
A Microfluidic Platform to Monitor Real-Time Effects of Extracellular Vesicle Exchange between Co-Cultured Cells across Selectively Permeable Barriers

 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
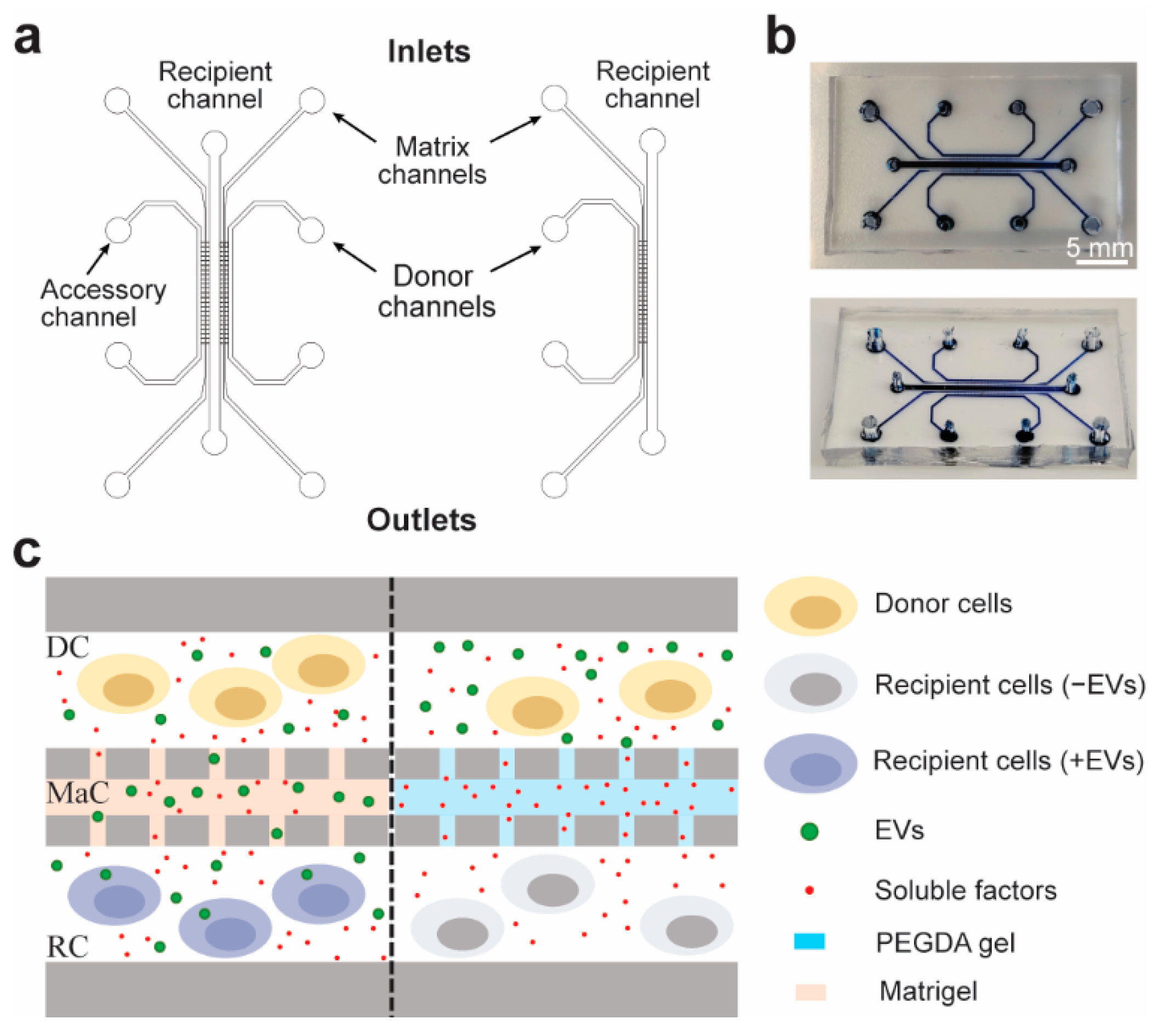
2.1. Microfluidic Design
2.2. Production and Optimization of Microfluidic Chips
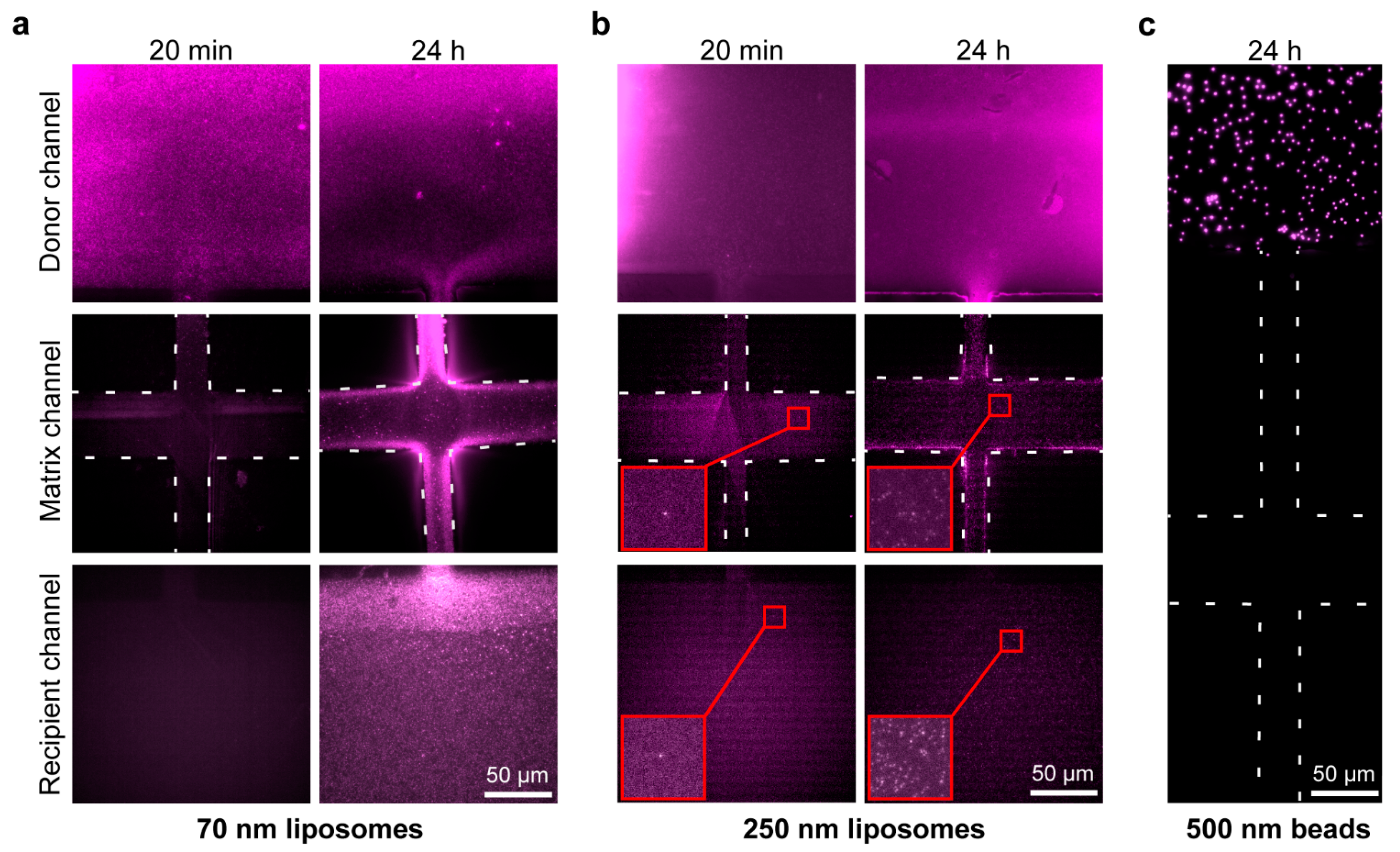
2.3. Matrigel Diffusion Characterization
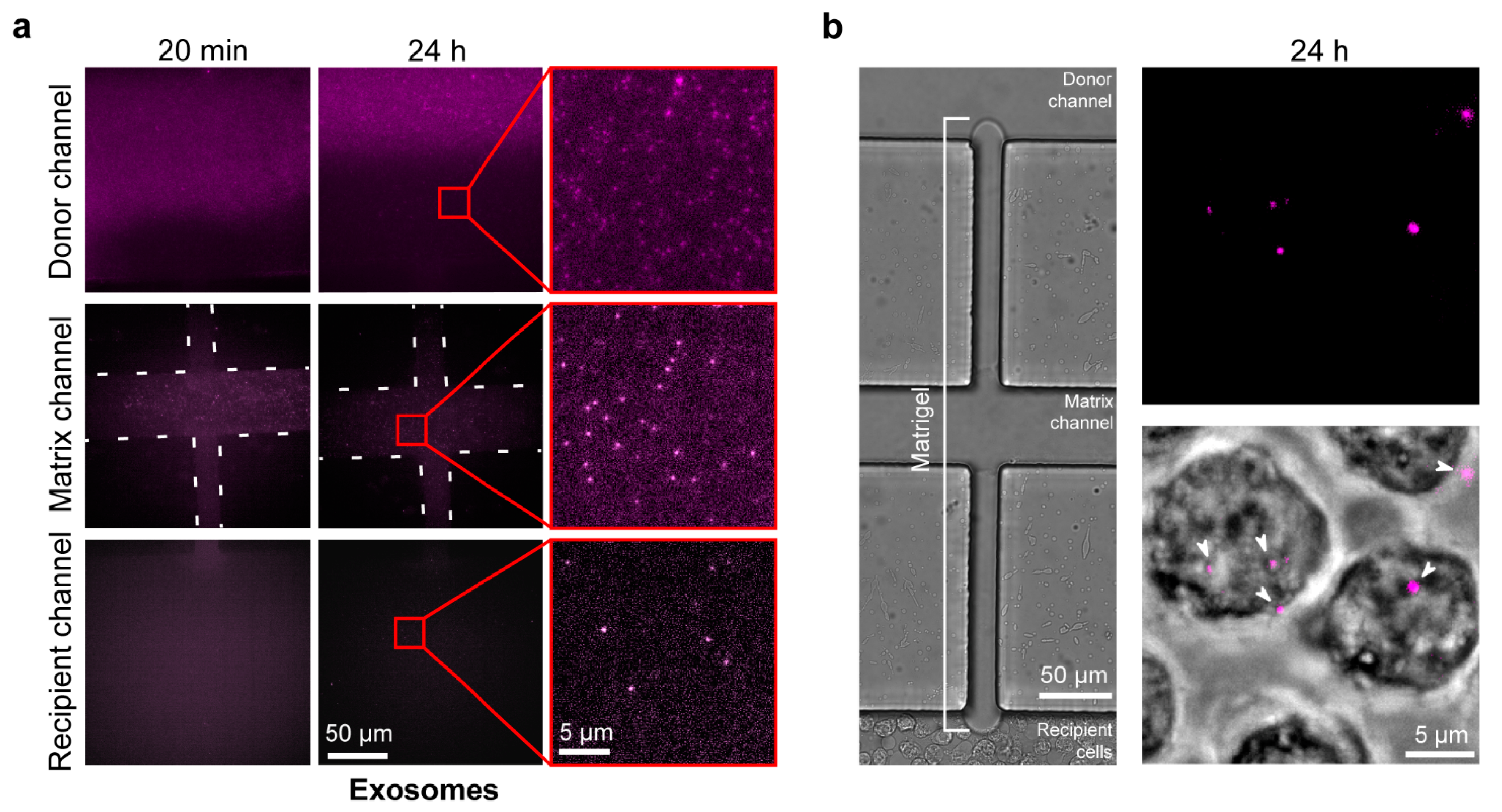
2.4. Exosome Diffusion in the Microfluidic Chip
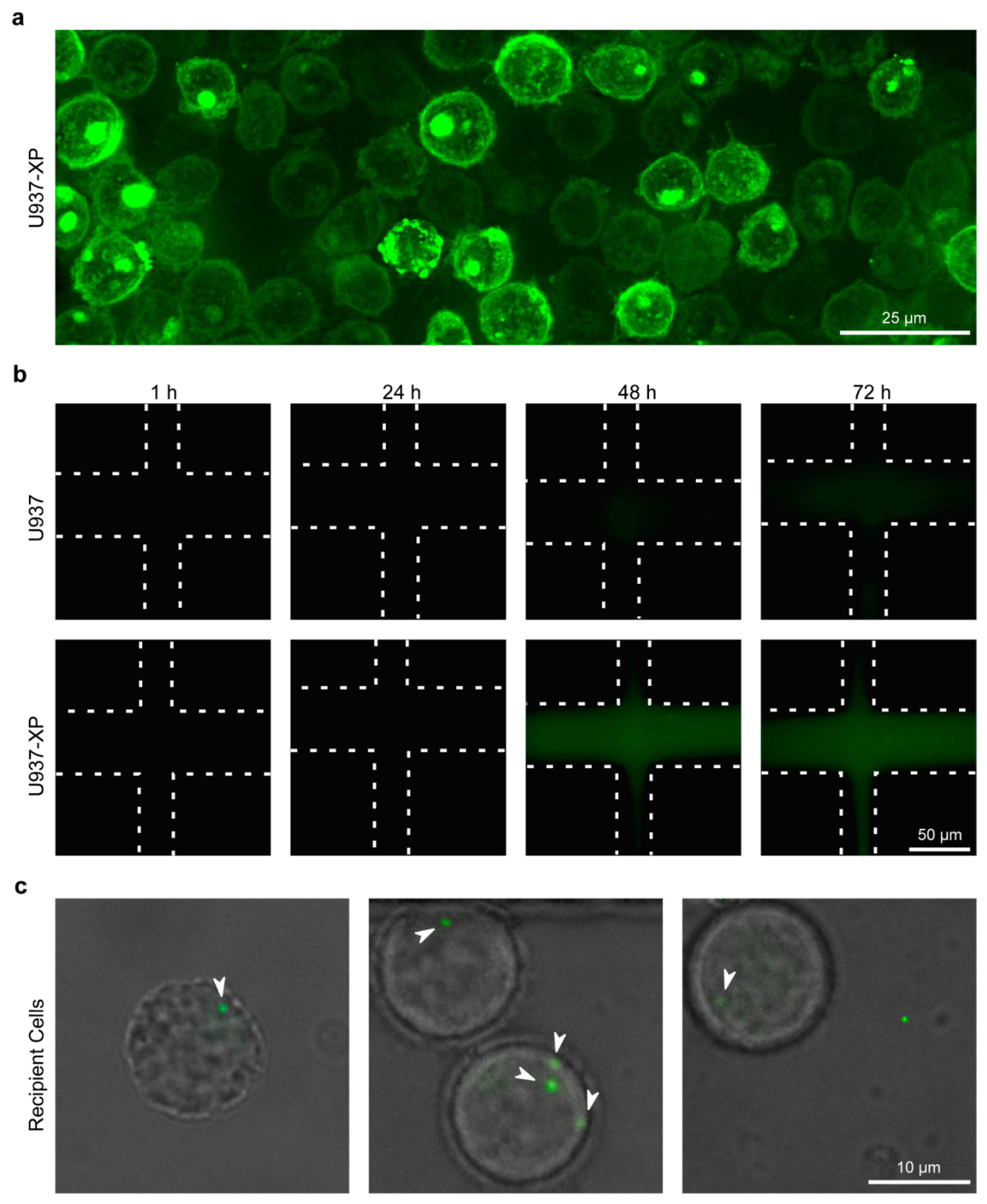
2.5. Fluorescent EV-Producing Stable Cell Line Generation
2.6. Live EV Secretion, Diffusion, and Uptake
3. Discussion
4. Materials and Methods
4.1. Chip Design
4.2. Chip Production/Optimization
4.3. Liposome Preparation
4.4. Cell Line and Maintenance
4.5. Puromycin Kill Curve
4.6. U937-XP Cell Line Generation
4.7. Extracellular Vesicle (EV) Purification
4.8. EV Fluorescence Measurement and Analysis of the EV Pellets
4.9. Vesicle and Cell Injections into Chips
4.10. Fluorescent Imaging
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- EL Andaloussi, S.; Mäger, I.; Breakefield, X.O.; Wood, M.J.A. Extracellular Vesicles: Biology and Emerging Therapeutic Opportunities. Nat. Rev. Drug Discov. 2013, 12, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Chuo, S.T.-Y.; Chien, J.C.-Y.; Lai, C.P.-K. Imaging Extracellular Vesicles: Current and Emerging Methods. J. Biomed. Sci. 2018, 25, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olanrewaju, A.A.; Hakami, R.M. The Messenger Apps of the Cell: Extracellular Vesicles as Regulatory Messengers of Microglial Function in the CNS. J. Neuroimmune Pharmacol. 