
Natural Compounds as Promising Adjuvant Agents in The Treatment of Gliomas




Abstract
1. Introduction
1.1. Glioma
1.2. Glioblastoma
2. Natural Compounds in the Therapy of Glioma
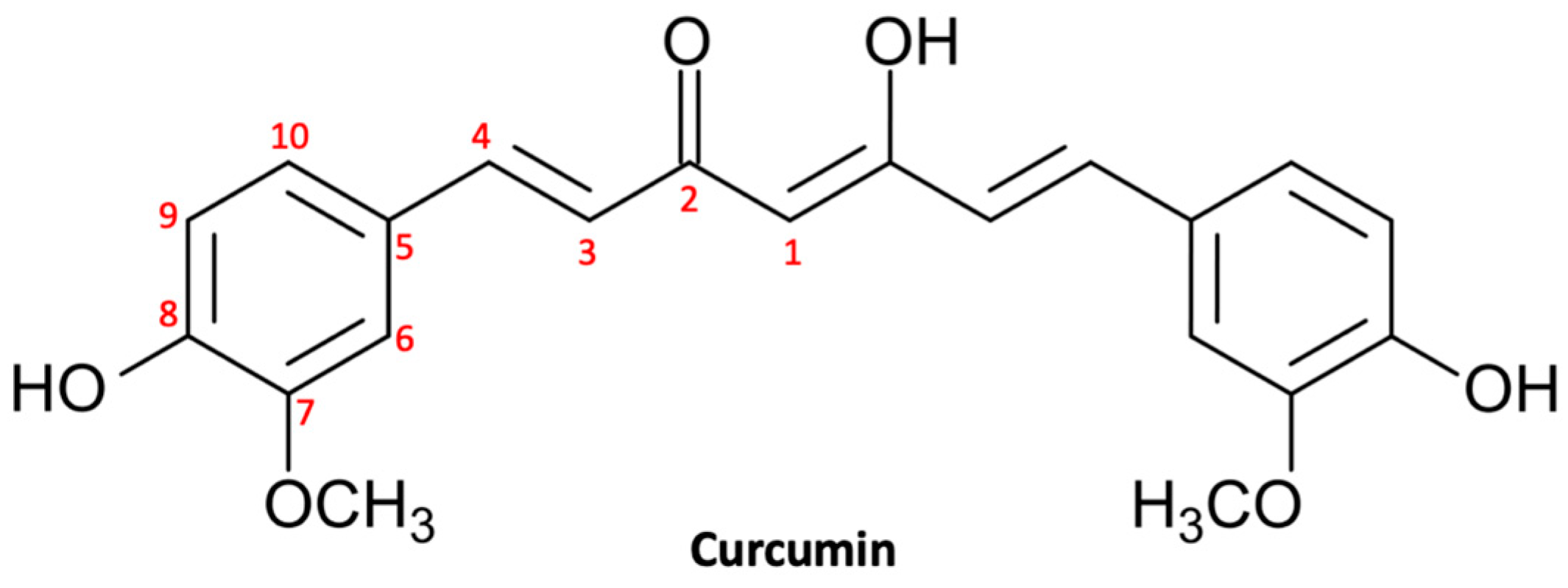
2.1. Curcuminoids
2.1.1. Curcumin
Curcumin Pharmacokinetics
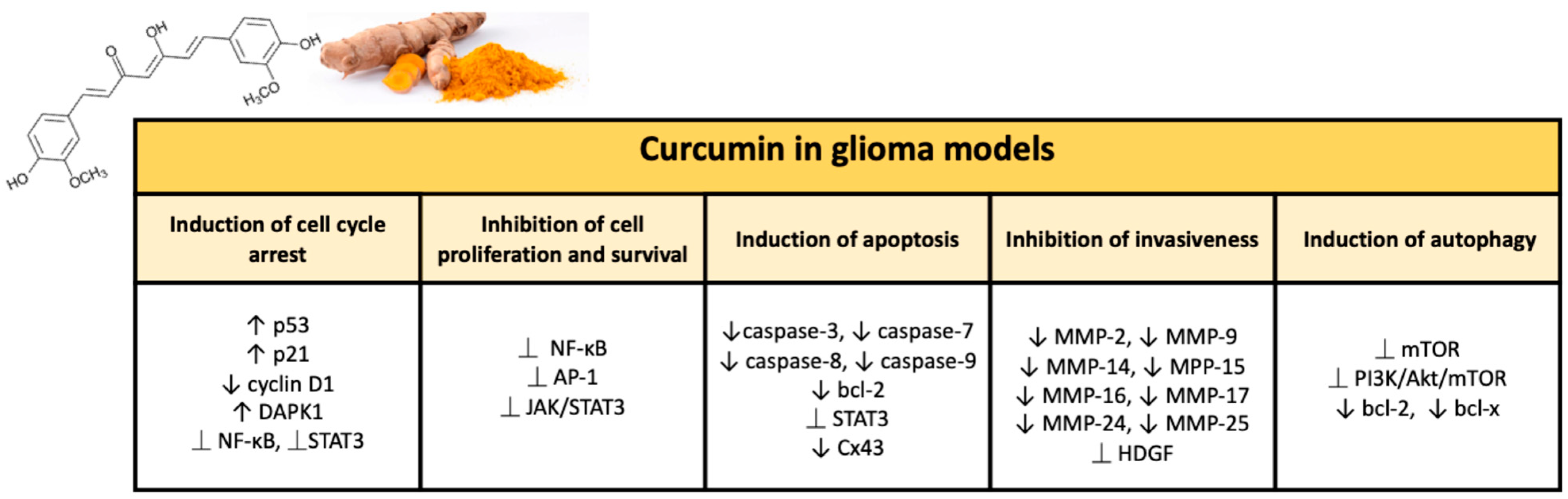
Antitumor Properties of Curcumin in Glioma
Inhibition of Cell Proliferation and Survival
Induction of Cell Cycle Arrest
Induction of Autophagy
Promotion of Apoptosis
Inhibition of Invasiveness
2.2. Flavonoids
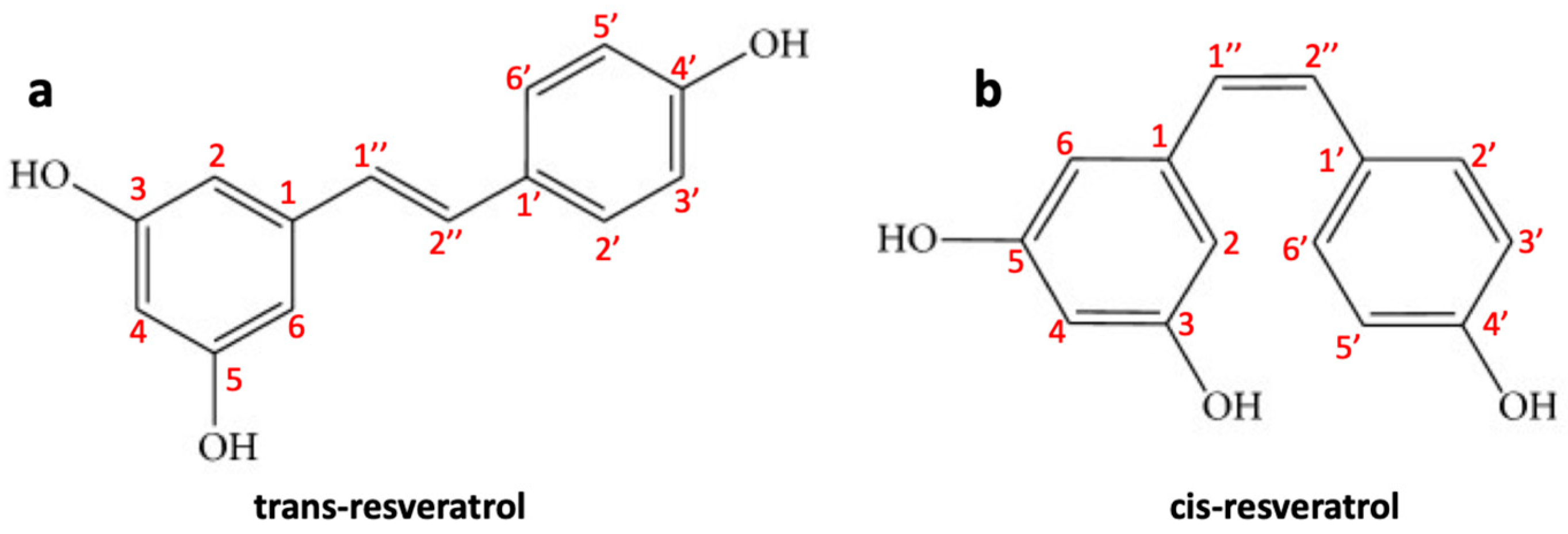
2.2.1. Resveratrol
Pharmacokinetics RES
Antitumor Properties of RES in Glioma
Cell Cycle Regulation and Carcinogenesis
Inhibition of Angiogenesis and Tumor Growth
Promotion of Apoptosis
RES and Cellular Senescence
Sensitization to Anticancer Drugs (Such As Temozolomide)
Radiosensitization
RES and Resistance Proteins
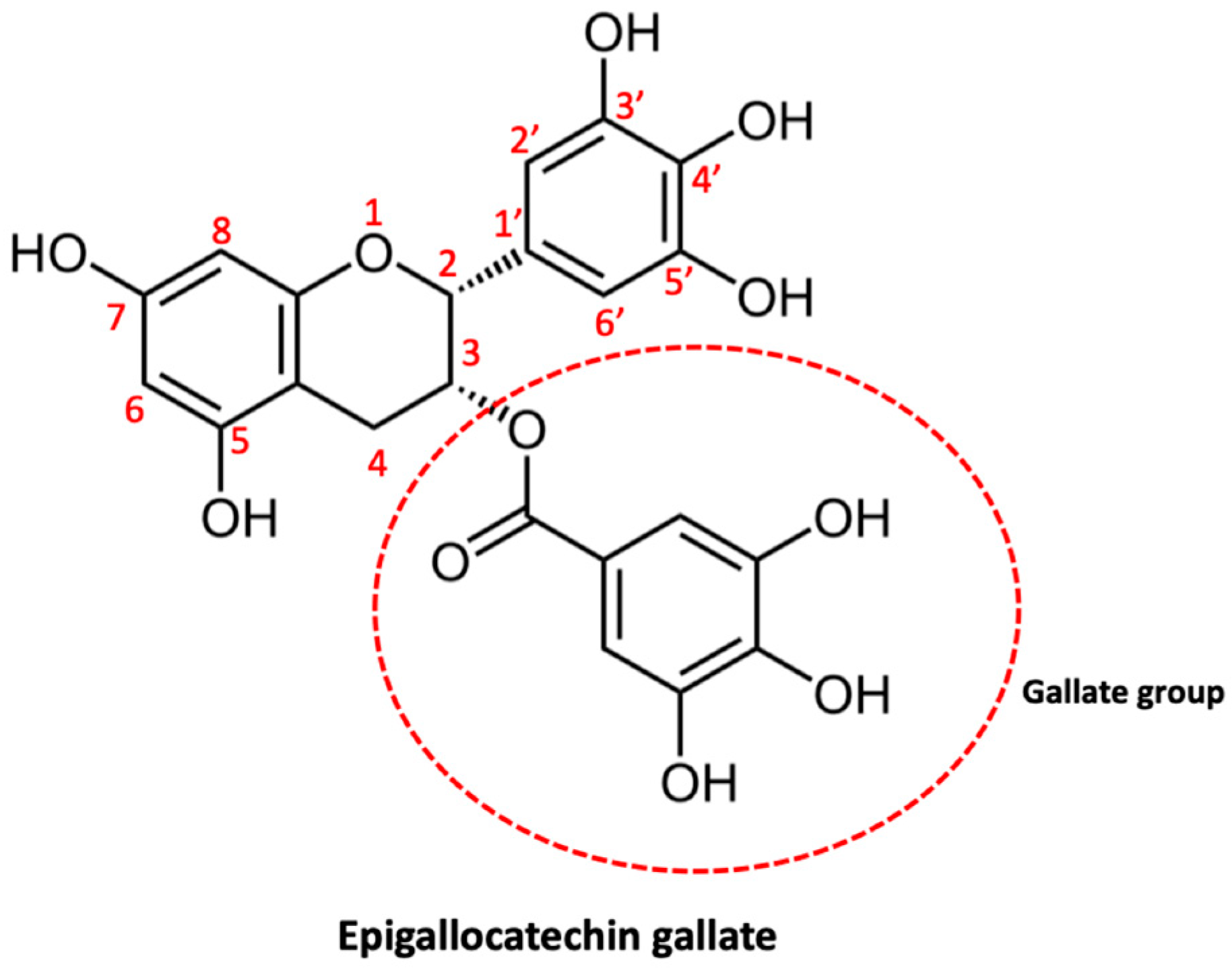
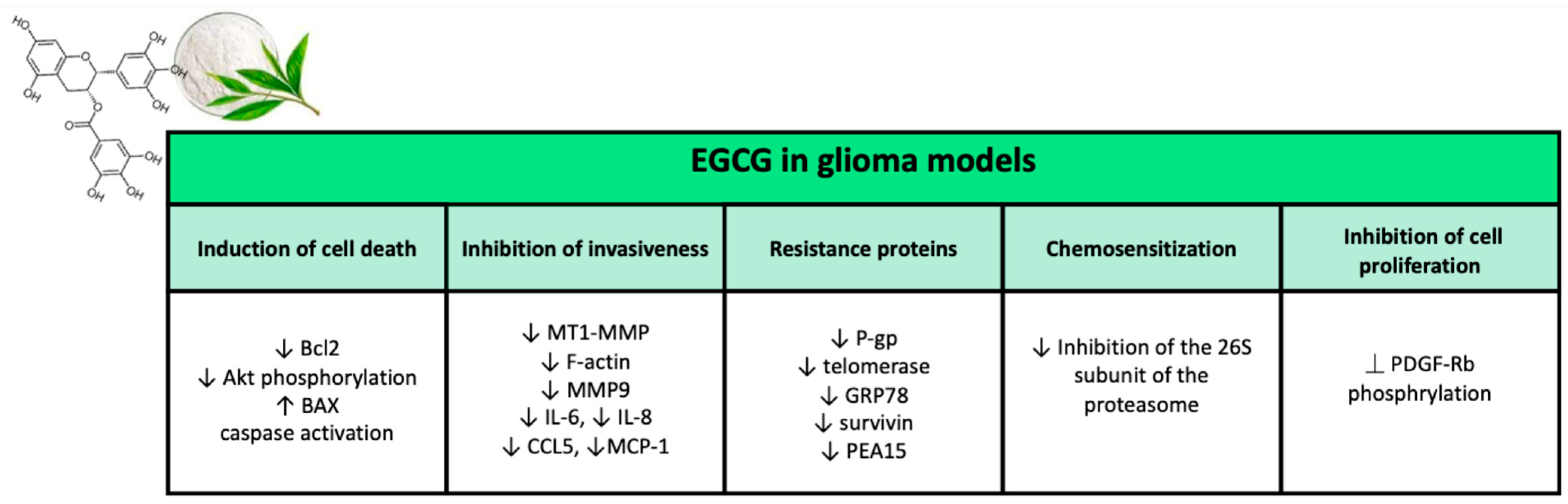
2.2.2. Epigallocatechin Gallate
EGCG Pharmacokinetics
Antitumor Properties of EGCG in Glioma
Inhibition of Cell Proliferation
Induction of Cell Death
Inhibition of Invasiveness
Chemosensitization
Radiosensitization
EGCG and Resistance Proteins
3. Challenges and Considerations in the Use of Natural Substances in the Treatment of Glioma
4. Concluding Remarks and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| RES | Resveratrol |
| EGCG | Epigallocatechin-3- gallate |
| CUR | Curcumin |
| BBB | Blood–brain barrier |
| WHO | World Health Organization |
| CNS | Central nervous system |
| TMZ | Temozolomide |
| GCS | Glioblastoma cancer stem cells |
| ATG | Autophagy |
| NPs | Nanoparticles |
References
- Islam, S.M.S.; Purnat, T.D.; Phuong, N.T.A.; Mwingira, U.; Schacht, K.; Fröschl, G. Non-Communicable Diseases (NCDs) in developing countries: A symposium report. Glob. Health 2014, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bondy, M.L.; Scheurer, M.E.; Malmer, B.; Barnholtz-Sloan, J.S.; Davis, F.G.; Il’Yasova, D.; Kruchko, C.; McCarthy, B.J.; Rajaraman, P.; Schwartzbaum, J.A.; et al. Brain tumor epidemiology: Consensus from the Brain Tumor Epidemiology Consortium. Cancer 2008, 113, 1953–1968. [Google Scholar] [CrossRef] [PubMed]
- di Martino, E.; Smith, L.; Bradley, S.H.; Hemphill, S.; Wright, J.; Renzi, C.; Bergin, R.; Emery, J.; Neal, R.D. Incidence trends for twelve cancers in younger adults—A rapid review. Br. J. Cancer 2022, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012. Neuro-Oncology 2019, 21, v1–v100. [Google Scholar] [CrossRef] [PubMed]
- Minniti, G.; Filippi, A.R.; Osti, M.F.; Ricardi, U. Radiation therapy for older patients with brain tumors. Radiat. Oncol. 2017, 12, 101. [Google Scholar] [CrossRef] [PubMed]
- Megari, K. Quality of life in chronic disease patients. Health Psychol. Res. 2013, 1, e27. [Google Scholar] [CrossRef] [PubMed]
- Komori, T. The 2016 WHO Classification of Tumours of the Central Nervous System: The Major Points of Revision. Neurol. Med.-Chir. 2017, 57, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee, S.U. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9. [Google Scholar] [PubMed]
- Lin, D.; Wang, M.; Chen, Y.; Gong, J.; Chen, L.; Shi, X.; Lan, F.; Chen, Z.; Xiong, T.; Sun, H.; et al. Trends in Intracranial Glioma Incidence and Mortality in the United States, 1975. Front. Oncol. 2021, 11, 11. [Google Scholar] [CrossRef] [PubMed]
- Wesseling, P.; Kros, J.M.; Jeuken, J.W. The pathological diagnosis of diffuse gliomas: Towards a smart synthesis of microscopic and molecular information in a multidisciplinary context. Diagn. Histopathol. 2011, 17, 486–494. [Google Scholar] [CrossRef]
- Zacher, A.; Kaulich, K.; Stepanow, S.; Wolter, M.; Köhrer, K.; Felsberg, J.; Malzkorn, B.; Reifenberger, G. Molecular Diagnostics of Gliomas Using Next Generation Sequencing of a Glioma-Tailored Gene Panel. Brain Pathol. 2017, 27, 146–159. [Google Scholar] [CrossRef] [PubMed]
- Wesseling, P.; Bent, M.V.D.; Perry, A. Oligodendroglioma: Pathology, molecular mechanisms and markers. Acta Neuropathol. 2015, 129, 809–827. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hu, G.; Quan, X. Analysis of the factors affecting the prognosis of glioma patients. Open Med. 2019, 14, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Tobin, M.K.; Geraghty, J.R.; Engelhard, H.H.; Linninger, A.A.; Mehta, A.I. Intramedullary spinal cord tumors: A review of current and future treatment strategies. Neurosurg. Focus 2015, 39, E14. [Google Scholar] [CrossRef] [PubMed]
- Girardi, F.; Allemani, C.; Coleman, M.P. Global trends in survival from astrocytic tumors in adolescents and young adults: A systematic review. JNCI Cancer Spectr. 2020, 4, pkaa049. [Google Scholar] [CrossRef] [PubMed]
- Claus, E.B.; Walsh, K.; Wiencke, J.K.; Molinaro, A.M.; Wiemels, J.L.; Schildkraut, J.M.; Bondy, M.L.; Berger, M.S.; Jenkins, R.B.; Wrensch, M. Survival and low-grade glioma: The emergence of genetic information. Neurosurg. Focus 2015, 38, E6. [Google Scholar] [CrossRef] [PubMed]
- Iwadate, Y.; Matsutani, T.; Hara, A.; Hirono, S.; Ikegami, S.; Kobayashi, M.; Ito, D.; Kawauchi, D.; Horiguchi, K.; Tamiya, A.; et al. Eighty percent survival rate at 15 years for 1p/19q co-deleted oligodendroglioma treated with upfront chemotherapy irrespective of tumor grade. J. Neuro-Oncol. 2018, 141, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Śledzińska, P.; Bebyn, M.G.; Furtak, J.; Kowalewski, J.; Lewandowska, M.A. Prognostic and Predictive Biomarkers in Gliomas. Int. J. Mol. Sci. 2021, 22, 10373. [Google Scholar] [CrossRef]
- Tamimi, A.F.; Juweid, M. Chapter 8—Epidemiology and Outcome of Glioblastoma. In Glioblastoma; De Vleeschouwer, S., Ed.; Exon Publications[M01]: Brisbane, Australia, 2017. [Google Scholar]
- Rodriguez, C.M.V. The Role of Cannabinoid Receptors in Retinoblastoma Progression. Doctoral Dissertation, University of Puerto Rico Medical Sciences, San Juan, Puerto Rico, 2021. [Google Scholar]
- Seker-Polat, F.; Degirmenci, N.P.; Solaroglu, I.; Bagci-Onder, T. Tumor Cell Infiltration into the Brain in Glioblastoma: From Mechanisms to Clinical Perspectives. Cancers 2022, 14, 443. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Van Straten, D.; Broekman, M.L.; Préat, V.; Schiffelers, R.M. Nanocarrier-based drug combination therapy for glioblastoma. Theranostics 2020, 10, 1355–1372. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Xu, R.; Xu, H.; Wang, G.; Shen, X.; Jiang, H. Extracranial metastases of high-grade glioma: The clinical characteristics and mechanism. World J. Surg. Oncol. 2017, 15, 181. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20, S2–S8. [Google Scholar] [CrossRef] [PubMed]
- Attia, N.; Mashal, M.; Pemminati, S.; Omole, A.; Edmondson, C.; Jones, W.; Priyadarshini, P.; Mughal, T.; Aziz, P.; Zenick, B.; et al. Cell-Based Therapy for the Treatment of Glioblastoma: An Update from Preclinical to Clinical Studies. Cells 2021, 11, 116. [Google Scholar] [CrossRef]
- Mao, H.; LeBrun, D.; Yang, J.; Zhu, V.F.; Li, M. Deregulated Signaling Pathways in Glioblastoma Multiforme: Molecular Mechanisms and Therapeutic Targets. Cancer Investig. 2012, 30, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Crespo, I.; Vital, A.L.; Gonzalez-Tablas, M.; Patino, M.D.C.; Otero, A.; Lopes, M.C.; de Oliveira, C.; Domingues, P.; Orfao, A.; Tabernero, M.D. Molecular and Genomic Alterations in Glioblastoma Multiforme. Am. J. Pathol. 2015, 185, 1820–1833. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Jin, Y.; Fan, Z. The Mechanism of Warburg Effect-Induced Chemoresistance in Cancer. Front. Oncol. 2021, 11, 3408. [Google Scholar] [CrossRef]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef] [PubMed]
- Rocca, E.; Anjum, R.L. Complexity, Reductionism and the Biomedical Model. In Rethinking Causality, Complexity and Evidence for the Unique Patient; Springer: Cham, Switzerland, 2020; pp. 75–94. [Google Scholar]
- Javaid, A.; Ullah, I.; Masud, H.; Basit, A.; Ahmad, W.; Butt, Z.A.; Qasim, M. Predictors of poor treatment outcomes in multi-drug-resistant tuberculosis patients: A retrospective cohort study. Clin. Microbiol. Infect. 2018, 24, 612–617. [Google Scholar] [CrossRef] [PubMed]
