
Mitochondria as the Target of Hepatotoxicity and Drug-Induced Liver Injury: Molecular Mechanisms and Detection Methods



Abstract
1. Introduction
2. Mitochondria and the Liver
2.1. Mitochondrial Functions
2.2. Liver Metabolic Functions and Mitochondrial Activity in Hepatocytes
2.2.1. Gluconeogenesis
2.2.2. De Novo Lipogenesis
2.2.3. Urea Cycle
2.2.4. Lipolysis
2.2.5. One-Carbon Metabolism
2.3. Oxidative Phosphorylation
2.4. Mitochondrial Permeability Transition Pore/Mitochondrial Outer Membrane Polarization and Cell Death
3. Mechanisms of Liver Injury
3.1. Cholestatic Injury
3.2. Hepatocellular Injury
3.2.1. Direct Hepatotoxicity
3.2.2. Immunological Hepatotoxicity
3.2.3. Metabolism-Related Hepatotoxicity
3.2.4. Mitochondria-Mediated Hepatotoxicity
4. Drug-Induced Mitochondrial Dysfunction and Liver Injury
4.1. Mitochondrial Permeability Transition Pore Opening
4.2. Alterations of Oxidative Phosphorylation and Electron Transport Chain
4.3. Alterations of Mitochondrial Fatty Acids β-Oxidation
5. Factors Influencing Drug-Induced Hepatic Mitochondrial Dysfunction
6. Experimental Models and Methods to Study Hepatic Mitochondrial Toxicity
6.1. Experimental Systems and Models to Study Mitochondrial Dysfunction and Related Hepatotoxicity
6.2. Experimental Methods and Assays to Study Mitochondrial Dysfunction and Related Hepatotoxicity
7. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Δψm | mitochondrial membrane potential |
| ADP | adenosine diphosphate |
| ALP | alkaline phosphatase |
| ALT (ALAT) | alanine aminotransferase |
| AOP | adverse outcome pathway |
| APAF1 | apoptotic peptidase activating factor 1 |
| AST (ASAT) | aspartate aminotransferase |
| ATP | adenosine triphosphate |
| Bcl-2 | B cell lymphoma 2 |
| BODIPYTM 493/503 | 4,4-Difluoro-1,3,5,7,8-Pentamethyl-4-Bora-3a,4a-Diaza-s-Indacene |
| BRET | bioluminescence energy transfer |
| ChREBP | carbohydrate-response element binding protein |
| CO2 | carbon dioxide |
| CoA | coenzyme A |
| CPT1 | carnitine palmitoyltransferase 1 |
| CYP | cytochrome P450 |
| DAMP | danger-associated molecular pattern |
| DCF | dichlorofluorescein |
| DILI | drug-induced liver injury |
| DiOC6(3) | 3,3′-dihexyloxacarbocyanine iodide |
| ETC | electron transport chain |
| FADH2 | flavin adenine dinucleotide |
| FDA | Food and Drug Administration |
| FRET | Förster resonance energy transfer |
| GGT | gamma-glutamyl transpeptidase |
| GPx | glutathione peroxidase |
| GSH | glutathione |
| GSTM1 | Glutathione S-Transferase Mu 1 |
| GSTT1 | Glutathione S-Transferase Theta 1 |
| H2O2 | hydrogen peroxide |
| H2S | hydrogen disulfide |
| HIF-1 | hypoxia-inducible factor-1 |
| HO· | hydroxyl radical |
| HPLC | high-performance liquid chromatography |
| IMI | Innovative Medicines Initiative |
| JC-1 | 1,1′,3,3′-tetraethyl-5,5′,6,6′-tetrachloroimidacarbocyanine iodide |
| JNK | c-Jun N terminal protein kinase |
| LC-MS | liquid chromatography-mass spectrometry |
| LDH | lactate dehydrogenase |
| MDR | multidrug resistance |
| MnSOD | manganese superoxide dismutase |
| MOMP | mitochondrial outer membrane polarization |
| MPTP | mitochondrial permeability transition pore |
| mtDNA | mitochondrial DNA |
| MTHFD1/2/1L | methylenetetrahydrofolate dehydrogenase 1/2/1L |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| NADH, NAD+ | nicotinamide adenine dinucleotide |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NAFLD | non-alcoholic fatty liver disease |
| NMR | nuclear magnetic resonance |
| NRTIs | nucleotide reverse transcriptase inhibitors |
| NSAID | nonsteroidal anti-inflammatory drug |
| O2− | superoxide anion |
| OCR | oxygen consumption rate |
| OXPHOS | oxidative phosphorylation |
| PBR | peripheral benzodiazepine receptor |
| PCR | polymerase chain reaction |
| PPARα | proliferator-activated receptor alpha |
| Q | ubiquinone |
| Rh123 | rhodamine-123 |
| ROS | reactive oxygen species |
| SREBP-1c | sterol regulatory element-binding protein 1c |
| TCA | tricarboxylic acid |
| TMRE | tetramethyl rhodamine ethyl ester |
| TMRM | tetramethyl rhodamine methyl ester |
| TNF-α | tumor necrosis factor α |
| TNFR1 | tumor necrosis factor receptor 1 |
| TPP | tetraphenylphosphonium |
| TRAIL | tumor necrosis factor-related apoptosis-inducing ligand |
| XIAP | X-linked inhibitor of apoptosis protein |
References
- Weaver, R.J.; Blomme, E.A.; Chadwick, A.E.; Copple, I.M.; Gerets, H.H.J.; Goldring, C.E.; Guillouzo, A.; Hewitt, P.G.; Ingelman-Sundberg, M.; Jensen, K.G.; et al. Managing the Challenge of Drug-Induced Liver Injury: A Roadmap for the Development and Deployment of Preclinical Predictive Models. Nat. Rev. Drug Discov. 2020, 19, 131–148. [Google Scholar] [CrossRef] [PubMed]
- Larrey, D. Drug-Induced Liver Diseases. J. Hepatol. 2000, 32, 77–88. [Google Scholar] [CrossRef]
- Chen, M.; Vijay, V.; Shi, Q.; Liu, Z.; Fang, H.; Tong, W. FDA-Approved Drug Labeling for the Study of Drug-Induced Liver Injury. Drug Discov. Today 2011, 16, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Björnsson, E. The Natural History of Drug-Induced Liver Injury. Semin. Liver Dis. 2009, 29, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Pessayre, D.; Mansouri, A.; Berson, A.; Fromenty, B. Mitochondrial Involvement in Drug-Induced Liver Injury. Handb. Exp. Pharmacol. 2010, 11, 311–365. [Google Scholar] [CrossRef]
- Labbe, G.; Pessayre, D.; Fromenty, B. Drug-Induced Liver Injury through Mitochondrial Dysfunction: Mechanisms and Detection during Preclinical Safety Studies. Fundam. Clin. Pharmacol. 2008, 22, 335–353. [Google Scholar] [CrossRef]
- Masubuchi, Y.; Suda, C.; Horie, T. Involvement of Mitochondrial Permeability Transition in Acetaminophen-Induced Liver Injury in Mice. J. Hepatol. 2005, 42, 110–116. [Google Scholar] [CrossRef]
- Begriche, K.; Massart, J.; Robin, M.-A.; Borgne-Sanchez, A.; Fromenty, B. Drug-Induced Toxicity on Mitochondria and Lipid Metabolism: Mechanistic Diversity and Deleterious Consequences for the Liver. J. Hepatol. 2011, 54, 773–794. [Google Scholar] [CrossRef]
- Yang, D.; Oyaizu, Y.; Oyaizu, H.; Olsen, G.J.; Woese, C.R. Mitochondrial Origins. Proc. Natl. Acad. Sci. USA 1985, 82, 4443–4447. [Google Scholar] [CrossRef]
- Benard, G.; Bellance, N.; James, D.; Parrone, P.; Fernandez, H.; Letellier, T.; Rossignol, R. Mitochondrial Bioenergetics and Structural Network Organization. J. Cell Sci. 2007, 120, 838–848. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Haigis, M.C. The Multifaceted Contributions of Mitochondria to Cellular Metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Bock, F.J.; Tait, S.W.G. Mitochondria as Multifaceted Regulators of Cell Death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and Molecular Mechanisms of Mitochondrial Function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Ahn, C.S.; Metallo, C.M. Mitochondria as Biosynthetic Factories for Cancer Proliferation. Cancer Metab. 2015, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In Sickness and in Health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef]
- Trefts, E.; Gannon, M.; Wasserman, D.H. The Liver. Curr. Biol. 2017, 27, R1147–R1151. [Google Scholar] [CrossRef]
- Rui, L. Energy Metabolism in the Liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar] [CrossRef]
- She, P.; Shiota, M.; Shelton, K.D.; Chalkley, R.; Postic, C.; Magnuson, M.A. Phosphoenolpyruvate Carboxykinase Is Necessary for the Integration of Hepatic Energy Metabolism. Mol. Cell. Biol. 2000, 20, 6508–6517. [Google Scholar] [CrossRef]
- Morio, B.; Panthu, B.; Bassot, A.; Rieusset, J. Role of Mitochondria in Liver Metabolic Health and Diseases. Cell Calcium 2021, 94, 102336. [Google Scholar] [CrossRef]
- Postic, C.; Girard, J. The Role of the Lipogenic Pathway in the Development of Hepatic Steatosis. Diabetes Metab. 2008, 34, 643–648. [Google Scholar] [CrossRef]
- Engelking, L.R. Urea Cycle (Krebs-Henseleit Ornithine Cycle). In Textbook of Veterinary Physiological Chemistry; Elsevier: Amsterdam, The Netherlands, 2015; pp. 58–64. ISBN 978-0-12-391909-0. [Google Scholar]
- Poirier, Y.; Antonenkov, V.D.; Glumoff, T.; Hiltunen, J.K. Peroxisomal Beta-Oxidation—A Metabolic Pathway with Multiple Functions. Biochim. Biophys. Acta 2006, 1763, 1413–1426. [Google Scholar] [CrossRef] [PubMed]