2020, 15, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Alem, F.; Olanrewaju, A.A.; Omole, S.; Hobbs, H.E.; Ahsan, N.; Matulis, G.; Brantner, C.A.; Zhou, W.; Petricoin, E.F.; Liotta, L.A.; et al. Exosomes Originating from Infection with the Cytoplasmic Single-Stranded RNA Virus Rift Valley Fever Virus (RVFV) Protect Recipient Cells by Inducing RIG-I Mediated IFN-B Response That Leads to Activation of Autophagy. Cell Biosci. 2021, 11, 220. [Google Scholar] [CrossRef]
- Pegtel, D.M.; Gould, S.J. Exosomes. Annu. Rev. Biochem. 2019, 88, 487–514. [Google Scholar] [CrossRef]
- Doyle, L.M.; Wang, M.Z. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef] [Green Version]
- Pegtel, D.M.; Cosmopoulos, K.; Thorley-Lawson, D.A.; van Eijndhoven, M.A.J.; Hopmans, E.S.; Lindenberg, J.L.; de Gruijl, T.D.; Würdinger, T.; Middeldorp, J.M. Functional Delivery of Viral MiRNAs via Exosomes. Proc. Natl. Acad. Sci. USA 2010, 107, 6328–6333. [Google Scholar] [CrossRef] [Green Version]
- Prada, I.; Meldolesi, J. Binding and Fusion of Extracellular Vesicles to the Plasma Membrane of Their Cell Targets. Int. J. Mol. Sci. 2016, 17, 1296. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Yu, D. Exosomes in Cancer Development, Metastasis, and Immunity. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 455–468. [Google Scholar] [CrossRef]
- Malm, T.; Loppi, S.; Kanninen, K.M. Exosomes in Alzheimer’s Disease. Neurochem. Int. 2016, 97, 193–199. [Google Scholar] [CrossRef]