- Vodovotz, Y.; Barnard, N.; Hu, F.B.; Jakicic, J.; Lianov, L.; Loveland, D.; Buysse, D.; Szigethy, E.; Finkel, T.; Sowa, G.; et al. Prioritized Research for the Prevention, Treatment, and Reversal of Chronic Disease: Recommendations from the Lifestyle Medicine Research Summit. Front. Med. 2020, 7, 959. [Google Scholar] [CrossRef] [PubMed]
- Houlden, R.L.; Yen, H.H.; Mirrahimi, A. The Lifestyle History: A Neglected but Essential Component of the Medical History. Am. J. Lifestyle Med. 2018, 12, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Tagde, P.; Tagde, P.; Tagde, S.; Bhattacharya, T.; Garg, V.; Akter, R.; Rahman, H.; Najda, A.; Albadrani, G.M.; Sayed, A.A.; et al. Natural bioactive molecules: An alternative approach to the treatment and control of glioblastoma multiforme. Biomed. Pharmacother. 2021, 141, 111928. [Google Scholar] [CrossRef]
- Bracci, L.; Schiavoni, G.; Sistigu, A.; Belardelli, F. Immune-based mechanisms of cytotoxic chemotherapy: Implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ. 2014, 21, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Garcia, H.D.; Scheer, M.; Henssen, A.G. Current and Future Treatment Strategies for Rhabdomyosarcoma. Front. Oncol. 2019, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- D’Alessio, A.; Proietti, G.; Sica, G.; Scicchitano, B.M. Pathological and Molecular Features of Glioblastoma and Its Peritumoral Tissue. Cancers 2019, 11, 469. [Google Scholar] [CrossRef] [PubMed]
- Sestito, S.; Runfola, M.; Tonelli, M.; Chiellini, G.; Rapposelli, S. New Multitarget Approaches in the War Against Glioblastoma: A Mini-Perspective. Front. Pharmacol. 2018, 9, 874. [Google Scholar] [CrossRef]
- Riley, R.S.; Day, E.S. Gold nanoparticle-mediated photothermal therapy: Applications and opportunities for multimodal cancer treatment. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9, 1449. [Google Scholar] [CrossRef] [PubMed]
- Kotecha, R.; Takami, A.; Espinoza, J.L. Dietary phytochemicals and cancer chemoprevention: A review of the clinical evidence. Oncotarget 2016, 7, 52517–52529. [Google Scholar] [CrossRef] [PubMed]
- Benzie, I.F.; Wachtel-Galor, S. (Eds.) Herbal Medicine: Biomolecular and Clinical Aspects; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2011. [Google Scholar]
- Akter, R.; Najda, A.; Rahman, H.; Shah, M.; Wesołowska, S.; Hassan, S.S.U.; Mubin, S.; Bibi, P.; Saeeda, S. Potential Role of Natural Products to Combat Radiotherapy and Their Future Perspectives. Molecules 2021, 26, 5997. [Google Scholar] [CrossRef]
- Bellettato, C.M.; Scarpa, M. Possible strategies to cross the blood–brain barrier. Ital. J. Pediatr. 2018, 44, 127–133. [Google Scholar] [CrossRef]
- Kubczak, M.; Szustka, A.; Rogalińska, M. Molecular Targets of Natural Compounds with Anti-Cancer Properties. Int. J. Mol. Sci. 2021, 22, 13659. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-H.; Loo, J.C.Y.; Bebawy, M.; Luk, F.; Mason, R.S.; Rohanizadeh, R. Curcumin and its Derivatives: Their Application in Neuropharmacology and Neuroscience in the 21st Century. Curr. Neuropharmacol. 2013, 11, 338–378. [Google Scholar] [CrossRef] [PubMed]
- Giordano, A.; Tommonaro, G. Curcumin and cancer. Nutrients 2019, 11, 2376. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.C.; Patchva, S.; Koh, W.; Aggarwal, B.B. Discovery of curcumin, a component of golden spice, and its miraculous biological activities. Clin. Exp. Pharmacol. Physiol. 2012, 39, 283–299. [Google Scholar] [CrossRef]
- Zhang, H.A.; Kitts, D.D. Turmeric and its bioactive constituents trigger cell signaling mechanisms that protect against di-abetes and cardiovascular diseases. Mol. Cell. Biochem. 2021, 476, 3785–3814. [Google Scholar] [CrossRef] [PubMed]
- Sharifi-Rad, J.; El Rayess, Y.; Rizk, A.A.; Sadaka, C.; Zgheib, R.; Zam, W.; Sestito, S.; Rapposelli, S.; Neffe-Skocińska, K.; Zielińska, D.; et al. Turmeric and Its Major Compound Curcumin on Health: Bioactive Effects and Safety Profiles for Food, Pharmaceutical, Biotechnological and Medicinal Applications. Front. Pharmacol. 2020, 11, 1021. [Google Scholar] [CrossRef] [PubMed]
- Karthikeyan, A.; Senthil, N.; Min, T. Nanocurcumin: A Promising Candidate for Therapeutic Applications. Front. Pharmacol. 2020, 11, 487. [Google Scholar] [CrossRef] [PubMed]
- Jabczyk, M.; Nowak, J.; Hudzik, B.; Zubelewicz-Szkodzińska, B. Curcumin in Metabolic Health and Disease. Nutrients 2021, 13, 4440. [Google Scholar] [CrossRef] [PubMed]
- Benameur, T.; Giacomucci, G.; Panaro, M.A.; Ruggiero, M.; Trotta, T.; Monda, V.; Pizzolorusso, I.; Lofrumento, D.D.; Porro, C.; Messina, G. New Promising Therapeutic Avenues of Curcumin in Brain Diseases. Molecules 2021, 27, 236. [Google Scholar] [CrossRef] [PubMed]
- Labanca, F.; Ullah, H.; Khan, H.; Milella, L.; Xiao, J.; Dajic-Stevanovic, Z.; Jeandet, P. Therapeutic and Mechanistic effects of Curcumin in Huntington’s disease. Curr. Neuropharmacol. 2020, 18, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Shahcheraghi, S.H.; Zangui, M.; Lotfi, M.; Ghayour-Mobarhan, M.; Ghorbani, A.; Jaliani, H.Z.; Sadeghnia, H.R.; Sahebkar, A. Therapeutic Potential of Curcumin in the Treatment of Glioblastoma Multiforme. Curr. Pharm. Des. 2019, 25, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; McClements, D.J. Formulation of More Efficacious Curcumin Delivery Systems Using Colloid Science: Enhanced Solubility, Stability, and Bioavailability. Molecules 2020, 25, 2791. [Google Scholar] [CrossRef] [PubMed]
- Dulbecco, P.; Savarino, V. Therapeutic potential of curcumin in digestive diseases. World J. Gastroenterol. WJG 2013, 19, 9256. [Google Scholar] [CrossRef]
- Vancamelbeke, M.; Vermeire, S. The intestinal barrier: A fundamental role in health and disease. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 821–834. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.C.; Mittal, S. Antitumor Activity of Curcumin in Glioblastoma. Int. J. Mol. Sci. 2020, 21, 9435. [Google Scholar] [CrossRef] [PubMed]
- Ryskalin, L.; Biagioni, F.; Busceti, C.L.; Lazzeri, G.; Frati, A.; Fornai, F. The Multi-Faceted Effect of Curcumin in Glioblastoma from Rescuing Cell Clearance to Autophagy-Independent Effects. Molecules 2020, 25, 4839. [Google Scholar] [CrossRef]
- Maiti, P.; Dunbar, G.L. Use of Curcumin, a Natural Polyphenol for Targeting Molecular Pathways in Treating Age-Related Neurodegenerative Diseases. Int. J. Mol. Sci. 2018, 19, 1637. [Google Scholar] [CrossRef] [PubMed]
- Amalraj, A.; Pius, A.; Gopi, S.; Gopi, S. Biological activities of curcuminoids, other biomolecules from turmeric and their derivatives–A review. J. Tradit. Complementary Med. 2017, 7, 205–233. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Ghosh, S. The NF-κB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Puliyappadamba, V.T.; Hatanpaa, K.J.; Chakraborty, S.; Habib, A.A. The role of NF-κB in the pathogenesis of glioma. Mol. Cell. Oncol. 2014, 1, e963478. [Google Scholar] [CrossRef]
- Lee, Z.H.; Kwack, K.; Kim, K.K.; Lee, S.H.; Kim, H.-H. Activation of c-Jun N-Terminal Kinase and Activator Protein 1 by Receptor Activator of Nuclear Factor κB. Mol. Pharmacol. 2000, 58, 1536–1545. [Google Scholar] [CrossRef] [PubMed]
- Chandrika, G.; Natesh, K.; Ranade, D.; Chugh, A.; Shastry, P. Suppression of the invasive potential of Glioblastoma cells by m, TOR inhibitors involves modulation of NFκB and PKC-α signaling. Sci. Rep. 2016, 6, 22455. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Lal, N.; Nemaysh, V.; Luthra, P.M. Demethoxycurcumin mediated targeting of Mn, SOD leading to activation of apoptotic pathway and inhibition of Akt/NF-κB survival signalling in human glioma U87 MG cells. Toxicol. Appl. Pharmacol. 2018, 345, 75–93. [Google Scholar] [CrossRef] [PubMed]
- Fratantonio, D.; Molonia, M.S.; Bashllari, R.; Muscara’, C.; Ferlazzo, G.; Costa, G.; Saija, A.; Cimino, F.; Speciale, A. Curcumin potentiates the antitumor activity of Paclitaxel in rat glioma C6 cells. Phytomedicine 2019, 55, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Hesari, A.; Rezaei, M.; Rezaei, M.; Dashtiahangar, M.; Fathi, M.; Rad, J.G.; Momeni, F.; Avan, A.; Ghasemi, F. Effect of curcumin on glioblastoma cells. J. Cell. Physiol. 2018, 234, 10281–10288. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.; Kamarudin, M.; Naidu, R. Anticancer Mechanism of Curcumin on Human Glioblastoma. Nutrients 2021, 13, 950. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhu, J.; Lv, X.; Xing, J.; Liu, S.; Chen, C.; Xu, Y. Curcumin potentiates the potent antitumor activity of ACNU against glioblastoma by suppressing the PI3K/AKT and NF-κB/COX-2 signaling pathways. Onco Targets Ther. 2017, 10, 5471. [Google Scholar] [CrossRef]