- Veldhorst, M.A.; Westerterp-Plantenga, M.S.; Westerterp, K.R. Gluconeogenesis and Energy Expenditure after a High-Protein, Carbohydrate-Free Diet. Am. J. Clin. Nutr. 2009, 90, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, M.V.; Lodhi, I.J.; Yin, L.; Malapaka, R.R.V.; Xu, H.E.; Turk, J.; Semenkovich, C.F. Identification of a Physiologically Relevant Endogenous Ligand for PPARα in Liver. Cell 2009, 138, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S.; Seydoux, J.; Peters, J.M.; Gonzalez, F.J.; Desvergne, B.; Wahli, W. Peroxisome Proliferator-Activated Receptor Alpha Mediates the Adaptive Response to Fasting. J. Clin. Investig. 1999, 103, 1489–1498. [Google Scholar] [CrossRef]
- Tibbetts, A.S.; Appling, D.R. Compartmentalization of Mammalian Folate-Mediated One-Carbon Metabolism. Annu. Rev. Nutr. 2010, 30, 57–81. [Google Scholar] [CrossRef]
- Clare, C.E.; Brassington, A.H.; Kwong, W.Y.; Sinclair, K.D. One-Carbon Metabolism: Linking Nutritional Biochemistry to Epigenetic Programming of Long-Term Development. Annu. Rev. Anim. Biosci. 2019, 7, 263–287. [Google Scholar] [CrossRef]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial Electron Transport Chain: Oxidative Phosphorylation, Oxidant Production, and Methods of Measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef]
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial Electron Transport Chain, ROS Generation and Uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Liu, Y.; Fiskum, G.; Schubert, D. Generation of Reactive Oxygen Species by the Mitochondrial Electron Transport Chain. J. Neurochem. 2002, 80, 780–787. [Google Scholar] [CrossRef]
- Murphy, M.P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- D’Autréaux, B.; Toledano, M.B. ROS as Signalling Molecules: Mechanisms That Generate Specificity in ROS Homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Bienert, G.P.; Schjoerring, J.K.; Jahn, T.P. Membrane Transport of Hydrogen Peroxide. Biochim. Biophys. Acta BBA Biomembr. 2006, 1758, 994–1003. [Google Scholar] [CrossRef]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial Reactive Oxygen Species Trigger Hypoxia-Induced Transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef]
- Wang, C.; Youle, R.J. The Role of Mitochondria in Apoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [PubMed]
- Rankin, E.B.; Rha, J.; Selak, M.A.; Unger, T.L.; Keith, B.; Liu, Q.; Haase, V.H. Hypoxia-Inducible Factor 2 Regulates Hepatic Lipid Metabolism. Mol. Cell. Biol. 2009, 29, 4527–4538. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.-O.; Welch, T.P.; Gonzalez, F.J.; Copple, B.L. Reduced Liver Fibrosis in Hypoxia-Inducible Factor-1α-Deficient Mice. Am. J. Physiol.-Gastrointest. Liver Physiol. 2009, 296, G582–G592. [Google Scholar] [CrossRef]
- Li, S.; Yao, D.; Wang, L.; Wu, W.; Qiu, L.Q.; Yao, M.; Yao, N.; Zhang, H.; Yu, D.; Ni, Q. Expression Characteristics of HIF-1α and Its Clinical Values in Diagnosis and Prognosis of Hepatocellular Carcinoma. Hepat. Mon. 2011, 11, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Brookes, P.S. Mitochondrial H+ Leak and ROS Generation: An Odd Couple. Free Radic. Biol. Med. 2005, 38, 12–23. [Google Scholar] [CrossRef]
- Sanz, A.; Caro, P.; Ayala, V.; Portero-Otin, M.; Pamplona, R.; Barja, G. Methionine Restriction Decreases Mitochondrial Oxygen Radical Generation and Leak as Well as Oxidative Damage to Mitochondrial DNA and Proteins. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2006, 20, 1064–1073. [Google Scholar] [CrossRef] [PubMed]
- Velsor, L.W.; Kovacevic, M.; Goldstein, M.; Leitner, H.M.; Lewis, W.; Day, B.J. Mitochondrial Oxidative Stress in Human Hepatoma Cells Exposed to Stavudine. Toxicol. Appl. Pharmacol. 2004, 199, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Krauskopf, A.; Basso, E.; Petronilli, V.; Blachly-Dyson, E.; Blalchy-Dyson, E.; Di Lisa, F.; Forte, M.A. The Mitochondrial Permeability Transition from in Vitro Artifact to Disease Target. FEBS J. 2006, 273, 2077–2099. [Google Scholar] [CrossRef]
- Malhi, H.; Gores, G.J.; Lemasters, J.J. Apoptosis and Necrosis in the Liver: A Tale of Two Deaths? Hepatology 2006, 43, S31–S44. [Google Scholar] [CrossRef]
- Dorstyn, L.; Akey, C.W.; Kumar, S. New Insights into Apoptosome Structure and Function. Cell Death Differ. 2018, 25, 1194–1208. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.C.; Zong, W.-X.; Cheng, E.H.-Y.; Lindsten, T.; Panoutsakopoulou, V.; Ross, A.J.; Roth, K.A.; MacGregor, G.R.; Thompson, C.B.; Korsmeyer, S.J. Proapoptotic BAX and BAK: A Requisite Gateway to Mitochondrial Dysfunction and Death. Science 2001, 292, 727–730. [Google Scholar] [CrossRef]
- Salvador-Gallego, R.; Mund, M.; Cosentino, K.; Schneider, J.; Unsay, J.; Schraermeyer, U.; Engelhardt, J.; Ries, J.; García-Sáez, A.J. Bax Assembly into Rings and Arcs in Apoptotic Mitochondria Is Linked to Membrane Pores. EMBO J. 2016, 35, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Riley, J.S.; Tait, S.W. Mitochondrial DNA in Inflammation and Immunity. EMBO Rep. 2020, 21, e49799. [Google Scholar] [CrossRef]
- Neuman, M.G. Hepatotoxicity: Mechanisms of Liver Injury. In Liver Diseases; Radu-Ionita, F., Pyrsopoulos, N.T., Jinga, M., Tintoiu, I.C., Sun, Z., Bontas, E., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 75–84. ISBN 978-3-030-24431-6. [Google Scholar]
- Tarantino, G.; Di Minno, M.N.D.; Capone, D. Drug-Induced Liver Injury: Is It Somehow Foreseeable? World J. Gastroenterol. 2009, 15, 2817–2833. [Google Scholar] [CrossRef]
- Lee, W.M.; Senior, J.R. Recognizing Drug-Induced Liver Injury: Current Problems, Possible Solutions. Toxicol. Pathol. 2005, 33, 155–164. [Google Scholar] [CrossRef]
- Pauli-Magnus, C.; Meier, P.J. Hepatobiliary Transporters and Drug-Induced Cholestasis. Hepatology 2006, 44, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Malhi, H.; Gores, G.J. Cellular and Molecular Mechanisms of Liver Injury. Gastroenterology 2008, 134, 1641–1654. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; Gores, G.J.; Cederbaum, A.I.; Hinson, J.A.; Pessayre, D.; Lemasters, J.J. Mechanisms of Hepatotoxicity. Toxicol. Sci. Off. J. Soc. Toxicol. 2002, 65, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Woolbright, B.L.; Jaeschke, H. Novel Insight into Mechanisms of Cholestatic Liver Injury. World J. Gastroenterol. 2012, 18, 4985–4993. [Google Scholar] [CrossRef]
- Guicciardi, M.E.; Gores, G.J. Cholestatic Hepatocellular Injury: What Do We Know and How Should We Proceed. J. Hepatol. 2005, 42, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Nelson, L.J.; Gómez Del Moral, M.; Martínez-Naves, E.; Cubero, F.J. Dissecting the Molecular Pathophysiology of Drug-Induced Liver Injury. World J. Gastroenterol. 2018, 24, 1373–1385. [Google Scholar] [CrossRef] [PubMed]
- Faouzi, S.; Burckhardt, B.E.; Hanson, J.C.; Campe, C.B.; Schrum, L.W.; Rippe, R.A.; Maher, J.J. Anti-Fas Induces Hepatic Chemokines and Promotes Inflammation by an NF-Kappa B-Independent, Caspase-3-Dependent Pathway. J. Biol. Chem. 2001, 276, 49077–49082. [Google Scholar] [CrossRef]
- Uetrecht, J. Idiosyncratic Drug Reactions: Past, Present, and Future. Chem. Res. Toxicol. 2008, 21, 84–92. [Google Scholar] [CrossRef]
- Cho, T.; Uetrecht, J. How Reactive Metabolites Induce an Immune Response That Sometimes Leads to an Idiosyncratic Drug Reaction. Chem. Res. Toxicol. 2017, 30, 295–314. [Google Scholar] [CrossRef]
- Naisbitt, D.J.; Farrell, J.; Wong, G.; Depta, J.P.H.; Dodd, C.C.; Hopkins, J.E.; Gibney, C.A.; Chadwick, D.W.; Pichler, W.J.; Pirmohamed, M.; et al. Characterization of Drug-Specific T Cells in Lamotrigine Hypersensitivity. J. Allergy Clin. Immunol. 2003, 111, 1393–1403. [Google Scholar] [CrossRef]
- Pichler, W.J.; Adam, J.; Watkins, S.; Wuillemin, N.; Yun, J.; Yerly, D. Drug Hypersensitivity: How Drugs Stimulate T Cells via Pharmacological Interaction with Immune Receptors. Int. Arch. Allergy Immunol. 2015, 168, 13–24. [Google Scholar] [CrossRef]
- McDonnell, A.M.; Dang, C.H. Basic Review of the Cytochrome P450 System. J. Adv. Pract. Oncol. 2013, 4, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Weber, L.W.D.; Boll, M.; Stampfl, A. Hepatotoxicity and Mechanism of Action of Haloalkanes: Carbon Tetrachloride as a Toxicological Model. Crit. Rev. Toxicol. 2003, 33, 105–136. [Google Scholar] [CrossRef] [PubMed]