- Wu, X.; Zheng, T.; Zhang, B. Exosomes in Parkinson’s Disease. Neurosci. Bull. 2017, 33, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Gao, W.; Yao, K.; Ge, J. Roles of Exosomes Derived From Immune Cells in Cardiovascular Diseases. Front. Immunol. 2019, 10, 648. [Google Scholar] [CrossRef] [Green Version]
- Fleming, A.; Sampey, G.; Chung, M.-C.; Bailey, C.; van Hoek, M.L.; Kashanchi, F.; Hakami, R.M. The Carrying Pigeons of the Cell: Exosomes and Their Role in Infectious Diseases Caused by Human Pathogens. Pathog. Dis. 2014, 71, 109–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schorey, J.S.; Cheng, Y.; Singh, P.P.; Smith, V.L. Exosomes and Other Extracellular Vesicles in Host-Pathogen Interactions. EMBO Rep. 2015, 16, 24–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwab, A.; Meyering, S.S.; Lepene, B.; Iordanskiy, S.; van Hoek, M.L.; Hakami, R.M.; Kashanchi, F. Extracellular Vesicles from Infected Cells: Potential for Direct Pathogenesis. Front. Microbiol. 2015, 6, 1132. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Li, J.; Huang, B.; Liu, J.; Chen, X.; Chen, X.-M.; Xu, Y.-M.; Huang, L.-F.; Wang, X.-Z. Exosomes: Novel Biomarkers for Clinical Diagnosis. Sci. World J. 2015, 2015, 657086. [Google Scholar] [CrossRef]
- Yang, D.; Zhang, W.; Zhang, H.; Zhang, F.; Chen, L.; Ma, L.; Larcher, L.M.; Chen, S.; Liu, N.; Zhao, Q.; et al. Progress, Opportunity, and Perspective on Exosome Isolation—Efforts for Efficient Exosome-Based Theranostics. Theranostics 2020, 10, 3684–3707. [Google Scholar] [CrossRef]