- Weissenberger, J.; Priester, M.; Bernreuther, C.; Rakel, S.; Glatzel, M.; Seifert, V.; Kögel, D. Dietary Curcumin Attenuates Glioma Growth in a Syngeneic Mouse Model by Inhibition of the JAK1,2/STAT3 Signaling Pathway. Clin. Cancer Res. 2010, 16, 5781–5795. [Google Scholar] [CrossRef]
- Senft, C.; Polacin, M.; Priester, M.; Seifert, V.; Kögel, D.; Weissenberger, J. The nontoxic natural compound Curcumin exerts anti-proliferative, anti-migratory, and anti-invasive properties against malignant gliomas. BMC Cancer 2010, 10, 491. [Google Scholar] [CrossRef] [PubMed]
- Cianfanelli, V.; Fuoco, C.; Lorente, M.; Salazar, M.; Quondamatteo, F.; Gherardini, P.F.; De Zio, D.; Nazio, F.; Antonioli, M.; D’Orazio, M.; et al. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat. Cell Biol. 2015, 17, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Park, K.-S.; Yoon, S.-Y.; Park, S.-H.; Hwang, J.-H. Anti-Migration and Anti-Invasion Effects of Curcumin via Suppression of Fascin Expression in Glioblastoma Cells. Brain Tumor Res. Treat. 2019, 7, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Sun, G. Low-Dose DMC Significantly Enhances the Effect of TMZ on Glioma Cells by Targeting Multiple Signaling Pathways Both In Vivo and In Vitro. Neuro Mol. Med. 2015, 17, 431–442. [Google Scholar] [CrossRef]
- Juric, V.; Murphy, B. Cyclin-dependent kinase inhibitors in brain cancer: Current state and future directions. Cancer Drug Resist 2020, 3, 48–62. [Google Scholar] [CrossRef]
- Liu, E.; Wu, J.; Cao, W.; Zhang, J.; Liu, W.; Jiang, X.; Zhang, X. Curcumin induces G2/M cell cycle arrest in a p53-dependent manner and upregulates ING4 expression in human glioma. J. Neuro-Oncol. 2007, 85, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Su, C.C.; Wang, M.J.; Chiu, T.L. The anti-cancer efficacy of curcumin scrutinized through core signaling pathways in glio-blastoma. Int. J. Mol. Med. 2010, 26, 217–224. [Google Scholar] [PubMed]
- Nocito, M.C.; De Luca, A.; Prestia, F.; Avena, P.; La Padula, D.; Zavaglia, L.; Sirianni, R.; Casaburi, I.; Puoci, F.; Chimento, A.; et al. Antitumoral Activities of Curcumin and Recent Advances to Im, Prove Its Oral Bioavailability. Biomedicine 2021, 9, 1476. [Google Scholar]
- Shan, J.; Dudenhausen, E.; Kilberg, M.S. Induction of early growth response gene 1 (EGR1) by endoplasmic reticulum stress is mediated by the extracellular regulated kinase (ERK) arm of the MAPK pathways. Biochim. Biophys. Acta 2019, 1866, 371–381. [Google Scholar] [CrossRef]
- Choi, B.H.; Kim, C.G.; Bae, Y.S.; Lim, Y.; Lee, Y.H.; Shin, S.Y. p21Waf1/Cip1 expression by curcumin in U-87MG human glioma cells: Role of early growth response-1 expression. Cancer Res. 2008, 68, 1369–1377. [Google Scholar] [CrossRef]
- Wu, B.; Yao, H.; Wang, S.; Xu, R. DAPK1 modulates a curcumin-induced G2/M arrest and apoptosis by regulating STAT3, NF-κB, and caspase-3 activation. Biochem. Biophys. Res. Commun. 2013, 434, 75–80. [Google Scholar] [CrossRef]
- Lee, J.E.; Yoon, S.S.; Moon, E.Y. Curcumin-induced autophagy augments its antitumor effect against A172 human glio-blastoma cells. Biomol. Ther. 2019, 27, 484. [Google Scholar] [CrossRef]
- Tamaddoni, A.; Mohammadi, E.; Sedaghat, F.; Qujeq, D.; As’Habi, A. The anticancer effects of curcumin via targeting the mammalian target of rapamycin complex 1 (m, TORC1) signaling pathway. Pharmacol. Res. 2020, 156, 104798. [Google Scholar] [CrossRef]
- Su, J.; Zhou, X.; Yin, X.; Wang, L.; Zhao, Z.; Hou, Y.; Zheng, N.; Xia, J.; Wang, Z. The effects of curcumin on proliferation, apoptosis, invasion, and NEDD4 expression in pancreatic cancer. Biochem. Pharmacol. 2017, 140, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Gallot, Y.; Bohnert, K. Confounding Roles of ER Stress and the Unfolded Protein Response in Skeletal Muscle Atrophy. Int. J. Mol. Sci. 2021, 22, 2567. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Huo, X.; Liu, X. “m, TOR Signaling Pathway”: A Potential Target of Curcumin in the Treatment of Spinal Cord Injury. BioMed Res. Int. 2017, 2017, 1634801. [Google Scholar]
- Aoki, H.; Takada, Y.; Kondo, S.; Sawaya, R.; Aggarwal, B.B.; Kondo, Y. Evidence That Curcumin Suppresses the Growth of Malignant Gliomas in Vitro and in Vivo through Induction of Autophagy: Role of Akt and Extracellular Signal-Regulated Kinase Signaling Pathways. Mol. Pharmacol. 2007, 72, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Maiti, P.; Scott, J.; Sengupta, D.; Al-Gharaibeh, A.; Dunbar, G.L. Curcumin and solid lipid curcumin particles induce autophagy, but inhibit mitophagy and the PI3K-Akt/m, TOR pathway in cultured glioblastoma cells. Int. J. Mol. Sci. 2019, 20, 399. [Google Scholar] [CrossRef] [PubMed]
- Zanotto-Filho, A.; Braganhol, E.; Edelweiss, M.I.; Behr, G.A.; Zanin, R.; Schröder, R.; Simões-Pires, A.; Battastini, A.M.O.; Moreira, J.C.F. The curry spice curcumin selectively inhibits cancer cells growth in vitro and in preclinical model of glioblastoma. J. Nutr. Biochem. 2012, 23, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Zanotto-Filho, A.; Coradini, K.; Braganhol, E.; Schröder, R.; de Oliveira, C.M.; Simões-Pires, A.; Battastini, A.M.O.; Pohlmann, A.; Guterres, S.; Forcelini, C.M.; et al. Curcumin-loaded lipid-core nanocapsules as a strategy to improve pharmacological efficacy of curcumin in glioma treatment. Eur. J. Pharm. Biopharm. 2013, 83, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Escamilla-Ramírez, A.; Castillo-Rodríguez, R.A.; Zavala-Vega, S.; Jimenez-Farfan, D.; Anaya-Rubio, I.; Briseño, E.; Palencia, G.; Guevara, P.; Cruz-Salgado, A.; Sotelo, J.; et al. Autophagy as a Potential Therapy for Malignant Glioma. Pharmaceuticals 2020, 13, 156. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Saha, S.K.; Rahman, S.; Uddin, J.; Uddin, S.; Pang, M.-G.; Rhim, H.; Cho, S.-G. Molecular insights into therapeutic potential of autophagy modulation by natural products for cancer stem cells. Front. Cell Dev. Biol. 2020, 8, 283. [Google Scholar] [CrossRef] [PubMed]
- Mattei, V.; Santilli, F.; Martellucci, S.; Monache, S.D.; Fabrizi, J.; Colapietro, A.; Angelucci, A.; Festuccia, C. The Importance of Tumor Stem Cells in Glioblastoma Resistance to Therapy. Int. J. Mol. Sci. 2021, 22, 3863. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.K.; Jermakowicz, A.; Mookhtiar, A.K.; Nemeroff, C.B.; Schürer, S.C.; Ayad, N.G. Drug Repositioning in Glioblastoma: A Pathway Perspective. Front. Pharmacol. 2018, 9, 218. [Google Scholar] [CrossRef] [PubMed]
- Ryskalin, L.; Gaglione, A.; Limanaqi, F.; Biagioni, F.; Familiari, P.; Frati, A.; Esposito, V.; Fornai, F. The Autophagy Status of Cancer Stem Cells in Gliobastoma Multiforme: From Cancer Promotion to Therapeutic Strategies. Int. J. Mol. Sci. 2019, 20, 3824. [Google Scholar] [CrossRef] [PubMed]
- Klinger, N.V.; Mittal, S. Therapeutic Potential of Curcumin for the Treatment of Brain Tumors. Oxidative Med. Cell. Longev. 2016, 2016, 9324085. [Google Scholar] [CrossRef] [PubMed]
- Tao, Z.; Li, T.; Ma, H.; Yang, Y.; Zhang, C.; Hai, L.; Liu, P.; Yuan, F.; Li, J.; Yi, L.; et al. Autophagy suppresses self-renewal ability and tumorigenicity of glioma-initiating cells and promotes Notch1 degradation. Cell Death Dis. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, W.; Long, L.; Zheng, B.; Ji, W.; Yang, N.; Zhang, Q.; Liang, Z. Curcumin promotes differentiation of glioma-initiating cells by inducing autophagy. Cancer Sci. 2012, 103, 684–690. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Hsieh, I.-Y.; Huang, X.; Li, J.; Zhao, W. Glioblastoma Stem-Like Cells: Characteristics, Microenvironment, and Therapy. Front. Pharmacol. 2016, 7, 477. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, F.; Liao, W.; Yu, L.; Hu, Z.; Li, M.; Xia, H. Curcumin suppresses glioblastoma cell proliferation by p-AKT/m, TOR pathway and increases the PTEN expression. Arch. Biochem. Biophys. 2020, 689, 108412. [Google Scholar] [CrossRef] [PubMed]