- Jeong, D.H.; Lee, S.J.; Lee, J.H.; Bae, I.H.; Jeong, K.S.; Jang, J.J.; Lim, I.K.; Kim, M.R.; Lee, M.J.; Lee, Y.S. Subcellular Redistribution of Protein Kinase C Isozymes Is Associated with Rat Liver Cirrhotic Changes Induced by Carbon Tetrachloride or Thioacetamide. J. Gastroenterol. Hepatol. 2001, 16, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P.; Lemasters, J.J.; Han, D.; Boelsterli, U.A.; Kaplowitz, N. Mechanisms of Pathogenesis in Drug Hepatotoxicity Putting the Stress on Mitochondria. Mol. Interv. 2010, 10, 98–111. [Google Scholar] [CrossRef]
- Lucena, M.I.; García-Martín, E.; Andrade, R.J.; Martínez, C.; Stephens, C.; Ruiz, J.D.; Ulzurrun, E.; Fernandez, M.C.; Romero-Gomez, M.; Castiella, A.; et al. Mitochondrial Superoxide Dismutase and Glutathione Peroxidase in Idiosyncratic Drug-Induced Liver Injury. Hepatology 2010, 52, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Fromenty, B.; Pessayre, D. Inhibition of Mitochondrial Beta-Oxidation as a Mechanism of Hepatotoxicity. Pharmacol. Ther. 1995, 67, 101–154. [Google Scholar] [CrossRef]
- Li, S.; Guo, J.; Ying, Z.; Chen, S.; Yang, L.; Chen, K.; Long, Q.; Qin, D.; Pei, D.; Liu, X. Valproic Acid-Induced Hepatotoxicity in Alpers Syndrome Is Associated with Mitochondrial Permeability Transition Pore Opening-Dependent Apoptotic Sensitivity in an Induced Pluripotent Stem Cell Model. Hepatology 2015, 61, 1730–1739. [Google Scholar] [CrossRef]
- Aires, C.C.P.; Ijlst, L.; Stet, F.; Prip-Buus, C.; de Almeida, I.T.; Duran, M.; Wanders, R.J.A.; Silva, M.F.B. Inhibition of Hepatic Carnitine Palmitoyl-Transferase I (CPT IA) by Valproyl-CoA as a Possible Mechanism of Valproate-Induced Steatosis. Biochem. Pharmacol. 2010, 79, 792–799. [Google Scholar] [CrossRef]
- Silva, M.F.B.; Aires, C.C.P.; Luis, P.B.M.; Ruiter, J.P.N.; IJlst, L.; Duran, M.; Wanders, R.J.A.; Tavares de Almeida, I. Valproic Acid Metabolism and Its Effects on Mitochondrial Fatty Acid Oxidation: A Review. J. Inherit. Metab. Dis. 2008, 31, 205–216. [Google Scholar] [CrossRef]
- Jafarian, I.; Eskandari, M.R.; Mashayekhi, V.; Ahadpour, M.; Hosseini, M.-J. Toxicity of Valproic Acid in Isolated Rat Liver Mitochondria. Toxicol. Mech. Methods 2013, 23, 617–623. [Google Scholar] [CrossRef]
- Berson, A.; Descatoire, V.; Sutton, A.; Fau, D.; Maulny, B.; Vadrot, N.; Feldmann, G.; Berthon, B.; Tordjmann, T.; Pessayre, D. Toxicity of Alpidem, a Peripheral Benzodiazepine Receptor Ligand, but Not Zolpidem, in Rat Hepatocytes: Role of Mitochondrial Permeability Transition and Metabolic Activation. J. Pharmacol. Exp. Ther. 2001, 299, 793–800. [Google Scholar]
- Masubuchi, Y.; Nakayama, S.; Horie, T. Role of Mitochondrial Permeability Transition in Diclofenac-Induced Hepatocyte Injury in Rats. Hepatology 2002, 35, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Gáll, J.; Skrha, J.; Buchal, R.; Sedláčková, E.; Verébová, K.; Pláteník, J. Induction of the Mitochondrial Permeability Transition (MPT) by Micromolar Iron: Liberation of Calcium Is More Important than NAD(P)H Oxidation. Biochim. Biophys. Acta 2012, 1817, 1537–1549. [Google Scholar] [CrossRef]
- Hu, J.; Kholmukhamedov, A.; Lindsey, C.C.; Beeson, C.C.; Jaeschke, H.; Lemasters, J.J. Translocation of Iron from Lysosomes to Mitochondria during Acetaminophen-Induced Hepatocellular Injury: Protection by Starch-Desferal and Minocycline. Free Radic. Biol. Med. 2016, 97, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Ramshesh, V.K.; McGill, M.R.; Jaeschke, H.; Lemasters, J.J. Low Dose Acetaminophen Induces Reversible Mitochondrial Dysfunction Associated with Transient C-Jun N-Terminal Kinase Activation in Mouse Liver. Toxicol. Sci. Off. J. Soc. Toxicol. 2016, 150, 204–215. [Google Scholar] [CrossRef]
- Du, K.; Xie, Y.; McGill, M.R.; Jaeschke, H. Pathophysiological Significance of C-Jun N-Terminal Kinase in Acetaminophen Hepatotoxicity. Expert Opin. Drug Metab. Toxicol. 2015, 11, 1769–1779. [Google Scholar] [CrossRef]
- Ikeyama, Y.; Sato, T.; Takemura, A.; Sekine, S.; Ito, K. Hypoxia/Reoxygenation Exacerbates Drug-Induced Cytotoxicity by Opening Mitochondrial Permeability Transition Pore: Possible Application for Toxicity Screening. Toxicol. In Vitro Int. J. Publ. Assoc. BIBRA 2020, 67, 104889. [Google Scholar] [CrossRef]
- Balakirev, M.Y.; Zimmer, G. Mitochondrial Injury by Disulfiram: Two Different Mechanisms of the Mitochondrial Permeability Transition. Chem. Biol. Interact. 2001, 138, 299–311. [Google Scholar] [CrossRef]
- Berson, A.; Cazanave, S.; Descatoire, V.; Tinel, M.; Grodet, A.; Wolf, C.; Feldmann, G.; Pessayre, D. The Anti-Inflammatory Drug, Nimesulide (4-Nitro-2-Phenoxymethane-Sulfoanilide), Uncouples Mitochondria and Induces Mitochondrial Permeability Transition in Human Hepatoma Cells: Protection by Albumin. J. Pharmacol. Exp. Ther. 2006, 318, 444–454. [Google Scholar] [CrossRef]
- Rocha-Rodrigues, S.; Santos-Alves, E.; Coxito, P.M.; Marques-Aleixo, I.; Passos, E.; Guimarães, J.T.; Martins, M.J.; Oliveira, P.J.; Magalhães, J.; Ascensão, A. Combined Effects of Aging and in Vitro Non-Steroid Anti-Inflammatory Drugs on Kidney and Liver Mitochondrial Physiology. Life Sci. 2013, 93, 329–337. [Google Scholar] [CrossRef]
- Suzuki, Y.; Inoue, T.; Ra, C. NSAIDs, Mitochondria and Calcium Signaling: Special Focus on Aspirin/Salicylates. Pharmaceuticals 2010, 3, 1594–1613. [Google Scholar] [CrossRef] [PubMed]
- Bae, M.-A.; Song, B.J. Critical Role of C-Jun N-Terminal Protein Kinase Activation in Troglitazone-Induced Apoptosis of Human HepG2 Hepatoma Cells. Mol. Pharmacol. 2003, 63, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Masubuchi, Y.; Kano, S.; Horie, T. Mitochondrial Permeability Transition as a Potential Determinant of Hepatotoxicity of Antidiabetic Thiazolidinediones. Toxicology 2006, 222, 233–239. [Google Scholar] [CrossRef]
- Sato, T.; Segawa, M.; Sekine, S.; Ito, K. Mild Depolarization Is Involved in Troglitazone-Induced Liver Mitochondrial Membrane Permeability Transition via Mitochondrial IPLA2 Activation. J. Toxicol. Sci. 2019, 44, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Terada, H. Uncouplers of Oxidative Phosphorylation. Environ. Health Perspect. 1990, 87, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Sánchez, R.; Bravo, C.; Vásquez, C.; Ayala, G.; Silveira, L.H.; Martínez-Lavín, M. Inhibition and Uncoupling of Oxidative Phosphorylation by Nonsteroidal Anti-Inflammatory Drugs: Study in Mitochondria, Submitochondrial Particles, Cells, and Whole Heart. Biochem. Pharmacol. 1999, 57, 743–752. [Google Scholar] [CrossRef]
- Fromenty, B. Inhibition of Mitochondrial Fatty Acid Oxidation in Drug-Induced Hepatic Steatosis. Liver Res. 2019, 3, 157–169. [Google Scholar] [CrossRef]
- Spaniol, M.; Bracher, R.; Ha, H.R.; Follath, F.; Krähenbühl, S. Toxicity of Amiodarone and Amiodarone Analogues on Isolated Rat Liver Mitochondria. J. Hepatol. 2001, 35, 628–636. [Google Scholar] [CrossRef]
- Berson, A.; Fau, D.; Fornacciari, R.; Degove-Goddard, P.; Sutton, A.; Descatoire, V.; Haouzi, D.; Lettéron, P.; Moreau, A.; Feldmann, G.; et al. Mechanisms for Experimental Buprenorphine Hepatotoxicity: Major Role of Mitochondrial Dysfunction versus Metabolic Activation. J. Hepatol. 2001, 34, 261–269. [Google Scholar] [CrossRef]
- Berson, A.; Schmets, L.; Fisch, C.; Fau, D.; Wolf, C.; Fromenty, B.; Deschamps, D.; Pessayre, D. Inhibition by Nilutamide of the Mitochondrial Respiratory Chain and ATP Formation. Possible Contribution to the Adverse Effects of This Antiandrogen. J. Pharmacol. Exp. Ther. 1994, 270, 167–176. [Google Scholar]
- Burcham, P.C.; Harman, A.W. Acetaminophen Toxicity Results in Site-Specific Mitochondrial Damage in Isolated Mouse Hepatocytes. J. Biol. Chem. 1991, 266, 5049–5054. [Google Scholar] [CrossRef]
- Lee, K.K.; Imaizumi, N.; Chamberland, S.R.; Alder, N.N.; Boelsterli, U.A. Targeting Mitochondria with Methylene Blue Protects Mice against Acetaminophen-Induced Liver Injury. Hepatology 2015, 61, 326–336. [Google Scholar] [CrossRef]
- Kaufmann, P.; Török, M.; Hänni, A.; Roberts, P.; Gasser, R.; Krähenbühl, S. Mechanisms of Benzarone and Benzbromarone-Induced Hepatic Toxicity. Hepatology 2005, 41, 925–935. [Google Scholar] [CrossRef]
- Petrescu, I.; Tarba, C. Uncoupling Effects of Diclofenac and Aspirin in the Perfused Liver and Isolated Hepatic Mitochondria of Rat. Biochim. Biophys. Acta 1997, 1318, 385–394. [Google Scholar] [CrossRef]
- Browne, G.S.; Nelson, C.; Nguyen, T.; Ellis, B.A.; Day, R.O.; Williams, K.M. Stereoselective and Substrate-Dependent Inhibition of Hepatic Mitochondria Beta-Oxidation and Oxidative Phosphorylation by the Non-Steroidal Anti-Inflammatory Drugs Ibuprofen, Flurbiprofen, and Ketorolac. Biochem. Pharmacol. 1999, 57, 837–844. [Google Scholar] [CrossRef]
