, I. Autologous bone marrow transplantation for advanced stage mycosis fungoides. Bone Marrow Transpl. 1991, 7, 133–137. [Google Scholar]
- Kempf, W.; Pfaltz, K.; Vermeer, M.H.; Cozzio, A.; Ortiz-Romero, P.L.; Bagot, M.; Olsen, E.; Kim, Y.H.; Dummer, R.; Pimpinelli, N.; et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: Lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood 2011, 118, 4024–4035. [Google Scholar]
- Benner, M.F.; Willemze, R. Applicability and prognostic value of the new TNM classification system in 135 patients with primary cutaneous anaplastic large cell lymphoma. Arch. Dermatol. 2009, 145, 1399–1404. [Google Scholar]
- Liggett, W.H., Jr.; Sidransky, D. Role of the p16 tumor suppressor gene in cancer. J. Clin. Oncol. 1998, 16, 1197–1206. [Google Scholar]
- van Doorn, R.; Zoutman, W.H.; Dijkman, R.; de Menezes, R.X.; Commandeur, S.; Mulder, A.A.; van der Velden, P.A.; Vermeer, M.H.; Willemze, R.; Yan, P.S.; et al. Epigenetic profiling of cutaneous T-cell lymphoma: Promoter hypermethylation of multiple tumor suppressor genes including BCL7a, PTPRG, and p73. J. Clin. Oncol. 2005, 23, 3886–3896. [Google Scholar]
- Navas, I.C.; Algara, P.; Mateo, M.; Martínez, P.; García, C.; Rodriguez, J.L.; Vanaclocha, F.; Barrientos, N.; Iglesias, L.; Sánchez, L.; et al. p16(INK4a) is selectively silenced in the tumoral progression of mycosis fungoides. Lab. Investig. 2002, 82, 123–132. [Google Scholar]
- Navas, I.C.; Ortiz-Romero, P.L.; Villuendas, R.; Martínez, P.; García, C.; Gómez, E.; Rodriguez, J.L.; García, D.; Vanaclocha, F.; Iglesias, L.; et al. p16(INK4a) gene alterations are frequent in lesions of mycosis fungoides. Am. J. Pathol. 2000, 156, 1565–1572. [Google Scholar]
- Otterson, G.A.; Khleif, S.N.; Chen, W.; Coxon, A.B.; Kaye, F.J. CDKN2 gene silencing in lung cancer by DNA hypermethylation and kinetics of p16INK4 protein induction by 5-aza 2′deoxycytidine. Oncogene 1995, 11, 1211–1216. [Google Scholar]
- Hassler, M.R.; Klisaroska, A.; Kollmann, K.; Steiner, I.; Bilban, M.; Schiefer, A.I.; Sexl, V.; Egger, G. Antineoplastic activity of the DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine in anaplastic large cell lymphoma. Biochimie 2012, 94, 2297–2307. [Google Scholar] [PubMed] [Green Version]
- Masumoto, J.; Taniguchi, S.; Ayukawa, K.; Sarvotham, H.; Kishino, T.; Niikawa, N.; Hidaka, E.; Katsuyama, T.; Higuchi, T.; Sagara, J. ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. J. Biol. Chem. 1999, 274, 33835–33838. [Google Scholar] [PubMed] [Green Version]
- Protti, M.P.; De Monte, L. Dual Role of Inflammasome Adaptor ASC in Cancer. Front. Cell Dev. Biol. 2020, 8, 40. [Google Scholar] [PubMed] [Green Version]
- Ricote, M.; Li, A.C.; Willson, T.M.; Kelly, C.J.; Glass, C.K. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 1998, 391, 79–82. [Google Scholar]
- Jiang, C.; Ting, A.T.; Seed, B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature 1998, 391, 82–86. [Google Scholar]