- Sa, G.; Das, T. Anti cancer effects of curcumin: Cycle of life and death. Cell Div. 2008, 3, 14. [Google Scholar] [CrossRef] [PubMed]
- Khaw, A.K.; Hande, M.P.; Kalthur, G. Curcumin inhibits telomerase and induces telomere shortening and apoptosis in brain tumour cells. J. Cell. Biochem. 2013, 114, 1257–1270. [Google Scholar] [CrossRef] [PubMed]
- Kuttikrishnan, S.; Siveen, K.S.; Prabhu, K.S.; Khan, A.Q.; Ahmed, E.I.; Akhtar, S.; Ali, T.A.; Merhi, M.; Dermime, S.; Steinhoff, M.; et al. Curcumin Induces Apoptotic Cell Death via Inhibition of PI3-Kinase/AKT Pathway in B-Precursor Acute Lymphoblastic Leukemia. Front. Oncol. 2019, 9, 484. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tu, L.; Zhou, X.; Li, B. Curcumin-Mediated Induction of Apoptosis in Human Glioma CHME Cells. Med Sci. Monit. Basic Res. 2018, 24, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Garg, H.; Suri, P.; Gupta, J.C.; Talwar, G.P.; Dubey, S. Survivin: A unique target for tumor therapy. Cancer Cell Int. 2016, 16, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Conde, M.; Michen, S.; Wiedemuth, R.; Klink, B.; Schröck, E.; Schackert, G.; Temme, A. Chromosomal instability induced by increased BIRC5/Survivin levels affects tumorigenicity of glioma cells. BMC Cancer 2017, 17, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Yang, P.; Wang, K.; Liu, Y.; Liu, X.; Shan, X.; Huang, R.; Zhang, K.; Wang, J. Survivin is a prognostic indicator in glioblastoma and may be a target of micro, RNA. Oncol. Lett. 2019, 18, 359–367. [Google Scholar] [PubMed]
- Gersey, Z.C.; Rodriguez, G.A.; Barbarite, E.; Sanchez, A.; Walters, W.M.; Ohaeto, K.C.; Komotar, R.J.; Graham, R.M. Curcumin decreases malignant characteristics of glioblastoma stem cells via induction of reactive oxygen species. BMC Cancer 2017, 17, 99. [Google Scholar] [CrossRef]
- Thani, N.A.A.; Sallis, B.; Nuttall, R.; Schubert, F.R.; Ahsan, M.; Davies, D.; Purewal, S.; Cooper, A.; Rooprai, H.K. Induction of apoptosis and reduction of MMP gene expression in the U373 cell line by polyphenolics in Aronia melanocarpa and by curcumin. Oncol. Rep. 2012, 28, 1435–1442. [Google Scholar] [CrossRef]
- Huang, B.-R.; Tsai, C.-H.; Chen, C.-C.; Way, T.-D.; Kao, J.-Y.; Liu, Y.-S.; Lin, H.-Y.; Lai, S.-W.; Lu, D.-Y. Curcumin Promotes Connexin 43 Degradation and Temozolomide-Induced Apoptosis in Glioblastoma Cells. Am. J. Chin. Med. 2019, 47, 657–674. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, C.; Nair, S.M.; Escalon, E.; Melnick, S.J. Potentiation of Etoposide and Temozolomide Cytotoxicity by Curcumin and Turmeric Force in Brain Tumor Cell Lines. J. Complement. Integr. Med. 2012, 9, 20. [Google Scholar] [CrossRef] [PubMed]
- Bourboulia, D.; Stetler-Stevenson, W.G. Matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs): Positive and negative regulators in tumor cell adhesion. Semin. Cancer Biol. 2010, 20, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, S.M.; Amini, A.; Morris, D.L.; Pourgholami, M.H. Significance of vascular endothelial growth factor in growth and peritoneal dissemination of ovarian cancer. Cancer Metastasis Rev. 2011, 31, 143–162. [Google Scholar] [CrossRef]
- Tabouret, E.; Boudouresque, F.; Farina, P.; Barrié, M.; Bequet, C.; Sanson, M.; Chinot, O. MMP2 and MMP9 as candidate bi-omarkers to monitor bevacizumab therapy in high-grade glioma. Neuro-Oncology 2015, 17, 1174–1176. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ma, Y.; Wang, H.; Xu, L.; Yu, Y. MMP-2 expression and correlation with pathology and MRI of glioma. Oncol. Lett. 2019, 17, 1826–1832. [Google Scholar] [PubMed]
- Wang, X.; Deng, J.; Yuan, J.; Tang, X.; Wang, Y.; Chen, H.; Liu, Y.; Zhou, L. Curcumin exerts its tumor suppressive function via inhibition of NEDD4 oncoprotein in glioma cancer cells. Int. J. Oncol. 2017, 51, 467–477. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/β-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Zuccarini, M.; Giuliani, P.; Ziberi, S.; Carluccio, M.; Di Iorio, P.; Caciagli, F.; Ciccarelli, R. The Role of Wnt Signal in Glioblastoma Development and Progression: A Possible New Pharmacological Target for the Therapy of This Tumor. Genes 2018, 9, 105. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, H.; Nakamura, H.; Nishikawa, H.; Nishiguchi, S.; Iijima, H. Hepatoma-Derived Growth Factor: An Overview and Its Role as a Potential Therapeutic Target Molecule for Digestive Malignancies. Int. J. Mol. Sci. 2020, 21, 4216. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Luo, H.; Fu, H.; Huang, H.; Huang, H.; Luo, K.; Li, C.; Hu, R.; Zheng, C.; Lan, C.; et al. [Curcumin suppresses invasiveness and migration of human glioma cells in vitro by inhibiting HDGF/β-catenin complex]. J. South. Med. Univ. 2019, 39, 911–916. [Google Scholar]
- Câmara, J.S.; Albuquerque, B.R.; Aguiar, J.; Corrêa, R.C.G.; Gonçalves, J.L.; Granato, D.; Pereira, J.A.M.; Barros, L.; Ferreira, I.C.F.R. Food Bioactive Compounds and Emerging Techniques for Their Extraction: Polyphenols as a Case Study. Foods 2020, 10, 37. [Google Scholar] [CrossRef]
- Kopustinskiene, D.M.; Jakstas, V.; Savickas, A.; Bernatoniene, J. Flavonoids as anticancer agents. Nutrients 2020, 12, 457. [Google Scholar] [CrossRef]
- Zhai, K.; Mazurakova, A.; Koklesova, L.; Kubatka, P.; Büsselberg, D. Flavonoids Synergistically Enhance the Anti-Glioblasoma Effects of Chemotherapeutic Drugs. Biomolecules 2021, 11, 1841. [Google Scholar] [CrossRef]
- Perrone, D.; Fuggetta, M.P.; Ardito, F.; Cottarelli, A.; De Filippis, A.; Ravagnan, G.; De Maria, S.; Muzio, L.L. Resveratrol (3,5,4′-trihydroxystilbene) and its properties in oral diseases. Exp. Ther. Med. 2017, 14, 3–9. [Google Scholar] [CrossRef]
- Valletta, A.; Iozia, L.M.; Leonelli, F. Impact of Environmental Factors on Stilbene Biosynthesis. Plants 2021, 10, 90. [Google Scholar] [CrossRef] [PubMed]
- Tungmunnithum, D.; Thongboonyou, A.; Pholboon, A.; Yangsabai, A. Flavonoids and other phenolic compounds from me-dicinal plants for pharmaceutical and medical aspects: An overview. Medicines 2018, 5, 93. [Google Scholar] [CrossRef]
- Shahidi, F.; Ambigaipalan, P. Phenolics and polyphenolics in foods, beverages and spices: Antioxidant activity and health effects—A review. J. Funct. Foods 2015, 18, 820–897. [Google Scholar] [CrossRef]
- Ko, J.-H.; Sethi, G.; Um, J.-Y.; Shanmugam, M.K.; Arfuso, F.; Kumar, A.P.; Bishayee, A.; Ahn, K.S. The Role of Resveratrol in Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 2589. [Google Scholar] [CrossRef]
- Ali, H.; Dixit, S. Quercetin attenuates the development of 7, 12-dimethyl benz (a) anthracene (DMBA) and croton oil-induced skin cancer in mice. J. Biomed. Res. 2015, 29, 139–144. [Google Scholar]
- Harder, B.G.; Blomquist, M.R.; Wang, J.; Kim, A.J.; Woodworth, G.F.; Winkles, J.A.; Loftus, J.C.; Tran, N.L. Developments in Blood-Brain Barrier Penetrance and Drug Repurposing for Improved Treatment of Glioblastoma. Front. Oncol. 2018, 8, 462. [Google Scholar] [CrossRef] [PubMed]
- Arabzadeh, A.; Mortezazadeh, T.; Aryafar, T.; Gharepapagh, E.; Majdaeen, M.; Farhood, B. Therapeutic potentials of resveratrol in combination with radiotherapy and chemotherapy during glioblastoma treatment: A mechanistic review. Cancer Cell Int. 2021, 21, 391. [Google Scholar] [CrossRef]
- Jannin, B.; Menzel, M.; Berlot, J.-P.; Delmas, D.; Lançon, A.; Latruffe, N. Transport of resveratrol, a cancer chemopreventive agent, to cellular targets: Plasmatic protein binding and cell uptake. Biochem. Pharmacol. 2004, 68, 1113–1118. [Google Scholar] [CrossRef] [PubMed]
- Gambini, J.; Inglés, M.; Olaso, G.; Lopez-Grueso, R.; Bonet-Costa, V.; Gimeno-Mallench, L.; Mas-Bargues, C.; Abdelaziz, K.M.; Gomez-Cabrera, M.C.; Vina, J.; et al. Properties of Resveratrol: In Vitro and In Vivo Studies about Metabolism, Bioavailability, and Biological Effects in Animal Models and Humans. Oxid. Med. Cell. Longev. 2015, 2015, 837042. [Google Scholar] [CrossRef]
- Chaplin, A.; Carpéné, C.; Mercader, J. Resveratrol, Metabolic Syndrome, and Gut Microbiota. Nutrients 2018, 10, 1651. [Google Scholar] [CrossRef] [PubMed]