- Deschamps, D.; DeBeco, V.; Fisch, C.; Fromenty, B.; Guillouzo, A.; Pessayre, D. Inhibition by Perhexiline of Oxidative Phosphorylation and the Beta-Oxidation of Fatty Acids: Possible Role in Pseudoalcoholic Liver Lesions. Hepatology 1994, 19, 948–961. [Google Scholar] [CrossRef]
- Berson, A.; Renault, S.; Lettéron, P.; Robin, M.A.; Fromenty, B.; Fau, D.; Le Bot, M.A.; Riché, C.; Durand-Schneider, A.M.; Feldmann, G.; et al. Uncoupling of Rat and Human Mitochondria: A Possible Explanation for Tacrine-Induced Liver Dysfunction. Gastroenterology 1996, 110, 1878–1890. [Google Scholar] [CrossRef]
- Larosche, I.; Lettéron, P.; Fromenty, B.; Vadrot, N.; Abbey-Toby, A.; Feldmann, G.; Pessayre, D.; Mansouri, A. Tamoxifen Inhibits Topoisomerases, Depletes Mitochondrial DNA, and Triggers Steatosis in Mouse Liver. J. Pharmacol. Exp. Ther. 2007, 321, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Tuquet, C.; Dupont, J.; Mesneau, A.; Roussaux, J. Effects of Tamoxifen on the Electron Transport Chain of Isolated Rat Liver Mitochondria. Cell Biol. Toxicol. 2000, 16, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.-Y.; Wang, B.-L.; Zhao, J.; Yao, X.-M.; Gu, Y.; Li, Y. Protective Effect of Bicyclol on Tetracycline-Induced Fatty Liver in Mice. Toxicology 2009, 261, 112–118. [Google Scholar] [CrossRef]
- Nadanaciva, S.; Dykens, J.A.; Bernal, A.; Capaldi, R.A.; Will, Y. Mitochondrial Impairment by PPAR Agonists and Statins Identified via Immunocaptured OXPHOS Complex Activities and Respiration. Toxicol. Appl. Pharmacol. 2007, 223, 277–287. [Google Scholar] [CrossRef]
- Amacher, D.E.; Chalasani, N. Drug-Induced Hepatic Steatosis. Semin. Liver Dis. 2014, 34, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Bessone, F.; Dirchwolf, M.; Rodil, M.A.; Razori, M.V.; Roma, M.G. Review Article: Drug-Induced Liver Injury in the Context of Nonalcoholic Fatty Liver Disease—A Physiopathological and Clinical Integrated View. Aliment. Pharmacol. Ther. 2018, 48, 892–913. [Google Scholar] [CrossRef] [PubMed]
- Massart, J.; Begriche, K.; Buron, N.; Porceddu, M.; Borgne-Sanchez, A.; Fromenty, B. Drug-Induced Inhibition of Mitochondrial Fatty Acid Oxidation and Steatosis. Curr. Pathobiol. Rep. 2013, 1, 147–157. [Google Scholar] [CrossRef]
- Satapathy, S.K.; Kuwajima, V.; Nadelson, J.; Atiq, O.; Sanyal, A.J. Drug-Induced Fatty Liver Disease: An Overview of Pathogenesis and Management. Ann. Hepatol. 2015, 14, 789–806. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, R.; Kakar, S. Histological Patterns in Drug-Induced Liver Disease. J. Clin. Pathol. 2009, 62, 481–492. [Google Scholar] [CrossRef]
- Patel, V.; Sanyal, A.J. Drug-Induced Steatohepatitis. Clin. Liver Dis. 2013, 17, 533–546. [Google Scholar] [CrossRef]
- Seitz, H.K.; Bataller, R.; Cortez-Pinto, H.; Gao, B.; Gual, A.; Lackner, C.; Mathurin, P.; Mueller, S.; Szabo, G.; Tsukamoto, H. Alcoholic Liver Disease. Nat. Rev. Dis. Primers 2018, 4, 16. [Google Scholar] [CrossRef]
- Kennedy, J.A.; Unger, S.A.; Horowitz, J.D. Inhibition of Carnitine Palmitoyltransferase-1 in Rat Heart and Liver by Perhexiline and Amiodarone. Biochem. Pharmacol. 1996, 52, 273–280. [Google Scholar] [CrossRef]
- Silva, M.F.; Ruiter, J.P.; IJlst, L.; Allers, P.; ten Brink, H.J.; Jakobs, C.; Duran, M.; Tavares de Almeida, I.; Wanders, R.J. Synthesis and Intramitochondrial Levels of Valproyl-Coenzyme A Metabolites. Anal. Biochem. 2001, 290, 60–67. [Google Scholar] [CrossRef]
- Freneaux, E.; Fromenty, B.; Berson, A.; Labbe, G.; Degott, C.; Letteron, P.; Larrey, D.; Pessayre, D. Stereoselective and Nonstereoselective Effects of Ibuprofen Enantiomers on Mitochondrial Beta-Oxidation of Fatty Acids. J. Pharmacol. Exp. Ther. 1990, 255, 529–535. [Google Scholar] [PubMed]
- Deschamps, D.; Fisch, C.; Fromenty, B.; Berson, A.; Degott, C.; Pessayre, D. Inhibition by Salicylic Acid of the Activation and Thus Oxidation of Long Chain Fatty Acids. Possible Role in the Development of Reye’s Syndrome. J. Pharmacol. Exp. Ther. 1991, 259, 894–904. [Google Scholar] [PubMed]
- Fromenty, B.; Fisch, C.; Berson, A.; Letteron, P.; Larrey, D.; Pessayre, D. Dual Effect of Amiodarone on Mitochondrial Respiration. Initial Protonophoric Uncoupling Effect Followed by Inhibition of the Respiratory Chain at the Levels of Complex I and Complex II. J. Pharmacol. Exp. Ther. 1990, 255, 1377–1384. [Google Scholar] [PubMed]
- Fromenty, B.; Fisch, C.; Labbe, G.; Degott, C.; Deschamps, D.; Berson, A.; Letteron, P.; Pessayre, D. Amiodarone Inhibits the Mitochondrial Beta-Oxidation of Fatty Acids and Produces Microvesicular Steatosis of the Liver in Mice. J. Pharmacol. Exp. Ther. 1990, 255, 1371–1376. [Google Scholar]
- Lewis, W.; Levine, E.S.; Griniuviene, B.; Tankersley, K.O.; Colacino, J.M.; Sommadossi, J.P.; Watanabe, K.A.; Perrino, F.W. Fialuridine and Its Metabolites Inhibit DNA Polymerase Gamma at Sites of Multiple Adjacent Analog Incorporation, Decrease MtDNA Abundance, and Cause Mitochondrial Structural Defects in Cultured Hepatoblasts. Proc. Natl. Acad. Sci. USA 1996, 93, 3592–3597. [Google Scholar] [CrossRef]
- Walker, U.A.; Venhoff, N. Uridine in the Prevention and Treatment of NRTI-Related Mitochondrial Toxicity. Antivir. Ther. 2005, 10 (Suppl. S2), 117–123. [Google Scholar]
- Igoudjil, A.; Begriche, K.; Pessayre, D.; Fromenty, B. Mitochondrial, Metabolic and Genotoxic Effects of Antiretroviral Nucleoside Reverse-Transcriptase Inhibitors. Anti-Infect. Agents Med. Chem. 2006, 5, 273–292. [Google Scholar] [CrossRef]
- Li, M.; Mislak, A.C.; Foli, Y.; Agbosu, E.; Bose, V.; Bhandari, S.; Szymanski, M.R.; Shumate, C.K.; Yin, Y.W.; Anderson, K.S.; et al. The DNA Polymerase Gamma R953C Mutant Is Associated with Antiretroviral Therapy-Induced Mitochondrial Toxicity. Antimicrob. Agents Chemother. 2016, 60, 5608–5611. [Google Scholar] [CrossRef]
- Koczor, C.A.; Lewis, W. Nucleoside Reverse Transcriptase Inhibitor Toxicity and Mitochondrial DNA. Expert Opin. Drug Metab. Toxicol. 2010, 6, 1493–1504. [Google Scholar] [CrossRef]
- Pommier, Y.; Sun, Y.; Huang, S.-Y.N.; Nitiss, J.L. Roles of Eukaryotic Topoisomerases in Transcription, Replication and Genomic Stability. Nat. Rev. Mol. Cell Biol. 2016, 17, 703–721. [Google Scholar] [CrossRef]
- Young, M.J. Off-Target Effects of Drugs That Disrupt Human Mitochondrial DNA Maintenance. Front. Mol. Biosci. 2017, 4, 74. [Google Scholar] [CrossRef]
- Rachek, L.I.; Yuzefovych, L.V.; Ledoux, S.P.; Julie, N.L.; Wilson, G.L. Troglitazone, but Not Rosiglitazone, Damages Mitochondrial DNA and Induces Mitochondrial Dysfunction and Cell Death in Human Hepatocytes. Toxicol. Appl. Pharmacol. 2009, 240, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Cover, C.; Mansouri, A.; Knight, T.R.; Bajt, M.L.; Lemasters, J.J.; Pessayre, D.; Jaeschke, H. Peroxynitrite-Induced Mitochondrial and Endonuclease-Mediated Nuclear DNA Damage in Acetaminophen Hepatotoxicity. J. Pharmacol. Exp. Ther. 2005, 315, 879–887. [Google Scholar] [CrossRef]
- Chen, C.; Krausz, K.W.; Shah, Y.M.; Idle, J.R.; Gonzalez, F.J. Serum Metabolomics Reveals Irreversible Inhibition of Fatty Acid Beta-Oxidation through the Suppression of PPARalpha Activation as a Contributing Mechanism of Acetaminophen-Induced Hepatotoxicity. Chem. Res. Toxicol. 2009, 22, 699–707. [Google Scholar] [CrossRef]
- Le Dinh, T.; Freneaux, E.; Labbe, G.; Letteron, P.; Degott, C.; Geneve, J.; Berson, A.; Larrey, D.; Pessayre, D. Amineptine, a Tricyclic Antidepressant, Inhibits the Mitochondrial Oxidation of Fatty Acids and Produces Microvesicular Steatosis of the Liver in Mice. J. Pharmacol. Exp. Ther. 1988, 247, 745–750. [Google Scholar]
- Walker, U.A.; Bäuerle, J.; Laguno, M.; Murillas, J.; Mauss, S.; Schmutz, G.; Setzer, B.; Miquel, R.; Gatell, J.M.; Mallolas, J. Depletion of Mitochondrial DNA in Liver under Antiretroviral Therapy with Didanosine, Stavudine, or Zalcitabine. Hepatology 2004, 39, 311–317. [Google Scholar] [CrossRef]
- Ulrich, R.G.; Bacon, J.A.; Cramer, C.T.; Petrella, D.K.; Sun, E.L.; Meglasson, M.D.; Holmuhamedov, E. Disruption of Mitochondrial Activities in Rabbit and Human Hepatocytes by a Quinoxalinone Anxiolytic and Its Carboxylic Acid Metabolite. Toxicology 1998, 131, 33–47. [Google Scholar] [CrossRef]