- Gurunathan, S.; Kang, M.-H.; Jeyaraj, M.; Qasim, M.; Kim, J.-H. Review of the Isolation, Characterization, Biological Function, and Multifarious Therapeutic Approaches of Exosomes. Cells 2019, 8, 307. [Google Scholar] [CrossRef] [Green Version]
- Betzer, O.; Barnoy, E.; Sadan, T.; Elbaz, I.; Braverman, C.; Liu, Z.; Popovtzer, R. Advances in Imaging Strategies for in Vivo Tracking of Exosomes. WIREs Nanomed. Nanobiotechnol. 2020, 12, e1594. [Google Scholar] [CrossRef]
- Salunkhe, S.; Dheeraj; Basak, M.; Chitkara, D.; Mittal, A. Surface Functionalization of Exosomes for Target-Specific Delivery and in Vivo Imaging & Tracking: Strategies and Significance. J. Control. Release 2020, 326, 599–614. [Google Scholar] [CrossRef]
- Luoto, J.C.; Coelho-Rato, L.S.; Bengs, S.H.; Roininen, J.; Eriksson, J.E.; Sistonen, L.; Henriksson, E. In Vivo-Mimicking 3D Cultures Secrete Distinct Extracellular Vesicles upon Cancer Cell Invasion. bioRxiv 2020. [Google Scholar] [CrossRef]
- Zhu, Q.; Heon, M.; Zhao, Z.; He, M. Microfluidic Engineering of Exosomes: Editing Cellular Messages for Precision Therapeutics. Lab Chip 2018, 18, 1690–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, W.; Jeong, D.; Kim, J.; Cho, S.; Jang, S.C.; Han, C.; Kang, J.Y.; Gho, Y.S.; Park, J. Microfluidic Fabrication of Cell-Derived Nanovesicles as Endogenous RNA Carriers. Lab Chip 2014, 14, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.; Han, S.; Jeon, J.S.; Yamamoto, K.; Zervantonakis, I.K.; Sudo, R.; Kamm, R.D.; Chung, S. Microfluidic Assay for Simultaneous Culture of Multiple Cell Types on Surfaces or within Hydrogels. Nat. Protoc. 2012, 7, 1247–1259. [Google Scholar] [CrossRef] [Green Version]