- Shaito, A.; Posadino, A.M.; Younes, N.; Hasan, H.; Halabi, S.; Alhababi, D.; Al-Mohannadi, A.; Abdel-Rahman, W.M.; Eid, A.H.; Nasrallah, G.K.; et al. Potential Adverse Effects of Resveratrol: A Literature Review. Int. J. Mol. Sci. 2020, 21, 2084. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.; Cai, L.; Udeani, G.O.; Slowing, K.V.; Thomas, C.F.; Beecher, C.W.W.; Fong, H.H.S.; Farnsworth, N.R.; Kinghorn, A.D.; Mehta, R.G.; et al. Cancer Chemopreventive Activity of Resveratrol, a Natural Product Derived from Grapes. Science 1997, 275, 218–220. [Google Scholar] [CrossRef] [PubMed]
- Varoni, E.M.; Lo Faro, A.F.; Sharifi-Rad, J.; Iriti, M. Anticancer Molecular Mechanisms of Resveratrol. Front. Nutr. 2016, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, P.; Tyers, M. How Cells Coordinate Growth and Division. Curr. Biol. 2004, 14, R1014–R1027. [Google Scholar] [CrossRef] [PubMed]
- Richard, S.A. The Therapeutic Potential of Resveratrol in Gliomas. Adv. Biosci. Clin. Med. 2019, 7, 44–59. [Google Scholar] [CrossRef][Green Version]
- Filippi-Chiela, E.C.; Villodre, E.S.; Zamin, L.L.; Lenz, G. Autophagy Interplay with Apoptosis and Cell Cycle Regulation in the Growth Inhibiting Effect of Resveratrol in Glioma Cells. PLoS ONE 2011, 6, e20849. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Banerjee, S.; Acosta, E.P.; Lillard, J.W.; Singh, R. Resveratrol induces cell cycle arrest and apoptosis with docetaxel in prostate cancer cells via a p53/p21WAF1/CIP1 and p27KIP1 pathway. Oncotarget 2017, 8, 17216. [Google Scholar] [CrossRef] [PubMed]
- Fontecave, M.; Lepoivre, M.; Elleingand, E.; Gerez, C.; Guittet, O. Resveratrol, a remarkable inhibitor of ribonucleotide reductase. FEBS Letters 1998, 421, 277–279. [Google Scholar] [CrossRef]
- West, A.J.; Tsui, V.; Stylli, S.S.; Nguyen, H.; Morokoff, A.P.; Kaye, A.H.; Luwor, R.B. The role of interleukin 6 STAT3 signalling in glioblastoma. Oncol. Lett. 2018, 16, 4095–4104. [Google Scholar] [CrossRef] [PubMed]
- Attarha, S.; Roy, A.; Westermark, B.; Tchougounova, E. Mast cells modulate proliferation, migration and stemness of glioma cells through downregulation of GSK3β expression and inhibition of STAT3 activation. Cell. Signal. 2017, 37, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Patel, M.; Ruzevick, J.; Jackson, C.M.; Lim, M. STAT3 Activation in Glioblastoma: Biochemical and Therapeutic Implications. Cancers 2014, 6, 376–395. [Google Scholar] [CrossRef] [PubMed]
- Brescia, P.; Ortensi, B.; Fornasari, L.; Levi, D.; Broggi, G.; Pelicci, G. CD133 Is Essential for Glioblastoma Stem Cell Maintenance. Stem Cells 2013, 31, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Long, L.; Wang, W.; Liang, Z. Resveratrol, a potential radiation sensitizer for glioma stem cells both in vitro and in vivo. J. Pharmacol. Sci. 2015, 129, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, H.; Zhang, P.; Yu, L.-J.; Huang, T.-M.; Song, X.; Kong, Q.-Y.; Dong, J.-L.; Li, P.-N.; Liu, J. SHP2, SOCS3 and PIAS3 Expression Patterns in Medulloblastomas: Relevance to STAT3 Activation and Resveratrol-Suppressed STAT3 Signaling. Nutrients 2016, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Quintero-Fabián, S.; Arreola, R.; Becerril-Villanueva, E.; Torres-Romero, J.C.; Arana-Argáez, V.; Lara-Riegos, J.; Ramírez-Camacho, M.A.; Alvarez-Sánchez, M.E. Role of Matrix Metalloproteinases in Angiogenesis and Cancer. Front. Oncol. 2019, 9, 1370. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.J.; Ward, J.H.; Tan, C.; Grundy, R.G.; Rahman, R. Endothelial-like malignant glioma cells in dynamic three di-mensional culture identifies a role for VEGF and FGFR in a tumor-derived angiogenic response. Oncotarget 2015, 6, 22191. [Google Scholar] [CrossRef] [PubMed]
- Tseng, S.-H.; Lin, S.-M.; Chen, J.-C.; Su, Y.-H.; Huang, H.-Y.; Chen, C.-K.; Lin, P.-Y.; Chen, Y. Resveratrol Suppresses the Angiogenesis and Tumor Growth of Gliomas in Rats. Clin. Cancer Res. 2004, 10, 2190–2202. [Google Scholar] [CrossRef]
- Claes, A.; Idema, A.J.; Wesseling, P. Diffuse glioma growth: A guerilla war. Acta Neuropathol. 2007, 114, 443–458. [Google Scholar] [CrossRef]
- Ryu, J.; Ku, B.M.; Lee, Y.K.; Jeong, J.Y.; Kang, S.; Choi, J.; Yang, Y.; Lee, D.H.; Roh, G.S.; Kim, H.J.; et al. Resveratrol reduces TNF-α-induced U373MG human glioma cell invasion through regulating NF-κB activation and u, PA/u, PAR expression. Anticancer Res. 2011, 31, 4223–4230. [Google Scholar]
- Fink, S.L.; Cookson, B.T. Apoptosis, Pyroptosis, and Necrosis: Mechanistic Description of Dead and Dying Eukaryotic Cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef]
- Neophytou, C.M.; Trougakos, I.P.; Erin, N.; Papageorgis, P. Apoptosis Deregulation and the Development of Cancer Multi-Drug Resistance. Cancers 2021, 13, 4363. [Google Scholar] [CrossRef]
- Jiang, H.; Zhang, L.; Kuo, J.; Kuo, K.; Gautam, S.C.; Groc, L.; Rodriguez, A.I.; Koubi, D.; Hunter, T.J.; Corcoran, G.; et al. Resveratrol-induced apoptotic death in human U251 glioma cells. Mol. Cancer Ther. 2005, 4, 554–561. [Google Scholar] [CrossRef]
- Zhang, W.; Fei, Z.; Zhen, H.-N.; Zhang, J.-N.; Zhang, X. Resveratrol inhibits cell growth and induces apoptosis of rat C6 glioma cells. J. Neuro-Oncol. 2006, 81, 231–240. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Bhardwaj, A.; Aggarwal, R.S.; Seeram, N.P.; Shishodia, S.; Takada, Y. Role of resveratrol in prevention and therapy of cancer: Preclinical and clinical studies. Anticancer Res. 2004, 24, 2783–2840. [Google Scholar]
- Wright, C.; Iyer, A.K.V.; Yakisich, J.S.; Azad, N. Anti-Tumorigenic Effects of Resveratrol in Lung Cancer Cells Through Modulation of c-FLIP. Curr. Cancer Drug Targets 2017, 17, 669–680. [Google Scholar] [CrossRef]
- Jiang, H.; Shang, X.; Wu, H.; Gautam, S.C.; Al-Holou, S.; Li, C.; Kuo, J.; Zhang, L.; Chopp, M. Resveratrol downregulates PI3K/Akt/m, TOR signaling pathways in human U251 glioma cells. J. Exp. Ther. Oncol. 2009, 8, 25–33. [Google Scholar]
- Fang, J.-Y.; Li, Z.-H.; Li, Q.; Huang, W.-S.; Kang, L.; Wang, J.-P. Resveratrol affects protein kinase C activity and promotes apoptosis in human colon carcinoma cells. Asian Pac. J. Cancer Prev. 2012, 13, 6017–6022. [Google Scholar] [CrossRef]
- Burris, H.A., 3rd. Overcoming acquired resistance to anticancer therapy: Focus on the PI3K/AKT/m, TOR pathway. Cancer Chemother. Pharmacol. 2013, 71, 829–842. [Google Scholar] [CrossRef]
- Sanduja, S.; Blanco, F.F.; Young, L.E.; Kaza, V.; Dixon, D.A. The role of tristetraprolin in cancer and inflammation. Front. Biosci. 2012, 17, 174–188. [Google Scholar] [CrossRef]
- Ryu, J.; Yoon, N.A.; Seong, H.; Jeong, J.Y.; Kang, S.; Park, N.; Choi, J.; Lee, D.H.; Roh, G.S.; Kim, H.J.; et al. Resveratrol Induces Glioma Cell Apoptosis through Activation of Tristetraprolin. Mol. Cells 2015, 38, 991–997. [Google Scholar] [PubMed]
- Ewald, J.A.; Desotelle, J.A.; Wilding, G.; Jarrard, D.F. Therapy-Induced Senescence in Cancer. JNCI J. Natl. Cancer Inst. 2010, 102, 1536–1546. [Google Scholar] [CrossRef]
- Gao, Z.; Xu, M.S.; Barnett, T.L.; Xu, C.W. Resveratrol induces cellular senescence with attenuated mono-ubiquitination of histone H2B in glioma cells. Biochem. Biophys. Res. Commun. 2011, 407, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Pospelova, T.V.; Chitikova, Z.V.; Pospelov, V.A. An Integrated Approach for Monitoring Cell Senescence. Methods Mol. Biol. 2013, 965, 383–408. [Google Scholar] [PubMed]
- Vargas, J.E.; Filippi-Chiela, E.C.; Suhre, T.; Kipper, F.; Bonatto, D.; Lenz, G. Inhibition of HDAC increases the senescence induced by natural polyphenols in glioma cells. Biochem. Cell Biol. 2014, 92, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Sayd, S.; Thirant, C.; El-Habr, E.A.; Lipecka, J.; Dubois, L.G.; Bogeas, A.; Tahiri-Jouti, N.; Chneiweiss, H.; Junier, M.-P. Sirtuin-2 Activity is Required for Glioma Stem Cell Proliferation Arrest but not Necrosis Induced by Resveratrol. Stem Cell Rev. Rep. 2013, 10, 103–113. [Google Scholar] [CrossRef]
- Yang, Y.; Cui, J.; Xue, F.; Overstreet, A.-M.; Zhan, Y.; Shan, D.; Li, H.; Wang, Y.; Zhang, M.; Yu, C.; et al. Resveratrol Represses Pokemon Expression in Human Glioma Cells. Mol. Neurobiol. 2016, 53, 1266–1278. [Google Scholar] [CrossRef] [PubMed]