- Ulrich, R.G.; Bacon, J.A.; Brass, E.P.; Cramer, C.T.; Petrella, D.K.; Sun, E.L. Metabolic, Idiosyncratic Toxicity of Drugs: Overview of the Hepatic Toxicity Induced by the Anxiolytic, Panadiplon. Chem. Biol. Interact. 2001, 134, 251–270. [Google Scholar] [CrossRef]
- Gudbrandsen, O.A.; Rost, T.H.; Berge, R.K. Causes and Prevention of Tamoxifen-Induced Accumulation of Triacylglycerol in Rat Liver. J. Lipid Res. 2006, 47, 2223–2232. [Google Scholar] [CrossRef]
- Lelliott, C.J.; López, M.; Curtis, R.K.; Parker, N.; Laudes, M.; Yeo, G.; Jimenez-Liñan, M.; Grosse, J.; Saha, A.K.; Wiggins, D.; et al. Transcript and Metabolite Analysis of the Effects of Tamoxifen in Rat Liver Reveals Inhibition of Fatty Acid Synthesis in the Presence of Hepatic Steatosis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 1108–1119. [Google Scholar] [CrossRef]
- Choi, Y.-J.; Lee, C.-H.; Lee, K.-Y.; Jung, S.-H.; Lee, B.-H. Increased Hepatic Fatty Acid Uptake and Esterification Contribute to Tetracycline-Induced Steatosis in Mice. Toxicol. Sci. Off. J. Soc. Toxicol. 2015, 145, 273–282. [Google Scholar] [CrossRef]
- Szalowska, E.; van der Burg, B.; Man, H.-Y.; Hendriksen, P.J.M.; Peijnenburg, A.A.C.M. Model Steatogenic Compounds (Amiodarone, Valproic Acid, and Tetracycline) Alter Lipid Metabolism by Different Mechanisms in Mouse Liver Slices. PLoS ONE 2014, 9, e86795. [Google Scholar] [CrossRef]
- Amacher, D.E.; Martin, B.A. Tetracycline-Induced Steatosis in Primary Canine Hepatocyte Cultures. Fundam. Appl. Toxicol. Off. J. Soc. Toxicol. 1997, 40, 256–263. [Google Scholar] [CrossRef]
- Fromenty, B.; Freneaux, E.; Labbe, G.; Deschamps, D.; Larrey, D.; Letteron, P.; Pessayre, D. Tianeptine, a New Tricyclic Antidepressant Metabolized by Beta-Oxidation of Its Heptanoic Side Chain, Inhibits the Mitochondrial Oxidation of Medium and Short Chain Fatty Acids in Mice. Biochem. Pharmacol. 1989, 38, 3743–3751. [Google Scholar] [CrossRef]
- Fulgencio, J.P.; Kohl, C.; Girard, J.; Pégorier, J.P. Troglitazone Inhibits Fatty Acid Oxidation and Esterification, and Gluconeogenesis in Isolated Hepatocytes from Starved Rats. Diabetes 1996, 45, 1556–1562. [Google Scholar] [CrossRef] [PubMed]
- Inoue, I.; Takahashi, K.; Katayama, S.; Harada, Y.; Negishi, K.; Itabashi, A.; Ishii, J. Effect of Troglitazone (CS-045) and Bezafibrate on Glucose Tolerance, Liver Glycogen Synthase Activity, and Beta-Oxidation in Fructose-Fed Rats. Metabolism 1995, 44, 1626–1630. [Google Scholar] [CrossRef]
- Lewis, W.; Simpson, J.F.; Meyer, R.R. Cardiac Mitochondrial DNA Polymerase-Gamma Is Inhibited Competitively and Noncompetitively by Phosphorylated Zidovudine. Circ. Res. 1994, 74, 344–348. [Google Scholar] [CrossRef]
- Babatin, M.; Lee, S.S.; Pollak, P.T. Amiodarone Hepatotoxicity. Curr. Vasc. Pharmacol. 2008, 6, 228–236. [Google Scholar] [CrossRef]
- Adams, P.C.; Holt, D.W.; Storey, G.C.; Morley, A.R.; Callaghan, J.; Campbell, R.W. Amiodarone and Its Desethyl Metabolite: Tissue Distribution and Morphologic Changes during Long-Term Therapy. Circulation 1985, 72, 1064–1075. [Google Scholar] [CrossRef]
- Chabrol, B.; Mancini, J.; Chretien, D.; Rustin, P.; Munnich, A.; Pinsard, N. Valproate-Induced Hepatic Failure in a Case of Cytochrome c Oxidase Deficiency. Eur. J. Pediatr. 1994, 153, 133–135. [Google Scholar] [CrossRef]
- Njølstad, P.R.; Skjeldal, O.H.; Agsteribbe, E.; Huckriede, A.; Wannag, E.; Søvik, O.; Waaler, P.E. Medium Chain Acyl-CoA Dehydrogenase Deficiency and Fatal Valproate Toxicity. Pediatr. Neurol. 1997, 16, 160–162. [Google Scholar] [CrossRef]
- Krähenbühl, S.; Brandner, S.; Kleinle, S.; Liechti, S.; Straumann, D. Mitochondrial Diseases Represent a Risk Factor for Valproate-Induced Fulminant Liver Failure. Liver 2000, 20, 346–348. [Google Scholar] [CrossRef] [PubMed]
- Corsini, A.; Bortolini, M. Drug-Induced Liver Injury: The Role of Drug Metabolism and Transport: The Journal of Clinical Pharmacology. J. Clin. Pharmacol. 2013, 53, 463–474. [Google Scholar] [CrossRef]
- Andrade, R.J.; Robles, M.; Ulzurrun, E.; Lucena, M.I. Drug-Induced Liver Injury: Insights from Genetic Studies. Pharmacogenomics 2009, 10, 1467–1487. [Google Scholar] [CrossRef]
- Morgan, M.Y.; Reshef, R.; Shah, R.R.; Oates, N.S.; Smith, R.L.; Sherlock, S. Impaired Oxidation of Debrisoquine in Patients with Perhexiline Liver Injury. Gut 1984, 25, 1057–1064. [Google Scholar] [CrossRef]
- Ohnishi, T.; Ogawa, Y.; Saibara, T.; Nishioka, A.; Kariya, S.; Fukumoto, M.; Onishi, S.; Yoshida, S. CYP17 Polymorphism as a Risk Factor of Tamoxifen-Induced Hepatic Steatosis in Breast Cancer Patients. Oncol. Rep. 2005, 13, 485–489. [Google Scholar] [CrossRef]
- Lucena, M.I.; Andrade, R.J.; Martínez, C.; Ulzurrun, E.; García-Martín, E.; Borraz, Y.; Fernández, M.C.; Romero-Gomez, M.; Castiella, A.; Planas, R.; et al. Glutathione S-Transferase M1 and T1 Null Genotypes Increase Susceptibility to Idiosyncratic Drug-Induced Liver Injury. Hepatology 2008, 48, 588–596. [Google Scholar] [CrossRef]
- Simon, T.; Becquemont, L.; Mary-Krause, M.; de Waziers, I.; Beaune, P.; Funck-Brentano, C.; Jaillon, P. Combined Glutathione-S-Transferase M1 and T1 Genetic Polymorphism and Tacrine Hepatotoxicity. Clin. Pharmacol. Ther. 2000, 67, 432–437. [Google Scholar] [CrossRef]
- Usui, T.; Hashizume, T.; Katsumata, T.; Yokoi, T.; Komuro, S. In Vitro Investigation of the Glutathione Transferase M1 and T1 Null Genotypes as Risk Factors for Troglitazone-Induced Liver Injury. Drug Metab. Dispos. Biol. Fate Chem. 2011, 39, 1303–1310. [Google Scholar] [CrossRef]
- Chanhom, N.; Udomsinprasert, W.; Chaikledkaew, U.; Mahasirimongkol, S.; Wattanapokayakit, S.; Jittikoon, J. GSTM1 and GSTT1 Genetic Polymorphisms and Their Association with Antituberculosis Drug-Induced Liver Injury. Biomed. Rep. 2020, 12, 153–162. [Google Scholar] [CrossRef]
- Huang, Y.-S.; Su, W.-J.; Huang, Y.-H.; Chen, C.-Y.; Chang, F.-Y.; Lin, H.-C.; Lee, S.-D. Genetic Polymorphisms of Manganese Superoxide Dismutase, NAD(P)H:Quinone Oxidoreductase, Glutathione S-Transferase M1 and T1, and the Susceptibility to Drug-Induced Liver Injury. J. Hepatol. 2007, 47, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Degoul, F.; Sutton, A.; Mansouri, A.; Cepanec, C.; Degott, C.; Fromenty, B.; Beaugrand, M.; Valla, D.; Pessayre, D. Homozygosity for Alanine in the Mitochondrial Targeting Sequence of Superoxide Dismutase and Risk for Severe Alcoholic Liver Disease. Gastroenterology 2001, 120, 1468–1474. [Google Scholar] [CrossRef] [PubMed]
- Sutton, A.; Khoury, H.; Prip-Buus, C.; Cepanec, C.; Pessayre, D.; Degoul, F. The Ala16Val Genetic Dimorphism Modulates the Import of Human Manganese Superoxide Dismutase into Rat Liver Mitochondria. Pharmacogenetics 2003, 13, 145–157. [Google Scholar] [CrossRef]
- Boelsterli, U.A.; Hsiao, C.-J.J. The Heterozygous Sod2(+/−) Mouse: Modeling the Mitochondrial Role in Drug Toxicity. Drug Discov. Today 2008, 13, 982–988. [Google Scholar] [CrossRef] [PubMed]
- Alachkar, H.; Fulton, N.; Sanford, B.; Malnassy, G.; Mutonga, M.; Larson, R.A.; Bloomfield, C.D.; Marcucci, G.; Nakamura, Y.; Stock, W. Expression and Polymorphism (Rs4880) of Mitochondrial Superoxide Dismutase (SOD2) and Asparaginase Induced Hepatotoxicity in Adult Patients with Acute Lymphoblastic Leukemia. Pharmacogenom. J. 2017, 17, 274–279. [Google Scholar] [CrossRef]
- Boelsterli, U.A.; Lim, P.L.K. Mitochondrial Abnormalities—A Link to Idiosyncratic Drug Hepatotoxicity? Toxicol. Appl. Pharmacol. 2007, 220, 92–107. [Google Scholar] [CrossRef]
- Penman, S.L.; Carter, A.S.; Chadwick, A.E. Investigating the Importance of Individual Mitochondrial Genotype in Susceptibility to Drug-Induced Toxicity. Biochem. Soc. Trans. 2020, 48, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Larrey, D. Epidemiology and Individual Susceptibility to Adverse Drug Reactions Affecting the Liver. Semin. Liver Dis. 2002, 22, 145–155. [Google Scholar] [CrossRef]
- Bell, L.N.; Chalasani, N. Epidemiology of Idiosyncratic Drug-Induced Liver Injury. Semin. Liver Dis. 2009, 29, 337–347. [Google Scholar] [CrossRef]
- Lucena, M.I.; Andrade, R.J.; Fernández, M.C.; Pachkoria, K.; Pelaez, G.; Durán, J.A.; Villar, M.; Rodrigo, L.; Romero-Gomez, M.; Planas, R.; et al. Determinants of the Clinical Expression of Amoxicillin-Clavulanate Hepatotoxicity: A Prospective Series from Spain. Hepatology 2006, 44, 850–856. [Google Scholar] [CrossRef]