- Roberts, S.A.; Waziri, A.E.; Agrawal, N. Development of a Single-Cell Migration and Extravasation Platform through Selective Surface Modification. Anal. Chem. 2016, 88, 2770–2776. [Google Scholar] [CrossRef]
- Li, R.; Hebert, J.D.; Lee, T.A.; Xing, H.; Boussommier-Calleja, A.; Hynes, R.O.; Lauffenburger, D.A.; Kamm, R.D. Macrophage-Secreted TNFα and TGFβ1 Influence Migration Speed and Persistence of Cancer Cells in 3D Tissue Culture via Independent Pathways. Cancer Res. 2017, 77, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Kojima, R.; Bojar, D.; Rizzi, G.; Hamri, G.C.-E.; El-Baba, M.D.; Saxena, P.; Ausländer, S.; Tan, K.R.; Fussenegger, M. Designer Exosomes Produced by Implanted Cells Intracerebrally Deliver Therapeutic Cargo for Parkinson’s Disease Treatment. Nat. Commun. 2018, 9, 1305. [Google Scholar] [CrossRef] [Green Version]
- Tomasetti, L.; Liebl, R.; Wastl, D.S.; Breunig, M. Influence of PEGylation on Nanoparticle Mobility in Different Models of the Extracellular Matrix. Eur. J. Pharm. Biopharm. 2016, 108, 145–155. [Google Scholar] [CrossRef]
- Ishraq Bari, S.M.; Hossain, F.B.; Nestorova, G.G. Advances in Biosensors Technology for Detection and Characterization of Extracellular Vesicles. Sensors 2021, 21, 7645. [Google Scholar] [CrossRef]
- Sidhom, K.; Obi, P.O.; Saleem, A. A Review of Exosomal Isolation Methods: Is Size Exclusion Chromatography the Best Option? Int. J. Mol. Sci. 2020, 21, 6466. [Google Scholar] [CrossRef]
- Louten, J. Virus Structure and Classification. Essent. Hum. Virol. 2016, 19–29. [Google Scholar] [CrossRef]