- Cocetta, V.; Quagliariello, V.; Fiorica, F.; Berretta, M.; Montopoli, M. Resveratrol as Chemosensitizer Agent: State of Art and Future Perspectives. Int. J. Mol. Sci. 2021, 22, 2049. [Google Scholar] [CrossRef]
- Yuan, Y.; Xue, X.; Guo, R.B.; Sun, X.L.; Hu, G. Resveratrol enhances the antitumor effects of temozolomide in glioblastoma via ROS-dependent AMPK-TSC-m, TOR signaling pathway. CNS Neurosci. Ther. 2012, 18, 536–546. [Google Scholar] [CrossRef]
- Goffart, N.; Kroonen, J.; Rogister, B. Glioblastoma-initiating cells: Relationship with neural stem cells and the micro-environment. Cancers 2013, 5, 1049–1071. [Google Scholar] [CrossRef]
- Rivera, M.; Sukhdeo, K.; Yu, J.S. Ionizing Radiation in Glioblastoma Initiating Cells. Front. Oncol. 2013, 3, 74. [Google Scholar] [CrossRef]
- Özdemir, F.; Apaydın, E.; Önder, N.I.; Şen, M.; Ayrım, A.; Öğünç, Y.; Incesu, Z. Apoptotic effects of ε-viniferin in combination with cis-platin in C6 cells. Cytotechnology 2018, 70, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Kma, L. Synergistic Effect of Resveratrol and Radiotherapy in Control of Cancers. Asian Pac. J. Cancer Prev. 2013, 14, 6197–6208. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Miner, A.; Hennis, L.; Mittal, S. Mechanisms of temozolomide resistance in glioblastoma—A comprehensive review. Cancer Drug Resist. 2020, 4, 17–43. [Google Scholar] [CrossRef] [PubMed]
- Kitange, G.J.; Carlson, B.L.; Schroeder, M.A.; Grogan, P.T.; Lamont, J.D.; Decker, P.A.; Wu, W.; James, C.D.; Sarkaria, J.N. Induction of MGMT expression is associated with temozolomide resistance in glioblastoma xenografts. Neuro-Oncology 2009, 11, 281–291. [Google Scholar] [CrossRef]
- Huang, H.; Lin, H.; Zhang, X.; Li, J. Resveratrol reverses temozolomide resistance by downregulation of MGMT in T98G glioblastoma cells by the NF-κB-dependent pathway. Oncol. Rep. 2012, 27, 2050–2056. [Google Scholar] [PubMed]
- Yang, H.; Wang, J.; Bu, X.; Yang, B.; Wang, B.; Hu, S.; Yan, Z.; Gao, Y.; Han, S.; Qu, M. Resveratrol restores sensitivity of glioma cells to temozolamide through inhibiting the activation of Wnt signaling pathway. J. Cell. Physiol. 2019, 234, 6783–6800. [Google Scholar] [CrossRef] [PubMed]
- Filippi-Chiela, E.C.; Thomé, M.P.; E Silva, M.M.B.; Pelegrini, A.L.; Ledur, P.F.; Garicochea, B.; Zamin, L.L.; Lenz, G. Resveratrol abrogates the Temozolomide-induced G2 arrest leading to mitotic catastrophe and reinforces the Temozolomide-induced senescence in glioma cells. BMC Cancer 2013, 13, 147. [Google Scholar] [CrossRef] [PubMed]
- Cháirez-Ramírez, M.H.; de la Cruz-López, K.G.; García-Carrancá, A. Polyphenols as Antitumor Agents Targeting Key Players in Cancer-Driving Signaling Pathways. Front. Pharmacol. 2021, 12, 12. [Google Scholar] [CrossRef] [PubMed]
- Martinotti, S.; Ranzato, E.; Burlando, B. (−)-Epigallocatechin-3-gallate induces GRP78 accumulation in the ER and shifts mes-othelioma constitutive UPR into proapoptotic ER stress. J. Cell. Physiol. 2018, 233, 7082–7090. [Google Scholar] [CrossRef] [PubMed]
- Lang, M.; Henson, R.; Braconi, C.; Patel, T. Epigallocatechin-gallate modulates chemotherapy-induced apoptosis in human cholangiocarcinoma cells. Liver Int. 2009, 29, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Zeng, L.; Wang, J.; Zhang, X.; Ruan, Q.; Wang, J.; Cui, S.; Yang, D. Reversal of 5-fluorouracil resistance by EGCG is mediate by inactivation of TFAP2A/VEGF signaling pathway and down-regulation of MDR-1 and P-gp expression in gastric cancer. Oncotarget 2017, 8, 82842–82853. [Google Scholar] [CrossRef] [PubMed]
- Ullmann, U.; Haller, J.; Decourt, J.P.; Girault, N.; Girault, J.; Richard-Caudron, A.S.; Pineau, B.; Weber, P. A Single Ascending Dose Study of Epigallocatechin Gallate in Healthy Volunteers. J. Int. Med Res. 2003, 31, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Chow, H.-H.S.; Cai, Y.; Hakim, I.A.; Crowell, J.A.; Shahi, F.; Brooks, C.A.; Dorr, R.T.; Hara, Y.; Alberts, D.S. Pharmacokinetics and safety of green tea polyphenols after multiple-dose administration of epigallocatechin gallate and polyphenon E in healthy individuals. Clin. Cancer Res. 2003, 9, 3312–3319. [Google Scholar] [PubMed]
- Lin, L.-C.; Wang, M.-N.; Tseng, T.-Y.; Sung, A.J.-S.; Tsai, T.-H. Pharmacokinetics of (−)-Epigallocatechin-3-gallate in Conscious and Freely Moving Rats and Its Brain Regional Distribution. J. Agric. Food Chem. 2007, 55, 1517–1524. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, K.; Miyazawa, T. Chemiluminescence–high-performance liquid chromatographic determination of tea cate-chin, (−)-epigallocatechin 3-gallate, at picomole levels in rat and human plasma. Anal. Biochem. 1997, 248, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Almatroodi, S.A.; Almatroudi, A.; Khan, A.A.; Alhumaydhi, F.A.; Alsahli, M.A.; Rahmani, A.H. Potential Therapeutic Targets of Epigallocatechin Gallate (EGCG), the Most Abundant Catechin in Green Tea, and its Role in the Therapy of Various Types of Cancer. Molecules 2020, 25, 3146. [Google Scholar] [CrossRef] [PubMed]
- Sharma, C.; Nusri, Q.E.-A.; Begum, S.; Javed, E.; Rizvi, T.A.; Hussain, A. (-)-Epigallocatechin-3-Gallate Induces Apoptosis and Inhibits Invasion and Migration of Human Cervical Cancer Cells. Asian Pac. J. Cancer Prev. 2012, 13, 4815–4822. [Google Scholar] [CrossRef]
- Westermark, B.; Heldin, C.-H.; Nistér, M. Platelet-derived growth factor in human glioma. Glia 1995, 15, 257–263. [Google Scholar] [CrossRef]
- Sachinidis, A.; Seul, C.; Seewald, S.; Ahn, H.-Y.; Ko, Y.; Vetter, H. Green tea compounds inhibit tyrosine phosphorylation of PDGF β-receptor and transformation of A172 human glioblastoma. FEBS Lett. 2000, 471, 51–55. [Google Scholar] [CrossRef]
- Sakata, R.; Ueno, T.; Nakamura, T.; Sakamoto, M.; Torimura, T.; Sata, M. Green tea polyphenol epigallocatechin-3-gallate inhibits platelet-derived growth factor-induced proliferation of human hepatic stellate cell line LI. J. Hepatology 2004, 40, 52–59. [Google Scholar] [CrossRef]
- Le, C.T.; Leenders, W.; Molenaar, R.J.; Van Noorden, C.J.F. Effects of the Green Tea Polyphenol Epigallocatechin-3-Gallate on Glioma: A Critical Evaluation of the Literature. Nutr. Cancer 2018, 70, 317–333. [Google Scholar] [CrossRef] [PubMed]
- Golden, E.B.; Lam, P.Y.; Kardosh, A.; Gaffney, K.; Cadenas, E.; Louie, S.G.; Petasis, N.; Chen, T.C.; Schönthal, A.H. Green tea polyphenols block the anticancer effects of bortezomib and other boronic acid–based proteasome inhibitors. Blood 2009, 113, 5927–5937. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.C.; Wang, W.; Golden, E.B.; Thomas, S.; Sivakumar, W.; Hofman, F.M.; Louie, S.G.; Schönthal, A.H. Green tea epigallocatechin gallate enhances therapeutic efficacy of temozolomide in orthotopic mouse glioblastoma models. Cancer Lett. 2011, 302, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, S.; Ma, J.-W.; Li, H.-Y.; Ye, J.-C.; Xie, S.-M.; Du, B.; Zhong, X.-Y. EGCG inhibits properties of glioma stem-like cells and synergizes with temozolomide through downregulation of P-glycoprotein inhibition. J. Neuro-Oncol. 2015, 121, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Hira, V.V.V.; Ploegmakers, K.J.; Grevers, F.; Verbovšek, U.; Silvestre-Roig, C.; Aronica, E.; Tigchelaar, W.; Turnšek, T.L.; Molenaar, R.J.; Van Noorden, C.J.F. CD133+ and Nestin+ Glioma Stem-Like Cells Reside Around CD31+ Arterioles in Niches that Express SDF-1α, CXCR4, Osteopontin and Cathepsin K. J. Histochem. Cytochem. 2015, 63, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, S.; Hirano, H.; Wakimaru, N.; Sarker, K.P.; Kuratsu, J.I. Inhibitory effect of epigallocatechin-gallate on brain tumor cell lines in vitro. Neuro-Oncology 2001, 3, 22–28. [Google Scholar] [CrossRef]
- Das, A.; Banik, N.L.; Ray, S.K. Flavonoids activated caspases for apoptosis in human glioblastoma T98G and U87MG cells but not in human normal astrocytes. Cancer 2009, 116, 164–176. [Google Scholar] [CrossRef]
- Agarwal, A.; Sharma, V.; Tewari, R.; Koul, N.; Joseph, C.; Sen, E. Epigallocatechin-3-gallate exhibits anti-tumor effect by perturbing redox homeostasis, modulating the release of pro-inflammatory mediators and decreasing the invasiveness of glioblastoma cells. Mol. Med. Rep. 2008, 1, 511–515. [Google Scholar] [CrossRef]