- Wynne, H.A.; Cope, L.H.; Mutch, E.; Rawlins, M.D.; Woodhouse, K.W.; James, O.F. The Effect of Age upon Liver Volume and Apparent Liver Blood Flow in Healthy Man. Hepatology 1989, 9, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Björnsson, E. Risk Factors for Idiosyncratic Drug-Induced Liver Injury. Gastroenterology 2010, 138, 2246–2259. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.J.; Hilmer, S.N. Drug-Induced Liver Injury in Older Adults. Ther. Adv. Drug Saf. 2010, 1, 65–77. [Google Scholar] [CrossRef]
- Chen, M.; Suzuki, A.; Borlak, J.; Andrade, R.J.; Lucena, M.I. Drug-Induced Liver Injury: Interactions between Drug Properties and Host Factors. J. Hepatol. 2015, 63, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Waxman, D.J.; Holloway, M.G. Sex Differences in the Expression of Hepatic Drug Metabolizing Enzymes. Mol. Pharmacol. 2009, 76, 215–228. [Google Scholar] [CrossRef]
- Hunt, C.M.; Westerkam, W.R.; Stave, G.M. Effect of Age and Gender on the Activity of Human Hepatic CYP3A. Biochem. Pharmacol. 1992, 44, 275–283. [Google Scholar] [CrossRef]
- Forget, P.; Wittebole, X.; Laterre, P.-F. Therapeutic Dose of Acetaminophen May Induce Fulminant Hepatitis in the Presence of Risk Factors: A Report of Two Cases. Br. J. Anaesth. 2009, 103, 899–900. [Google Scholar] [CrossRef][Green Version]
- Tarantino, G.; Conca, P.; Basile, V.; Gentile, A.; Capone, D.; Polichetti, G.; Leo, E. A Prospective Study of Acute Drug-Induced Liver Injury in Patients Suffering from Non-Alcoholic Fatty Liver Disease. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2007, 37, 410–415. [Google Scholar] [CrossRef]
- Fromenty, B. Drug-Induced Liver Injury in Obesity. J. Hepatol. 2013, 58, 824–826. [Google Scholar] [CrossRef]
- Massart, J.; Begriche, K.; Moreau, C.; Fromenty, B. Role of Nonalcoholic Fatty Liver Disease as Risk Factor for Drug-Induced Hepatotoxicity. J. Clin. Transl. Res. 2017, 3, 212–232. [Google Scholar] [CrossRef]
- Allard, J.; Le Guillou, D.; Begriche, K.; Fromenty, B. Drug-Induced Liver Injury in Obesity and Nonalcoholic Fatty Liver Disease. Adv. Pharmacol. 2019, 85, 75–107. [Google Scholar] [CrossRef]
- Cahill, A.; Cunningham, C.C.; Adachi, M.; Ishii, H.; Bailey, S.M.; Fromenty, B.; Davies, A. Effects of Alcohol and Oxidative Stress on Liver Pathology: The Role of the Mitochondrion. Alcohol. Clin. Exp. Res. 2002, 26, 907–915. [Google Scholar] [CrossRef]
- Fromenty, B.; Grimbert, S.; Mansouri, A.; Beaugrand, M.; Erlinger, S.; Rötig, A.; Pessayre, D. Hepatic Mitochondrial DNA Deletion in Alcoholics: Association with Microvesicular Steatosis. Gastroenterology 1995, 108, 193–200. [Google Scholar] [CrossRef]
- Robin, M.-A.; Sauvage, I.; Grandperret, T.; Descatoire, V.; Pessayre, D.; Fromenty, B. Ethanol Increases Mitochondrial Cytochrome P450 2E1 in Mouse Liver and Rat Hepatocytes. FEBS Lett. 2005, 579, 6895–6902. [Google Scholar] [CrossRef]
- Anandatheerthavarada, H.K.; Addya, S.; Dwivedi, R.S.; Biswas, G.; Mullick, J.; Avadhani, N.G. Localization of Multiple Forms of Inducible Cytochromes P450 in Rat Liver Mitochondria: Immunological Characteristics and Patterns of Xenobiotic Substrate Metabolism. Arch. Biochem. Biophys. 1997, 339, 136–150. [Google Scholar] [CrossRef]
- Sepuri, N.B.V.; Yadav, S.; Anandatheerthavarada, H.K.; Avadhani, N.G. Mitochondrial Targeting of Intact CYP2B1 and CYP2E1 and N-Terminal Truncated CYP1A1 Proteins in Saccharomyces Cerevisiae—Role of Protein Kinase A in the Mitochondrial Targeting of CYP2E1. FEBS J. 2007, 274, 4615–4630. [Google Scholar] [CrossRef]
- Neuman, M.G.; Shear, N.H.; Jacobson-Brown, P.M.; Katz, G.G.; Neilson, H.K.; Malkiewicz, I.M.; Cameron, R.G.; Abbott, F. CYP2E1-Mediated Modulation of Valproic Acid-Induced Hepatocytotoxicity. Clin. Biochem. 2001, 34, 211–218. [Google Scholar] [CrossRef]
- Ji, C.; Chan, C.; Kaplowitz, N. Predominant Role of Sterol Response Element Binding Proteins (SREBP) Lipogenic Pathways in Hepatic Steatosis in the Murine Intragastric Ethanol Feeding Model. J. Hepatol. 2006, 45, 717–724. [Google Scholar] [CrossRef]
- Hakkola, J.; Hukkanen, J.; Turpeinen, M.; Pelkonen, O. Inhibition and Induction of CYP Enzymes in Humans: An Update. Arch. Toxicol. 2020, 94, 3671–3722. [Google Scholar] [CrossRef]
- Belay, E.D.; Bresee, J.S.; Holman, R.C.; Khan, A.S.; Shahriari, A.; Schonberger, L.B. Reye’s Syndrome in the United States from 1981 through 1997. N. Engl. J. Med. 1999, 340, 1377–1382. [Google Scholar] [CrossRef]
- Korenaga, M.; Wang, T.; Li, Y.; Showalter, L.A.; Chan, T.; Sun, J.; Weinman, S.A. Hepatitis C Virus Core Protein Inhibits Mitochondrial Electron Transport and Increases Reactive Oxygen Species (ROS) Production. J. Biol. Chem. 2005, 280, 37481–37488. [Google Scholar] [CrossRef]
- Piccoli, C.; Scrima, R.; D’Aprile, A.; Ripoli, M.; Lecce, L.; Boffoli, D.; Capitanio, N. Mitochondrial Dysfunction in Hepatitis C Virus Infection. Biochim. Biophys. Acta 2006, 1757, 1429–1437. [Google Scholar] [CrossRef][Green Version]
- Novak, D.; Lewis, J.H. Drug-Induced Liver Disease. Curr. Opin. Gastroenterol. 2003, 19, 203–215. [Google Scholar] [CrossRef]
- Smith, D.A. Species Differences in Metabolism and Pharmacokinetics: Are We Close to an Understanding? Drug Metab. Rev. 1991, 23, 355–373. [Google Scholar] [CrossRef]
- Xu, Z.; Kang, Q.; Yu, Z.; Tian, L.; Zhang, J.; Wang, T. Research on the Species Difference of the Hepatotoxicity of Medicine Based on Transcriptome. Front. Pharmacol. 2021, 12, 647084. [Google Scholar] [CrossRef]
- Lanza, I.R.; Nair, K.S. Functional Assessment of Isolated Mitochondria in Vitro. Methods Enzymol. 2009, 457, 349–372. [Google Scholar] [CrossRef]
- Frezza, C.; Cipolat, S.; Scorrano, L. Organelle Isolation: Functional Mitochondria from Mouse Liver, Muscle and Cultured Filroblasts. Nat. Protoc. 2007, 2, 287–295. [Google Scholar] [CrossRef]
- Schulz, S.; Lichtmannegger, J.; Schmitt, S.; Leitzinger, C.; Eberhagen, C.; Einer, C.; Kerth, J.; Aichler, M.; Zischka, H. A Protocol for the Parallel Isolation of Intact Mitochondria from Rat Liver, Kidney, Heart, and Brain. Methods Mol. Biol. 2015, 1295, 75–86. [Google Scholar] [CrossRef]
- Picard, M.; Taivassalo, T.; Ritchie, D.; Wright, K.J.; Thomas, M.M.; Romestaing, C.; Hepple, R.T. Mitochondrial Structure and Function Are Disrupted by Standard Isolation Methods. PLoS ONE 2011, 6, e18317. [Google Scholar] [CrossRef]
- Picard, M.; Ritchie, D.; Wright, K.J.; Romestaing, C.; Thomas, M.M.; Rowan, S.L.; Taivassalo, T.; Hepple, R.T. Mitochondrial Functional Impairment with Aging Is Exaggerated in Isolated Mitochondria Compared to Permeabilized Myofibers. Aging Cell 2010, 9, 1032–1046. [Google Scholar] [CrossRef]
- Castell, J.V.; Jover, R.; Martnez-Jimnez, C.P.; Gmez-Lechn, M.J. Hepatocyte Cell Lines: Their Use, Scope and Limitations in Drug Metabolism Studies. Expert Opin. Drug Metab. Toxicol. 2006, 2, 183–212. [Google Scholar] [CrossRef]
- Xie, Y.; McGill, M.R.; Du, K.; Dorko, K.; Kumer, S.C.; Schmitt, T.M.; Ding, W.-X.; Jaeschke, H. Mitochondrial Protein Adducts Formation and Mitochondrial Dysfunction during N-Acetyl-m-Aminophenol (AMAP)-Induced Hepatotoxicity in Primary Human Hepatocytes. Toxicol. Appl. Pharmacol. 2015, 289, 213–222. [Google Scholar] [CrossRef]
- Vinken, M. Primary Hepatocyte Cultures for Liver Disease Modeling. Curr. Opin. Toxicol. 2021, 25, 1–5. [Google Scholar] [CrossRef]
- Lecluyse, E.L.; Alexandre, E. Isolation and Culture of Primary Hepatocytes from Resected Human Liver Tissue. Methods Mol. Biol. 2010, 640, 57–82. [Google Scholar] [CrossRef]
- LeCluyse, E.L.; Alexandre, E.; Hamilton, G.A.; Viollon-Abadie, C.; Coon, D.J.; Jolley, S.; Richert, L. Isolation and Culture of Primary Human Hepatocytes. Methods Mol. Biol. 2005, 290, 207–229. [Google Scholar] [CrossRef]
- Heslop, J.A.; Rowe, C.; Walsh, J.; Sison-Young, R.; Jenkins, R.; Kamalian, L.; Kia, R.; Hay, D.; Jones, R.P.; Malik, H.Z.; et al. Mechanistic Evaluation of Primary Human Hepatocyte Culture Using Global Proteomic Analysis Reveals a Selective Dedifferentiation Profile. Arch. Toxicol. 2017, 91, 439–452. [Google Scholar] [CrossRef]
- Maes, M.; Vinken, M.; Jaeschke, H. Experimental Models of Hepatotoxicity Related to Acute Liver Failure. Toxicol. Appl. Pharmacol. 2016, 290, 86–97. [Google Scholar] [CrossRef]
- Liu, C.; Sekine, S.; Song, B.; Ito, K. Use of Primary Rat Hepatocytes for Prediction of Drug-Induced Mitochondrial Dysfunction. Curr. Protoc. Toxicol. 2017, 72, 14.16.1–14.16.10. [Google Scholar] [CrossRef]