- Gavira, J.A.; Cera-Manjarres, A.; Ortiz, K.; Mendez, J.; Jimenez-Torres, J.A.; Patiño-Lopez, L.D.; Torres-Lugo, M. Use of Cross-Linked Poly(Ethylene Glycol)-Based Hydrogels for Protein Crystallization. Cryst. Growth Des. 2014, 14, 3239–3248. [Google Scholar] [CrossRef] [PubMed]
- Yim, N.; Choi, C. Extracellular Vesicles as Novel Carriers for Therapeutic Molecules. BMB Rep. 2016, 49, 585. [Google Scholar] [CrossRef] [Green Version]
- Shen, B.; Wu, N.; Yang, J.-M.; Gould, S.J. Protein Targeting to Exosomes/Microvesicles by Plasma Membrane Anchors. J. Biol. Chem. 2011, 286, 14383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panagopoulou, M.S.; Wark, A.W.; Birch, D.J.S.; Gregory, C.D. Phenotypic Analysis of Extracellular Vesicles: A Review on the Applications of Fluorescence. J. Extracell. Vesicles 2020, 9, 1710020. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, T.L.; Russell, A.J.; Riley, P. Experimental Limitations of Extracellular Vesicle-Based Therapies for the Treatment of Myocardial Infarction. Trends Cardiovasc. Med. 2021, 31, 405–415. [Google Scholar] [CrossRef]
- Li, P.; Lu, X.; Hu, J.; Dai, M.; Yan, J.; Tan, H.; Yu, P.; Chen, X.; Zhang, C. Human Amniotic Fluid Derived-Exosomes Alleviate Hypoxic Encephalopathy by Enhancing Angiogenesis in Neonatal Mice after Hypoxia. Neurosci. Lett. 2022, 768, 136361. [Google Scholar] [CrossRef]
- Wang, Y.; Li, P.; Mao, S.; Mo, Z.; Cao, Z.; Luo, J.; Zhou, M.; Liu, X.; Zhang, S.; Yu, L. Exosome CTLA-4 Regulates PTEN/CD44 Signal Pathway in Spleen Deficiency Internal Environment to Promote Invasion and Metastasis of Hepatocellular Carcinoma. Front. Pharmacol. 2021, 12, 757194. [Google Scholar] [CrossRef]