- Sarraf, P.; Mueller, E.; Jones, D.; King, F.J.; DeAngelo, D.J.; Partridge, J.B.; Holden, S.A.; Chen, L.B.; Singer, S.; Fletcher, C.; et al. Differentiation and reversal of malignant changes in colon cancer through PPARgamma. Nat. Med. 1998, 4, 1046–1052. [Google Scholar]
- Ferrara, G.; Pancione, M.; Votino, C.; Quaglino, P.; Tomasini, C.; Santucci, M.; Pimpinelli, N.; Cusano, F.; Sabatino, L.; Colantuoni, V. A specific DNA methylation profile correlates with a high risk of disease progression in stage I classical (Alibert-Bazin type) mycosis fungoides. Br. J. Derm. 2014, 170, 1266–1275. [Google Scholar]
- Wu, J.; Salva, K.A.; Wood, G.S. c-CBL E3 ubiquitin ligase is overexpressed in cutaneous T-cell lymphoma: Its inhibition promotes activation-induced cell death. J. Investig. Derm. 2015, 135, 861–868. [Google Scholar]
- Jones, C.L.; Wain, E.M.; Chu, C.C.; Tosi, I.; Foster, R.; McKenzie, R.C.; Whittaker, S.J.; Mitchell, T.J. Downregulation of Fas gene expression in Sézary syndrome is associated with promoter hypermethylation. J. Investig. Derm. 2010, 130, 1116–1125. [Google Scholar]
- Zhu, Q.Q.; Ma, C.; Wang, Q.; Song, Y.; Lv, T. The role of TWIST1 in epithelial-mesenchymal transition and cancers. Tumour Biol. 2016, 37, 185–197. [Google Scholar]
- Kang, Y.; Massagué, J. Epithelial-mesenchymal transitions: Twist in development and metastasis. Cell 2004, 118, 277–279. [Google Scholar] [PubMed] [Green Version]
- Wong, H.K.; Gibson, H.; Hake, T.; Geyer, S.; Frederickson, J.; Marcucci, G.; Caligiuri, M.A.; Porcu, P.; Mishra, A. Promoter-Specific Hypomethylation Is Associated with Overexpression of PLS3, GATA6, and TWIST1 in the Sezary Syndrome. J. Investig. Derm. 2015, 135, 2084–2092. [Google Scholar]
- Häyrinen, M.J.; Uotila, P.M.; Sahi, H.; Haapasaari, K.M.; Teppo, H.R.; Soini, Y.; Lapela, M.; Vasala, K.; Turpeenniemi-Hujanen, T.; Ranki, A.; et al. Twist and Zeb1 expression identify mycosis fungoides patients with low risk of disease progression. J. Eur. Acad. Derm. Venereol. 2020, 34, e95–e98. [Google Scholar]
- Piazza, R.; Magistroni, V.; Mogavero, A.; Andreoni, F.; Ambrogio, C.; Chiarle, R.; Mologni, L.; Bachmann, P.S.; Lock, R.B.; Collini, P.; et al. Epigenetic silencing of the proapoptotic gene BIM in anaplastic large cell lymphoma through an MeCP2/SIN3a deacetylating complex. Neoplasia 2013, 15, 511–522. [Google Scholar]
- Yi, S.; Sun, J.; Qiu, L.; Fu, W.; Wang, A.; Liu, X.; Yang, Y.; Kadin, M.E.; Tu, P.; Wang, Y. Dual Role of EZH2 in Cutaneous Anaplastic Large Cell Lymphoma: Promoting Tumor Cell Survival and Regulating Tumor Microenvironment. J. Investig. Derm. 2018, 138, 1126–1136. [Google Scholar]
- Tsubota, S.; Kishida, S.; Shimamura, T.; Ohira, M.; Yamashita, S.; Cao, D.; Kiyonari, S.; Ushijima, T.; Kadomatsu, K. PRC2-Mediated Transcriptomic Alterations at the Embryonic Stage Govern Tumorigenesis and Clinical Outcome in MYCN-Driven Neuroblastoma. Cancer Res. 2017, 77, 5259–5271. [Google Scholar]