- Yamamoto, C.; Takemura, A.; Ishii, S.; Doi, A.; Saito, I.; Yamada, H.; Sakai, Y.; Matsunaga, T.; Ito, K. A Novel Perfusion Culture System for Screening Mitochondrial Toxicity in Primary Mouse Hepatocytes. J. Toxicol. Sci. 2022, 47, 13–18. [Google Scholar] [CrossRef]
- Pinti, M.; Troiano, L.; Nasi, M.; Ferraresi, R.; Dobrucki, J.; Cossarizza, A. Hepatoma HepG2 Cells as a Model for in Vitro Studies on Mitochondrial Toxicity of Antiviral Drugs: Which Correlation with the Patient? J. Biol. Regul. Homeost. Agents 2003, 17, 166–171. [Google Scholar]
- Cui, L.; Yoon, S.; Schinazi, R.F.; Sommadossi, J.P. Cellular and Molecular Events Leading to Mitochondrial Toxicity of 1-(2-Deoxy-2-Fluoro-1-Beta-D-Arabinofuranosyl)-5-Iodouracil in Human Liver Cells. J. Clin. Investig. 1995, 95, 555–563. [Google Scholar] [CrossRef][Green Version]
- Rodríguez-Enríquez, S.; Juárez, O.; Rodríguez-Zavala, J.S.; Moreno-Sánchez, R. Multisite Control of the Crabtree Effect in Ascites Hepatoma Cells. Eur. J. Biochem. 2001, 268, 2512–2519. [Google Scholar] [CrossRef] [PubMed]
- Marroquin, L.D.; Hynes, J.; Dykens, J.A.; Jamieson, J.D.; Will, Y. Circumventing the Crabtree Effect: Replacing Media Glucose with Galactose Increases Susceptibility of HepG2 Cells to Mitochondrial Toxicants. Toxicol. Sci. Off. J. Soc. Toxicol. 2007, 97, 539–547. [Google Scholar] [CrossRef]
- Will, Y.; Dykens, J. Mitochondrial Toxicity Assessment in Industry—A Decade of Technology Development and Insight. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1061–1067. [Google Scholar] [CrossRef]
- Rana, P.; Aleo, M.D.; Gosink, M.; Will, Y. Evaluation of in Vitro Mitochondrial Toxicity Assays and Physicochemical Properties for Prediction of Organ Toxicity Using 228 Pharmaceutical Drugs. Chem. Res. Toxicol. 2019, 32, 156–167. [Google Scholar] [CrossRef]
- Bhakuni, G.S.; Bedi, O.; Bariwal, J.; Deshmukh, R.; Kumar, P. Animal Models of Hepatotoxicity. Inflamm. Res. 2016, 65, 13–24. [Google Scholar] [CrossRef]
- Pan, Y.; Cao, M.; You, D.; Qin, G.; Liu, Z. Research Progress on the Animal Models of Drug-Induced Liver Injury: Current Status and Further Perspectives. BioMed Res. Int. 2019, 2019, 1283824. [Google Scholar] [CrossRef] [PubMed]
- Ong, M.M.K.; Latchoumycandane, C.; Boelsterli, U.A. Troglitazone-Induced Hepatic Necrosis in an Animal Model of Silent Genetic Mitochondrial Abnormalities. Toxicol. Sci. Off. J. Soc. Toxicol. 2007, 97, 205–213. [Google Scholar] [CrossRef]
- Williams, M.D.; Van Remmen, H.; Conrad, C.C.; Huang, T.T.; Epstein, C.J.; Richardson, A. Increased Oxidative Damage Is Correlated to Altered Mitochondrial Function in Heterozygous Manganese Superoxide Dismutase Knockout Mice. J. Biol. Chem. 1998, 273, 28510–28515. [Google Scholar] [CrossRef]
- Ong, M.M.K.; Wang, A.S.; Leow, K.Y.; Khoo, Y.M.; Boelsterli, U.A. Nimesulide-Induced Hepatic Mitochondrial Injury in Heterozygous Sod2(+/−) Mice. Free Radic. Biol. Med. 2006, 40, 420–429. [Google Scholar] [CrossRef]
- Li, W.; Zhang, C.; Sun, X. Mitochondrial Ca2+ Retention Capacity Assay and Ca2+-Triggered Mitochondrial Swelling Assay. J. Vis. Exp. JoVE 2018, 135, e56236. [Google Scholar] [CrossRef]
- Wong, R.; Steenbergen, C.; Murphy, E. Mitochondrial Permeability Transition Pore and Calcium Handling. In Mitochondrial Bioenergetics; Methods in Molecular Biology; Palmeira, C.M., Moreno, A.J., Eds.; Humana Press: Totowa, NJ, USA, 2012; Volume 810, pp. 235–242. ISBN 978-1-61779-381-3. [Google Scholar]
- Bhosale, G.; Duchen, M.R. Investigating the Mitochondrial Permeability Transition Pore in Disease Phenotypes and Drug Screening. Curr. Protoc. Pharmacol. 2019, 85, e59. [Google Scholar] [CrossRef]
- Javadov, S.; Chapa-Dubocq, X.; Makarov, V. Different Approaches to Modeling Analysis of Mitochondrial Swelling. Mitochondrion 2018, 38, 58–70. [Google Scholar] [CrossRef]
- Arrázola, M.S.; Inestrosa, N.C. Monitoring Mitochondrial Membranes Permeability in Live Neurons and Mitochondrial Swelling through Electron Microscopy Analysis. Methods Mol. Biol. 2015, 1254, 87–97. [Google Scholar] [CrossRef]
- Elustondo, P.A.; Nichols, M.; Negoda, A.; Thirumaran, A.; Zakharian, E.; Robertson, G.S.; Pavlov, E.V. Mitochondrial Permeability Transition Pore Induction Is Linked to Formation of the Complex of ATPase C-Subunit, Polyhydroxybutyrate and Inorganic Polyphosphate. Cell Death Discov. 2016, 2, 16070. [Google Scholar] [CrossRef]
- Huynh, F.K.; Green, M.F.; Koves, T.R.; Hirschey, M.D. Measurement of Fatty Acid Oxidation Rates in Animal Tissues and Cell Lines. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2014; Volume 542, pp. 391–405. ISBN 978-0-12-416618-9. [Google Scholar]
- Ma, Y.; Wang, W.; Devarakonda, T.; Zhou, H.; Wang, X.-Y.; Salloum, F.N.; Spiegel, S.; Fang, X. Functional Analysis of Molecular and Pharmacological Modulators of Mitochondrial Fatty Acid Oxidation. Sci. Rep. 2020, 10, 1450. [Google Scholar] [CrossRef]
- Mehlem, A.; Hagberg, C.E.; Muhl, L.; Eriksson, U.; Falkevall, A. Imaging of Neutral Lipids by Oil Red O for Analyzing the Metabolic Status in Health and Disease. Nat. Protoc. 2013, 8, 1149–1154. [Google Scholar] [CrossRef]
- Koopman, R.; Schaart, G.; Hesselink, M.K. Optimisation of Oil Red O Staining Permits Combination with Immunofluorescence and Automated Quantification of Lipids. Histochem. Cell Biol. 2001, 116, 63–68. [Google Scholar] [CrossRef]
- Fukumoto, S.; Fujimoto, T. Deformation of Lipid Droplets in Fixed Samples. Histochem. Cell Biol. 2002, 118, 423–428. [Google Scholar] [CrossRef]
- Greenspan, P.; Mayer, E.P.; Fowler, S.D. Nile Red: A Selective Fluorescent Stain for Intracellular Lipid Droplets. J. Cell Biol. 1985, 100, 965–973. [Google Scholar] [CrossRef]
- Mirejovsky, D.; Patel, A.S.; Rodriguez, D.D.; Hunt, T.J. Lipid Adsorption onto Hydrogel Contact Lens Materials. Advantages of Nile Red over Oil Red O in Visualization of Lipids. Optom. Vis. Sci. Off. Publ. Am. Acad. Optom. 1991, 68, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Fam, T.K.; Klymchenko, A.S.; Collot, M. Recent Advances in Fluorescent Probes for Lipid Droplets. Materials 2018, 11, 1768. [Google Scholar] [CrossRef]
- Ohsaki, Y.; Shinohara, Y.; Suzuki, M.; Fujimoto, T. A Pitfall in Using BODIPY Dyes to Label Lipid Droplets for Fluorescence Microscopy. Histochem. Cell Biol. 2010, 133, 477–480. [Google Scholar] [CrossRef]
- Fuchs, B.; Süss, R.; Teuber, K.; Eibisch, M.; Schiller, J. Lipid Analysis by Thin-Layer Chromatography—A Review of the Current State. J. Chromatogr. A 2011, 1218, 2754–2774. [Google Scholar] [CrossRef]
- Cheng, Y.-S.; Zheng, Y.; VanderGheynst, J.S. Rapid Quantitative Analysis of Lipids Using a Colorimetric Method in a Microplate Format. Lipids 2011, 46, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Han, J.; Wang, Z.; Liu, J.; Wei, J.; Xiong, S.; Zhao, Z. Mass Spectrometry Methodology in Lipid Analysis. Int. J. Mol. Sci. 2014, 15, 10492–10507. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Graham, B.H. Measurement of Mitochondrial Oxygen Consumption Using a Clark Electrode. In Mitochondrial Disorders; Methods in Molecular Biology; Wong, L.-J.C., Ed.; Humana Press: Totowa, NJ, USA, 2012; Volume 837, pp. 63–72. ISBN 978-1-61779-503-9. [Google Scholar]
- Barrientos, A.; Fontanesi, F.; Díaz, F. Evaluation of the Mitochondrial Respiratory Chain and Oxidative Phosphorylation System Using Polarography and Spectrophotometric Enzyme Assays. Curr. Protoc. Hum. Genet. 2009, 63, 19. [Google Scholar] [CrossRef] [PubMed]
- Horan, M.P.; Pichaud, N.; Ballard, J.W.O. Review: Quantifying Mitochondrial Dysfunction in Complex Diseases of Aging. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2012, 67, 1022–1035. [Google Scholar] [CrossRef]
- Espinosa, J.A.; Pohan, G.; Arkin, M.R.; Markossian, S. Real-Time Assessment of Mitochondrial Toxicity in HepG2 Cells Using the Seahorse Extracellular Flux Analyzer. Curr. Protoc. 2021, 1, e75. [Google Scholar] [CrossRef]
- Oliveira, J.M.A. Techniques to Investigate Neuronal Mitochondrial Function and Its Pharmacological Modulation. Curr. Drug Targets 2011, 12, 762–773. [Google Scholar] [CrossRef]
- Chinopoulos, C.; Kiss, G.; Kawamata, H.; Starkov, A.A. Measurement of ADP-ATP Exchange in Relation to Mitochondrial Transmembrane Potential and Oxygen Consumption. Methods Enzymol. 2014, 542, 333–348. [Google Scholar] [CrossRef]