- Zhang, Y.; Tan, X.; Lu, Y. Exosomal Transfer of Circ_0006174 Contributes to the Chemoresistance of Doxorubicin in Colorectal Cancer by Depending on the MiR-1205/CCND2 Axis. J. Physiol. Biochem. 2022, 78, 39–50. [Google Scholar] [CrossRef]
- Hu, N.; Zeng, X.; Tang, F.; Xiong, S. Exosomal Long Non-Coding RNA LIPCAR Derived from OxLDL-Treated THP-1 Cells Regulates the Proliferation of Human Umbilical Vein Endothelial Cells and Human Vascular Smooth Muscle Cells. Biochem. Biophys. Res. Commun. 2021, 575, 65–72. [Google Scholar] [CrossRef]
- Wang, L.; Chen, J.; Lu, C. Circular RNA Foxo3 Enhances Progression of Ovarian Carcinoma Cells. Aging 2021, 13, 22432–22443. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Wang, M.; Li, Q. Exosomal MiR-4488 and MiR-1273g-5p Inhibit the Epithelial-Mesenchymal Transition of Transforming Growth Factor Β2-Mediated Retinal Pigment Epithelial Cells by Targeting ATP-Binding Cassette A4. Bioengineered 2021, 12, 9693–9706. [Google Scholar] [CrossRef] [PubMed]
- Fang, G.; Lu, H.; Aboulkheyr Es, H.; Wang, D.; Liu, Y.; Warkiani, M.E.; Lin, G.; Jin, D. Unidirectional Intercellular Communication on a Microfluidic Chip. Biosens. Bioelectron. 2021, 175, 112833. [Google Scholar] [CrossRef]
- Boos, J.A.; Misun, P.M.; Brunoldi, G.; Furer, L.A.; Aengenheister, L.; Modena, M.; Rousset, N.; Buerki-Thurnherr, T.; Hierlemann, A. Microfluidic Co-Culture Platform to Recapitulate the Maternal–Placental–Embryonic Axis. Adv. Biol. 2021, 5, 2100609. [Google Scholar] [CrossRef]