- McLaughlin, N.; Annabi, B.; Bouzeghrane, M.; Temme, A.; Bahary, J.-P.; Moumdjian, R.; Béliveau, R. The Survivin-mediated radioresistant phenotype of glioblastomas is regulated by Rho, A and inhibited by the green tea polyphenol (−)-epigallocatechin-3-gallate. Brain Res. 2006, 1071, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hatoum, A.; Mohammed, R.; Zakieh, O. The unique invasiveness of glioblastoma and possible drug targets on extracellular matrix. Cancer Manag. Res. 2019, 11, 1843–1855. [Google Scholar] [CrossRef] [PubMed]
- Demeule, M.; Brossard, M.; Pagé, M.; Gingras, D.; Béliveau, R. Matrix metalloproteinase inhibition by green tea catechins. Biochim. Biophys. Acta Protein Struct. Mol. Enzym. 2000, 1478, 51–60. [Google Scholar] [CrossRef]
- Annabi, B.; Lachambre, M.-P.; Bousquet-Gagnon, N.; Pagé, M.; Gingras, D.; Béliveau, R. Green tea polyphenol (−)-epigallocatechin 3-gallate inhibits MMP-2 secretion and MT1-MMP-driven migration in glioblastoma cells. Biochim. Biophys. Acta 2002, 1542, 209–220. [Google Scholar] [CrossRef]
- Mook, O.R.F.; Frederiks, W.M.; Van Noorden, C.J.F. The role of gelatinases in colorectal cancer progression and metastasis. Biochim. Biophys. Acta 2004, 1705, 69–89. [Google Scholar] [CrossRef] [PubMed]
- Annabi, B.; Thibeault, S.; Moumdjian, R.; Béliveau, R. Hyaluronan Cell Surface Binding Is Induced by Type I Collagen and Regulated by Caveolae in Glioma Cells. J. Biol. Chem. 2004, 279, 21888–21896. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, K.N.; Murai, T.; Nishinakamura, H.; Kawashima, H.; Saya, H.; Miyasaka, M. Hyaluronan Oligosaccharides Induce CD44 Cleavage and Promote Cell Migration in CD44-expressing Tumor Cells. J. Biol. Chem. 2003, 278, 32259–32265. [Google Scholar] [CrossRef] [PubMed]
- Annabi, B.; Bouzeghrane, M.; Moumdjian, R.; Moghrabi, A.; Béliveau, R. Probing the infiltrating character of brain tumors: Inhibition of Rho, A/ROK-mediated CD44 cell surface shedding from glioma cells by the green tea catechin EGCg. J. Neurochem. 2005, 94, 906–916. [Google Scholar] [CrossRef]
- Pan, H.; Wang, H.; Zhu, L.; Mao, L.; Qiao, L.; Su, X. The Role of Nrf2 in Migration and Invasion of Human Glioma Cell U. World Neurosurg. 2013, 80, 363–370. [Google Scholar] [CrossRef]
- Dinarello, C.A. The paradox of pro-inflammatory cytokines in cancer. Cancer Metastasis Rev. 2006, 25, 307–313. [Google Scholar] [CrossRef]
- Loeffler, S.; Fayard, B.; Weis, J.; Weissenberger, J. Interleukin-6 induces transcriptional activation of vascular endothelial growth factor (VEGF) in astrocytes in vivo and regulates VEGF promoter activity in glioblastoma cells via direct interaction between STAT3 and Sp. Int. J. Cancer 2005, 115, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Park, S.S.; Park, S.K.; Lim, J.H.; Choi, Y.H.; Kim, W.J.; Moon, S.K. Esculetin inhibits cell proliferation through the Ras/ERK1/2 pathway in human colon cancer cells. Oncol. Rep. 2011, 25, 223–230. [Google Scholar] [PubMed]
- Herbener, V.J.; Burster, T.; Goreth, A.; Pruss, M.; Von Von Bandemer, H.; Baisch, T.; Fitzel, R.; Siegelin, M.D.; Karpel-Massler, G.; Debatin, K.-M.; et al. Considering the Experimental Use of Temozolomide in Glioblastoma Research. Biomedicine 2020, 8, 151. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Su, Y.S.; Wang, W.; Kardosh, A.; Liebes, L.F.; Hofman, F.M.; Schönthal, A.H.; Chen, T.C. Enhancement of glioblastoma cell killing by combination treatment with temozolomide and tamoxifen or hypericin. Neurosurg. Focus 2006, 20, E20. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Siegelin, M.; Habel, A.; Gaiser, T. Epigalocatechin-3-gallate (EGCG) downregulates PEA15 and thereby augments TRAIL-mediated apoptosis in malignant glioma. Neurosci. Lett. 2008, 448, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Lamb, H.K.; Brady, C.; Lefkove, B.; Bonner, M.Y.; Thompson, P.; Lovat, P.E.; Arbiser, J.L.; Hawkins, A.R.; Redfern, C.P.F. Inducing apoptosis of cancer cells using small-molecule plant compounds that bind to GRP. Br. J. Cancer 2013, 109, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Thomas, F.; Holly, J.M.; Persad, R.; Bahl, A.; Perks, C.M. Green Tea Extract (Epigallocatechin-3-Gallate) Reduces Efficacy of Radiotherapy on Prostate Cancer Cells. Urology 2011, 78, 475.e15–475.e15-21. [Google Scholar] [CrossRef] [PubMed]
- Waghray, D.; Zhang, Q. Inhibit or evade multidrug resistance P-glycoprotein in cancer treatment: Miniperspective. J. Med. Chem. 2017, 61, 5108–5121. [Google Scholar] [CrossRef] [PubMed]
- Ferrandon, S.; Malleval, C.; El Hamdani, B.; Battiston-Montagne, P.; I Bolbos, R.; Langlois, J.-B.; Manas, P.; Gryaznov, S.M.; Alphonse, G.; Honnorat, J.; et al. Telomerase inhibition improves tumor response to radiotherapy in a murine orthotopic model of human glioblastoma. Mol. Cancer 2015, 14, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Sadava, D.; Whitlock, E.; Kane, S.E. The green tea polyphenol, epigallocatechin-3-gallate inhibits telomerase and induces apoptosis in drug-resistant lung cancer cells. Biochem. Biophys. Res. Commun. 2007, 360, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Shervington, A.; Pawar, V.; Menon, S.; Thakkar, D.; Patel, R. The sensitization of glioma cells to cisplatin and tamoxifen by the use of catechin. Mol. Biol. Rep. 2009, 36, 1181–1186. [Google Scholar] [CrossRef]
- Lee, A.S. GRP78 Induction in Cancer: Therapeutic and Prognostic Implications. Figure Cancer Res. 2007, 67, 3496–3499. [Google Scholar] [CrossRef] [PubMed]
- Pyrko, P.; Schönthal, A.H.; Hofman, F.M.; Chen, T.C.; Lee, A.S. The unfolded protein response regulator GRP78/Bi, P as a novel target for increasing chemosensitivity in malignant gliomas. Cancer Res. 2007, 67, 9809–9816. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, R.; Devi, A.; Mishra, S. Molecular docking and molecular dynamics studies reveal structural basis of inhibition and selectivity of inhibitors EGCG and OSU-03012 toward glucose regulated protein-78 (GRP78) overexpressed in glioblas-toma. J. Mol. Model. 2015, 21, 272. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, A.; Zhai, G.G.; Zhang, M.; Malhotra, R.; E Latham, D.; A Delaney, M.; Robe, P.; Nestler, U.; Song, Q.; Loeffler, J. Survivin enhances radiation resistance in primary human glioblastoma cells via caspase-independent mechanisms. Oncogene 2004, 23, 7494–7506. [Google Scholar] [CrossRef]
- Zhai, K.; Siddiqui, M.; Abdellatif, B.; Liskova, A.; Kubatka, P.; Büsselberg, D. Natural Compounds in Glioblastoma Therapy: Preclinical Insights, Mechanistic Pathways, and Outlook. Cancers 2021, 13, 2317. [Google Scholar] [CrossRef]
- Sadgrove, N.J.; Jones, G.L. From Petri Dish to Patient: Bioavailability Estimation and Mechanism of Action for Antimicrobial and Immunomodulatory Natural Products. Front. Microbiol. 2019, 10, 2470. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.M.; Dahlin, J.L.; Bisson, J.; Graham, J.; Pauli, G.F.; Walters, M.A. The essential medicinal chemistry of curcumin: Miniperspective. J. Med. Chem. 2017, 60, 1620–1637. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, Z.; Yang, S.; Yin, T.; Zhang, Y.; Qin, Y.; Weinreb, R.N.; Sun, X. Tissue Distribution of trans-Resveratrol and Its Metabolites after Oral Administration in Human Eyes. J. Ophthalmol. 2017, 2017, 1–12. [Google Scholar]
- Krupkova, O.; Ferguson, S.J.; Wuertz-Kozak, K. Stability of (−)-epigallocatechin gallate and its activity in liquid formulations and delivery systems. J. Nutr. Biochem. 2016, 37, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Fernández, V.A.; Toledano, L.A.; Lozano, N.P.; Tapia, E.N.; Roig, M.D.G.; Fornell, R.D.L.T.; Algar Óscar, G. Bioavailability of Epigallocatechin Gallate Administered with Different Nutritional Strategies in Healthy Volunteers. Antioxidants 2020, 9, 440. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Ballesta, M.; Gil-Izquierdo, Á.; García-Viguera, C.; Domínguez-Perles, R. Nanoparticles and Controlled Delivery for Bioactive Compounds: Outlining Challenges for New “Smart-Foods” for Health. Foods 2018, 7, 72. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, Z.; Mohammadinejad, R.; Ashrafizadeh, M. Drug delivery systems for resveratrol, a non-flavonoid polyphenol: Emerging evidence in last decades. J. Drug Deliv. Sci. Technol. 2019, 51, 591–604. [Google Scholar] [CrossRef]