- Wu, X.; Liu, D.; Tao, D.; Xiang, W.; Xiao, X.; Wang, M.; Wang, L.; Luo, G.; Li, Y.; Zeng, F.; et al. BRD4 Regulates EZH2 Transcription through Upregulation of C-MYC and Represents a Novel Therapeutic Target in Bladder Cancer. Mol. Cancer 2016, 15, 1029–1042. [Google Scholar]
- Weilemann, A.; Grau, M.; Erdmann, T.; Merkel, O.; Sobhiafshar, U.; Anagnostopoulos, I.; Hummel, M.; Siegert, A.; Hayford, C.; Madle, H.; et al. Essential role of IRF4 and MYC signaling for survival of anaplastic large cell lymphoma. Blood 2015, 125, 124–132. [Google Scholar]
- Miettinen, M.; McCue, P.A.; Sarlomo-Rikala, M.; Rys, J.; Czapiewski, P.; Wazny, K.; Langfort, R.; Waloszczyk, P.; Biernat, W.; Lasota, J.; et al. GATA3: A multispecific but potentially useful marker in surgical pathology: A systematic analysis of 2500 epithelial and nonepithelial tumors. Am. J. Surg. Pathol. 2014, 38, 13–22. [Google Scholar]
- Zhang, W.; Wang, Z.; Luo, Y.; Zhong, D.; Luo, Y.; Zhou, D. GATA3 expression correlates with poor prognosis and tumor-associated macrophage infiltration in peripheral T cell lymphoma. Oncotarget 2016, 7, 65284–65294. [Google Scholar]
- Joosten, M.; Seitz, V.; Zimmermann, K.; Sommerfeld, A.; Berg, E.; Lenze, D.; Leser, U.; Stein, H.; Hummel, M. Histone acetylation and DNA demethylation of T cells result in an anaplastic large cell lymphoma-like phenotype. Haematologica 2013, 98, 247–254. [Google Scholar] [PubMed] [Green Version]
- Litvinov, I.V.; Netchiporouk, E.; Cordeiro, B.; Doré, M.A.; Moreau, L.; Pehr, K.; Gilbert, M.; Zhou, Y.; Sasseville, D.; Kupper, T.S. The Use of Transcriptional Profiling to Improve Personalized Diagnosis and Management of Cutaneous T-cell Lymphoma (CTCL). Clin. Cancer Res. 2015, 21, 2820–2829. [Google Scholar]
- Santiago, L.; Daniels, G.; Wang, D.; Deng, F.M.; Lee, P. Wnt signaling pathway protein LEF1 in cancer, as a biomarker for prognosis and a target for treatment. Am. J. Cancer Res. 2017, 7, 1389–1406. [Google Scholar] [PubMed]
- Sandoval, J.; Díaz-Lagares, A.; Salgado, R.; Servitje, O.; Climent, F.; Ortiz-Romero, P.L.; Pérez-Ferriols, A.; Garcia-Muret, M.P.; Estrach, T.; Garcia, M.; et al. MicroRNA expression profiling and DNA methylation signature for deregulated microRNA in cutaneous T-cell lymphoma. J. Investig. Derm. 2015, 135, 1128–1137. [Google Scholar]
- Gallardo, F.; Sandoval, J.; Díaz-Lagares, A.; Garcia, R.; D’Altri, T.; González, J.; Alegre, V.; Servitje, O.; Crujeiras, A.B.; Stefánsson, Ó.-A.; et al. Notch1 Pathway Activation Results from the Epigenetic Abrogation of Notch-Related MicroRNAs in Mycosis Fungoides. J. Investig. Derm. 2015, 135, 3144–3152. [Google Scholar]
- Ransohoff, K.J.; Jaju, P.D.; Tang, J.Y.; Carbone, M.; Leachman, S.; Sarin, K.Y. Familial skin cancer syndromes: Increased melanoma risk. J. Am. Acad. Derm. 2016, 74, 423–434. [Google Scholar]
- Stravodimou, A.; Mazzoccoli, G.; Voutsadakis, I.A. Peroxisome proliferator-activated receptor gamma and regulations by the ubiquitin-proteasome system in pancreatic cancer. PPAR Res. 2012, 2012, 367450. [Google Scholar] [PubMed] [Green Version]