- Shimasaki, T.; Yamamoto, S.; Omura, R.; Ito, K.; Nishide, Y.; Yamada, H.; Ohtomo, K.; Ishisaka, T.; Okano, K.; Ogawa, T.; et al. Novel Platform for Regulation of Extracellular Vesicles and Metabolites Secretion from Cells Using a Multi-Linkable Horizontal Co-Culture Plate. Micromachines 2021, 12, 1431. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mason, H.G.; Bush, J.; Agrawal, N.; Hakami, R.M.; Veneziano, R. A Microfluidic Platform to Monitor Real-Time Effects of Extracellular Vesicle Exchange between Co-Cultured Cells across Selectively Permeable Barriers. Int. J. Mol. Sci. 2022, 23, 3534. https://doi.org/10.3390/ijms23073534
Mason HG, Bush J, Agrawal N, Hakami RM, Veneziano R. A Microfluidic Platform to Monitor Real-Time Effects of Extracellular Vesicle Exchange between Co-Cultured Cells across Selectively Permeable Barriers. International Journal of Molecular Sciences. 2022; 23(7):3534. https://doi.org/10.3390/ijms23073534
Chicago/Turabian StyleMason, Hunter G., Joshua Bush, Nitin Agrawal, Ramin M. Hakami, and Remi Veneziano. 2022. "A Microfluidic Platform to Monitor Real-Time Effects of Extracellular Vesicle Exchange between Co-Cultured Cells across Selectively Permeable Barriers" International Journal of Molecular Sciences 23, no. 7: 3534. https://doi.org/10.3390/ijms23073534
APA StyleMason, H. G., Bush, J., Agrawal, N., Hakami, R. M., & Veneziano, R. (2022). A Microfluidic Platform to Monitor Real-Time Effects of Extracellular Vesicle Exchange between Co-Cultured Cells across Selectively Permeable Barriers. International Journal of Molecular Sciences, 23(7), 3534. https://doi.org/10.3390/ijms23073534