- Figueiró, F.; Bernardi, A.; Frozza, R.L.; Terroso, T.; Zanotto-Filho, A.; Jandrey, E.H.F.; Moreira, J.C.F.; Salbego, C.G.; Edelweiss, M.I.; Pohlmann, A.; et al. Resveratrol-Loaded Lipid-Core Nanocapsules Treatment Reduces In Vitro and In Vivo Glioma Growth. J. Biomed. Nanotechnol. 2013, 9, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Peñalva, R.; Morales, J.; González-Navarro, C.J.; Larrañeta, E.; Quincoces, G.; Peñuelas, I.; Irache, J.M. Increased Oral Bioavailability of Resveratrol by Its Encapsulation in Casein Nanoparticles. Int. J. Mol. Sci. 2018, 19, 2816. [Google Scholar] [CrossRef] [PubMed]
- Dende, C.; Meena, J.; Nagarajan, P.; Nagaraj, V.A.; Panda, A.K.; Padmanaban, G. Nanocurcumin is superior to native curcumin in preventing degenerative changes in Experimental Cerebral Malaria. Sci. Rep. 2017, 7, 10062. [Google Scholar] [CrossRef]
- Wang, Y.; Ying, X.; Xu, H.; Yan, H.; Li, X.; Tang, H. The functional curcumin liposomes induce apoptosis in C6 glioblastoma cells and C6 glioblastoma stem cells in vitro and in animals. Int. J. Nanomed. 2017, 12, 1369–1384. [Google Scholar] [CrossRef]
- Zhang, Q.; Polyakov, N.E.; Chistyachenko, Y.S.; Khvostov, M.; Frolova, T.S.; Tolstikova, T.G.; Dushkin, A.V.; Su, W. Preparation of curcumin self-micelle solid dispersion with enhanced bioavailability and cytotoxic activity by mechanochemistry. Drug Deliv. 2018, 25, 198–209. [Google Scholar] [CrossRef]
- Hu, B.; Ting, Y.; Zeng, X.; Huang, Q. Cellular uptake and cytotoxicity of chitosan–caseinophosphopeptides nanocomplexes loaded with epigallocatechin gallate. Carbohydr. Polym. 2012, 89, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Lu, C.; Leszek, J.; Zhang, J. Design and Development of Nanomaterial-Based Drug Carriers to Overcome the Blood–Brain Barrier by Using Different Transport Mechanisms. Int. J. Mol. Sci. 2021, 22, 10118. [Google Scholar] [CrossRef] [PubMed]
- Gosselet, F.; Loiola, R.A.; Roig, A.; Rosell, A.; Culot, M. Central nervous system delivery of molecules across the blood-brain barrier. Neurochem. Int. 2021, 144, 104952. [Google Scholar] [CrossRef] [PubMed]
- Persano, F.; Leporatti, S. Current Overview of Inorganic Nanoparticles for the Treatment of Central Nervous System (CNS) Diseases. Curr. Nanomater. 2020, 5, 92–110. [Google Scholar] [CrossRef]
- Georgieva, J.V.; Hoekstra, D.; Zuhorn, I.S. Smuggling Drugs into the Brain: An Overview of Ligands Targeting Transcytosis for Drug Delivery across the Blood–Brain Barrier. Pharmaceutics 2014, 6, 557–583. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, T.; Maishu, S.P.; Akter, R.; Rahman, H.; Akhtar, M.F.; Saleem, A.; Bin-Jumah, M.; Kamel, M.; Abdel-Latif, M.A.; Abdel-Daim, M.M. A Review on Natural Sources Derived Protein Nanoparticles as Anticancer Agents. Curr. Top. Med. Chem. 2021, 21, 1014–1026. [Google Scholar] [CrossRef] [PubMed]
- Dützmann, S.; Schiborr, C.; Kocher, A.; Pilatus, U.; Hattingen, E.; Weissenberger, J.; Geßler, F.; Quick-Weller, J.; Franz, K.; Seifert, V.; et al. Intratumoral Concentrations and Effects of Orally Administered Micellar Curcuminoids in Glioblastoma Patients. Nutr. Cancer 2016, 68, 943–948. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups of Natural Compounds | Natural Compound | Cell Lines/Model | Effect | Anti-Cancer Mechanism Proposed | Reference |
|---|---|---|---|---|---|
| Curcumin | U87MG, U373, T67, T98G, and C6 cell lines | inhibition of cell survival | suppression of NF-κB and inhibition of the AP-1 signaling pathway | [66,67,68] | |
| Curcumin | Tu-2449, Tu-9648, and Tu-251 glioma cell lines | inhibition of invasiveness | inhibition of the JAK/STAT3 pathway | [71] | |
| Curcumin | human primary (A-172, MZ-18) and recurrent glioblastoma lines (MZ-54, MZ-256, MZ-304) | inhibition of cell proliferation | decrease in intracellular STAT3 levels | [72] | |
| Curcumin | U251 glioma cells | induction of cell cycle arrest in G2/M | increased p53 protein levels | [77] | |
| Curcumin | U87MG cells | promoting cell cycle arrest | downregulation of cyclin D1 with upregulation of p21 | [79] | |
| Curcuminoids | Curcumin | U251 | induction of cell cycle arrest in G2/M | increased expression of the DAPK1 protein | [82] |
| Curcumin | U87-MG and U373-MG cell lines | promoting of cell cycle arrest in G2/M | promotion of mTOR dependent ATG | [88] | |
| Curcumin | Rat F98 and mouse GL261 | induction of ATG | activation of the mTOR-dependent ATG pathway | [89] | |
| Curcumin | C6 and U251MG cell lines | induction of arrest in G2/M and autophagy | inhibition of constitutive activation of the PI3K/Akt/mTOR pathway | [91] | |
| Curcumin | GSCs | suppression of stem-like features with stimulation of ATG-dependent differentiation of GSCs | induction of mTOR-dependent ATG | [99] | |
| Curcumin | U51, U87, and U235 cell lines | induction of apoptosis | STAT3 inhibition | [109] | |
| Curcumin | U87MG and T98G cell lines | improved cytotoxic and apoptotic promoting action of TMZ and etoposide | downregulation of mRNA encoding p53 and upregulation of BAX-Bcl2 | [112] | |
| Curcumin | U373 cell line | inhibition of invasiveness | reduction in the expression of MMP-2, 9, 14, 15, 16, 17, 24, and 25 | [110] | |
| Curcumin | U251 and LN229 cell lines | reduced distance of invasion, migration and proliferation | inhibition of HDGF | [121] | |
| RES | U87 cell line | induction of S-G2/M cell cycle arrest | reduction in cyclin D1 | [141] | |
| RES | Daoy, UW228-2, and UW228-3 medulloblastoma cell lines | cell growth suppression | STAT3 downregulation | [149] | |
| RES | RT-2 glioma cell line | suppression of angiogenesis | inhibition of VEGF expression | [152] | |
| RES | U373MG human glioma cell | reduction in cellular invasiveness | suppression of activation of the NF-κB factor | [154] | |
| RES | U251, U87, and C6 cell lines | induction of apoptosis | induction of caspase-3 activation | [157,158] | |
| RES | U87MG cell line | induction of apoptosis and inhibition of cell growth | increased expression of TTP | [165] | |
| RES | SHG44 cell line | enhanced the antiproliferative effects of TMZ | activation of AMPK, inhibition of mTOR signaling, and downregulation of the antiapoptotic protein Bcl-2 | [174] | |
| RES | T98G cell line | improved efficacy of TMZ therapy | reduced expression of the MGMT protein with suppression of the activation of the transcription factor NF-kB | [180] | |
| Flavonoids | EGCG | Spheroids of A172 cells | suppression of spheroid formation | inhibition of phosphorylation of PDGF-BB tyrosine residues | [194] |
| EGCG | U87MG cell line | induction of apoptosis | reduced levels of Bcl-2 and phosphorylated Akt and increased levels of BAX, activated caspases and increased ROS | [204] | |
| EGCG | U87 cell line | reduction in the invasiveness of glioma cells | inhibition of MMP2 | [208] | |
| EGCG | U87 cell line | inhibition of the invasiveness | reduction in levels of IL-6, IL-8, CCL5, and MCP-1 | [214] | |
| EGCG | 1321N1 and U87-MG cell lines | improved cytotoxic effect of cisplatin and tamoxifen | suppression of telomerase | [226] | |
| EGCG | U87 and A172 cell lines | promotion of TRAIL-mediated apoptosis | reduction PEA15 levels | [220] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Persano, F.; Gigli, G.; Leporatti, S. Natural Compounds as Promising Adjuvant Agents in The Treatment of Gliomas. Int. J. Mol. Sci. 2022, 23, 3360. https://doi.org/10.3390/ijms23063360
Persano F, Gigli G, Leporatti S. Natural Compounds as Promising Adjuvant Agents in The Treatment of Gliomas. International Journal of Molecular Sciences. 2022; 23(6):3360. https://doi.org/10.3390/ijms23063360
Chicago/Turabian StylePersano, Francesca, Giuseppe Gigli, and Stefano Leporatti. 2022. "Natural Compounds as Promising Adjuvant Agents in The Treatment of Gliomas" International Journal of Molecular Sciences 23, no. 6: 3360. https://doi.org/10.3390/ijms23063360
APA StylePersano, F., Gigli, G., & Leporatti, S. (2022). Natural Compounds as Promising Adjuvant Agents in The Treatment of Gliomas. International Journal of Molecular Sciences, 23(6), 3360. https://doi.org/10.3390/ijms23063360