- Zand, H.; Rhimipour, A.; Bakhshayesh, M.; Shafiee, M.; Nour Mohammadi, I.; Salimi, S. Involvement of PPAR-gamma and p53 in DHA-induced apoptosis in Reh cells. Mol. Cell. Biochem. 2007, 304, 71–77. [Google Scholar]
- Brlek, P.; Bukovac, A.; Kafka, A.; Pećina-Šlaus, N. TWIST1 upregulation affects E-cadherin expression in brain metastases. Clin. Transl. Oncol. 2021, 23, 1085–1095. [Google Scholar]
- Yan, W.; Cao, Q.J.; Arenas, R.B.; Bentley, B.; Shao, R. GATA3 inhibits breast cancer metastasis through the reversal of epithelial-mesenchymal transition. J. Biol. Chem. 2010, 285, 14042–14051. [Google Scholar]
- Duvic, M.; Vu, J. Update on the treatment of cutaneous T-cell lymphoma (CTCL): Focus on vorinostat. Biologics 2007, 1, 377–392. [Google Scholar] [PubMed]
- Duvic, M.; Talpur, R.; Ni, X.; Zhang, C.; Hazarika, P.; Kelly, C.; Chiao, J.H.; Reilly, J.F.; Ricker, J.L.; Richon, V.M.; et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 2007, 109, 31–39. [Google Scholar] [PubMed]
- Piekarz, R.L.; Frye, R.; Turner, M.; Wright, J.J.; Allen, S.L.; Kirschbaum, M.H.; Zain, J.; Prince, H.M.; Leonard, J.P.; Geskin, L.J.; et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J. Clin. Oncol. 2009, 27, 5410–5417. [Google Scholar] [PubMed] [Green Version]
- Foss, F.; Advani, R.; Duvic, M.; Hymes, K.B.; Intragumtornchai, T.; Lekhakula, A.; Shpilberg, O.; Lerner, A.; Belt, R.J.; Jacobsen, E.D.; et al. A Phase II trial of Belinostat (PXD101) in patients with relapsed or refractory peripheral or cutaneous T-cell lymphoma. Br. J. Haematol. 2015, 168, 811–819. [Google Scholar] [PubMed]
- Chebly, A.; Prochazkova-Carlotti, M.; Idrissi, Y.; Bresson-Bepoldin, L.; Poglio, S.; Farra, C.; Beylot-Barry, M.; Merlio, J.P.; Tomb, R.; Chevret, E. Targeting Epigenetic Modifiers Can Reduce the Clonogenic Capacities of Sézary Cells. Front. Oncol. 2021, 11, 775253. [Google Scholar]
- Chebly, A.; Ropio, J.; Peloponese, J.M.; Poglio, S.; Prochazkova-Carlotti, M.; Cherrier, F.; Ferrer, J.; Idrissi, Y.; Segal-Bendirdjian, E.; Chouery, E.; et al. Exploring hTERT promoter methylation in cutaneous T-cell lymphomas. Mol. Oncol. 2021. [Google Scholar] [CrossRef]
- Chevret, E.; Andrique, L.; Prochazkova-Carlotti, M.; Ferrer, J.; Cappellen, D.; Laharanne, E.; Idrissi, Y.; Boettiger, A.; Sahraoui, W.; Ruiz, F.; et al. Telomerase functions beyond telomere maintenance in primary cutaneous T-cell lymphoma. Blood 2014, 123, 1850–1859. [Google Scholar]
- Ragheb, R.; Venton, G.; Chelbi, R.; Bonnet, N.; Le Treut, T.; Ivanov, V.; Mercier, C.; Poulin, P.; Beaufils, N.; Gabert, J.; et al. Vorinostat and Mithramycin A in combination therapy as an interesting strategy for the treatment of Sézary T lymphoma: A transcriptomic approach. Arch. Derm. Res. 2017, 309, 611–623. [Google Scholar]
- Chaidos, A.; Caputo, V.; Karadimitris, A. Inhibition of bromodomain and extra-terminal proteins (BET) as a potential therapeutic approach in haematological malignancies: Emerging preclinical and clinical evidence. Ther. Adv. Hematol. 2015, 6, 128–141. [Google Scholar]
- Zhao, L.; Okhovat, J.P.; Hong, E.K.; Kim, Y.H.; Wood, G.S. Preclinical Studies Support Combined Inhibition of BET Family Proteins and Histone Deacetylases as Epigenetic Therapy for Cutaneous T-Cell Lymphoma. Neoplasia 2019, 21, 82–92. [Google Scholar]
- Dueñas-Gonzalez, A.; Vega, M.T.; Martinez-Baños, D.; García-Hidalgo, L.; Sobrevilla, P. Response to hydralazine-valproate in a patient with mycosis fungoides. Case Rep. Med. 2010, 2010, 657579. [Google Scholar] [PubMed] [Green Version]
- Wang, L.; Chang, J.; Varghese, D.; Dellinger, M.; Kumar, S.; Best, A.M.; Ruiz, J.; Bruick, R.; Peña-Llopis, S.; Xu, J.; et al. A small molecule modulates Jumonji histone demethylase activity and selectively inhibits cancer growth. Nat. Commun. 2013, 4, 2035. [Google Scholar] [PubMed]
- Van Rechem, C.; Black, J.C.; Boukhali, M.; Aryee, M.J.; Gräslund, S.; Haas, W.; Benes, C.H.; Whetstine, J.R. Lysine demethylase KDM4A associates with translation machinery and regulates protein synthesis. Cancer Discov. 2015, 5, 255–263. [Google Scholar] [PubMed] [Green Version]
- Olsen, E.A.; Kim, Y.H.; Kuzel, T.M.; Pacheco, T.R.; Foss, F.M.; Parker, S.; Frankel, S.R.; Chen, C.; Ricker, J.L.; Arduino, J.M.; et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J. Clin. Oncol. 2007, 25, 3109–3115. [Google Scholar]
- Lesokhin, A.M.; Ansell, S.M.; Armand, P.; Scott, E.C.; Halwani, A.; Gutierrez, M.; Millenson, M.M.; Cohen, A.D.; Schuster, S.J.; Lebovic, D.; et al. Nivolumab in Patients With Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. J. Clin. Oncol. 2016, 34, 2698–2704. [Google Scholar]
- Terranova-Barberio, M.; Pawlowska, N.; Dhawan, M.; Moasser, M.; Chien, A.J.; Melisko, M.E.; Rugo, H.; Rahimi, R.; Deal, T.; Daud, A.; et al. Exhausted T cell signature predicts immunotherapy response in ER-positive breast cancer. Nat. Commun. 2020, 11, 3584. [Google Scholar]
- Topper, M.J.; Vaz, M.; Chiappinelli, K.B.; DeStefano Shields, C.E.; Niknafs, N.; Yen, R.C.; Wenzel, A.; Hicks, J.; Ballew, M.; Stone, M.; et al. Epigenetic Therapy Ties MYC Depletion to Reversing Immune Evasion and Treating Lung Cancer. Cell 2017, 171, 1284–1300.e21. [Google Scholar]


{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hara, N.; Sawada, Y. Epigenetics of Cutaneous T-Cell Lymphomas. Int. J. Mol. Sci. 2022, 23, 3538. https://doi.org/10.3390/ijms23073538
Hara N, Sawada Y. Epigenetics of Cutaneous T-Cell Lymphomas. International Journal of Molecular Sciences. 2022; 23(7):3538. https://doi.org/10.3390/ijms23073538
Chicago/Turabian StyleHara, Natsumi, and Yu Sawada. 2022. "Epigenetics of Cutaneous T-Cell Lymphomas" International Journal of Molecular Sciences 23, no. 7: 3538. https://doi.org/10.3390/ijms23073538
APA StyleHara, N., & Sawada, Y. (2022). Epigenetics of Cutaneous T-Cell Lymphomas. International Journal of Molecular Sciences, 23(7), 3538. https://doi.org/10.3390/ijms23073538