- Blacker, T.S.; Duchen, M.R. Investigating Mitochondrial Redox State Using NADH and NADPH Autofluorescence. Free Radic. Biol. Med. 2016, 100, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Heikal, A.A.; Webb, W.W. Two-Photon Fluorescence Spectroscopy and Microscopy of NAD(P)H and Flavoprotein. Biophys. J. 2002, 82, 2811–2825. [Google Scholar] [CrossRef]
- Ogikubo, S.; Nakabayashi, T.; Adachi, T.; Islam, M.S.; Yoshizawa, T.; Kinjo, M.; Ohta, N. Intracellular PH Sensing Using Autofluorescence Lifetime Microscopy. J. Phys. Chem. B 2011, 115, 10385–10390. [Google Scholar] [CrossRef] [PubMed]
- Bilan, D.S.; Matlashov, M.E.; Gorokhovatsky, A.Y.; Schultz, C.; Enikolopov, G.; Belousov, V.V. Genetically Encoded Fluorescent Indicator for Imaging NAD(+)/NADH Ratio Changes in Different Cellular Compartments. Biochim. Biophys. Acta 2014, 1840, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Bilan, D.S.; Belousov, V.V. Genetically Encoded Probes for NAD+/NADH Monitoring. Free Radic. Biol. Med. 2016, 100, 32–42. [Google Scholar] [CrossRef]
- Hung, Y.P.; Albeck, J.G.; Tantama, M.; Yellen, G. Imaging Cytosolic NADH-NAD(+) Redox State with a Genetically Encoded Fluorescent Biosensor. Cell Metab. 2011, 14, 545–554. [Google Scholar] [CrossRef]
- Tao, R.; Zhao, Y.; Chu, H.; Wang, A.; Zhu, J.; Chen, X.; Zou, Y.; Shi, M.; Liu, R.; Su, N.; et al. Genetically Encoded Fluorescent Sensors Reveal Dynamic Regulation of NADPH Metabolism. Nat. Methods 2017, 14, 720–728. [Google Scholar] [CrossRef]
- Rottenberg, H.; Wu, S. Quantitative Assay by Flow Cytometry of the Mitochondrial Membrane Potential in Intact Cells. Biochim. Biophys. Acta 1998, 1404, 393–404. [Google Scholar] [CrossRef]
- Sakamuru, S.; Attene-Ramos, M.S.; Xia, M. Mitochondrial Membrane Potential Assay. Methods Mol. Biol. 2016, 1473, 17–22. [Google Scholar] [CrossRef]
- Perry, S.W.; Norman, J.P.; Barbieri, J.; Brown, E.B.; Gelbard, H.A. Mitochondrial Membrane Potential Probes and the Proton Gradient: A Practical Usage Guide. BioTechniques 2011, 50, 98–115. [Google Scholar] [CrossRef]
- Sakamuru, S.; Li, X.; Attene-Ramos, M.S.; Huang, R.; Lu, J.; Shou, L.; Shen, M.; Tice, R.R.; Austin, C.P.; Xia, M. Application of a Homogenous Membrane Potential Assay to Assess Mitochondrial Function. Physiol. Genom. 2012, 44, 495–503. [Google Scholar] [CrossRef]
- Li, X.; Zhao, Y.; Yin, J.; Lin, W. Organic Fluorescent Probes for Detecting Mitochondrial Membrane Potential. Coord. Chem. Rev. 2020, 420, 213419. [Google Scholar] [CrossRef]
- Frazier, A.E.; Vincent, A.E.; Turnbull, D.M.; Thorburn, D.R.; Taylor, R.W. Assessment of Mitochondrial Respiratory Chain Enzymes in Cells and Tissues. Methods Cell Biol. 2020, 155, 121–156. [Google Scholar] [CrossRef] [PubMed]
- Spinazzi, M.; Casarin, A.; Pertegato, V.; Salviati, L.; Angelini, C. Assessment of Mitochondrial Respiratory Chain Enzymatic Activities on Tissues and Cultured Cells. Nat. Protoc. 2012, 7, 1235–1246. [Google Scholar] [CrossRef]
- Brand, M.D.; Nicholls, D.G. Assessing Mitochondrial Dysfunction in Cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Kalyanaraman, B.; Darley-Usmar, V.; Davies, K.J.A.; Dennery, P.A.; Forman, H.J.; Grisham, M.B.; Mann, G.E.; Moore, K.; Roberts, L.J.; Ischiropoulos, H. Measuring Reactive Oxygen and Nitrogen Species with Fluorescent Probes: Challenges and Limitations. Free Radic. Biol. Med. 2012, 52, 1–6. [Google Scholar] [CrossRef]
- Karlsson, M.; Kurz, T.; Brunk, U.T.; Nilsson, S.E.; Frennesson, C.I. What Does the Commonly Used DCF Test for Oxidative Stress Really Show? Biochem. J. 2010, 428, 183–190. [Google Scholar] [CrossRef]
- Winterbourn, C.C. The Challenges of Using Fluorescent Probes to Detect and Quantify Specific Reactive Oxygen Species in Living Cells. Biochim. Biophys. Acta 2014, 1840, 730–738. [Google Scholar] [CrossRef]
- Hempel, S.L.; Buettner, G.R.; O’Malley, Y.Q.; Wessels, D.A.; Flaherty, D.M. Dihydrofluorescein Diacetate Is Superior for Detecting Intracellular Oxidants: Comparison with 2′,7′-Dichlorodihydrofluorescein Diacetate, 5(and 6)-Carboxy-2′,7′-Dichlorodihydrofluorescein Diacetate, and Dihydrorhodamine 123. Free Radic. Biol. Med. 1999, 27, 146–159. [Google Scholar] [CrossRef]
- Chen, X.; Zhong, Z.; Xu, Z.; Chen, L.; Wang, Y. 2′,7′-Dichlorodihydrofluorescein as a Fluorescent Probe for Reactive Oxygen Species Measurement: Forty Years of Application and Controversy. Free Radic. Res. 2010, 44, 587–604. [Google Scholar] [CrossRef]
- Zielonka, J.; Kalyanaraman, B. Hydroethidine- and MitoSOX-Derived Red Fluorescence Is Not a Reliable Indicator of Intracellular Superoxide Formation: Another Inconvenient Truth. Free Radic. Biol. Med. 2010, 48, 983–1001. [Google Scholar] [CrossRef] [PubMed]
- Woolley, J.F.; Stanicka, J.; Cotter, T.G. Recent Advances in Reactive Oxygen Species Measurement in Biological Systems. Trends Biochem. Sci. 2013, 38, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Booth, D.M.; Joseph, S.K.; Hajnóczky, G. Subcellular ROS Imaging Methods: Relevance for the Study of Calcium Signaling. Cell Calcium 2016, 60, 65–73. [Google Scholar] [CrossRef]
- Hanson, G.T.; Aggeler, R.; Oglesbee, D.; Cannon, M.; Capaldi, R.A.; Tsien, R.Y.; Remington, S.J. Investigating Mitochondrial Redox Potential with Redox-Sensitive Green Fluorescent Protein Indicators. J. Biol. Chem. 2004, 279, 13044–13053. [Google Scholar] [CrossRef]
- Sipos, I.; Tretter, L.; Adam-Vizi, V. Quantitative Relationship between Inhibition of Respiratory Complexes and Formation of Reactive Oxygen Species in Isolated Nerve Terminals. J. Neurochem. 2003, 84, 112–118. [Google Scholar] [CrossRef]
- Lushchak, O.V.; Piroddi, M.; Galli, F.; Lushchak, V.I. Aconitase Post-Translational Modification as a Key in Linkage between Krebs Cycle, Iron Homeostasis, Redox Signaling, and Metabolism of Reactive Oxygen Species. Redox Rep. Commun. Free Radic. Res. 2014, 19, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, M.; Dane, E.; Conley, J.; Tantama, M. Imaging Adenosine Triphosphate (ATP). Biol. Bull. 2016, 231, 73–84. [Google Scholar] [CrossRef]
- Morciano, G.; Sarti, A.C.; Marchi, S.; Missiroli, S.; Falzoni, S.; Raffaghello, L.; Pistoia, V.; Giorgi, C.; Di Virgilio, F.; Pinton, P. Use of Luciferase Probes to Measure ATP in Living Cells and Animals. Nat. Protoc. 2017, 12, 1542–1562. [Google Scholar] [CrossRef]
- Petty, R.D.; Sutherland, L.A.; Hunter, E.M.; Cree, I.A. Comparison of MTT and ATP-Based Assays for the Measurement of Viable Cell Number. J. Biolumin. Chemilumin. 1995, 10, 29–34. [Google Scholar] [CrossRef]
- Yoshida, T.; Kakizuka, A.; Imamura, H. BTeam, a Novel BRET-Based Biosensor for the Accurate Quantification of ATP Concentration within Living Cells. Sci. Rep. 2016, 6, 39618. [Google Scholar] [CrossRef]
- Tantama, M.; Martínez-François, J.R.; Mongeon, R.; Yellen, G. Imaging Energy Status in Live Cells with a Fluorescent Biosensor of the Intracellular ATP-to-ADP Ratio. Nat. Commun. 2013, 4, 2550. [Google Scholar] [CrossRef] [PubMed]
- Nakano, M.; Imamura, H.; Nagai, T.; Noji, H. Ca2+ Regulation of Mitochondrial ATP Synthesis Visualized at the Single Cell Level. ACS Chem. Biol. 2011, 6, 709–715. [Google Scholar] [CrossRef]
- De Michele, R.; Carimi, F.; Frommer, W.B. Mitochondrial Biosensors. Int. J. Biochem. Cell Biol. 2014, 48, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Pozzan, T.; Rudolf, R. Measurements of Mitochondrial Calcium in Vivo. Biochim. Biophys. Acta 2009, 1787, 1317–1323. [Google Scholar] [CrossRef]
- McKenzie, M.; Lim, S.C.; Duchen, M.R. Simultaneous Measurement of Mitochondrial Calcium and Mitochondrial Membrane Potential in Live Cells by Fluorescent Microscopy. J. Vis. Exp. JoVE 2017, 119, e55166. [Google Scholar] [CrossRef]
- Pendin, D.; Greotti, E.; Filadi, R.; Pozzan, T. Spying on Organelle Ca2+ in Living Cells: The Mitochondrial Point of View. J. Endocrinol. Investig. 2015, 38, 39–45. [Google Scholar] [CrossRef]
- Miyawaki, A.; Griesbeck, O.; Heim, R.; Tsien, R.Y. Dynamic and Quantitative Ca2+ Measurements Using Improved Cameleons. Proc. Natl. Acad. Sci. USA 1999, 96, 2135–2140. [Google Scholar] [CrossRef]
- Rizzuto, R.; Simpson, A.W.; Brini, M.; Pozzan, T. Rapid Changes of Mitochondrial Ca2+ Revealed by Specifically Targeted Recombinant Aequorin. Nature 1992, 358, 325–327. [Google Scholar] [CrossRef]
- Bonora, M.; Giorgi, C.; Bononi, A.; Marchi, S.; Patergnani, S.; Rimessi, A.; Rizzuto, R.; Pinton, P. Subcellular Calcium Measurements in Mammalian Cells Using Jellyfish Photoprotein Aequorin-Based Probes. Nat. Protoc. 2013, 8, 2105–2118. [Google Scholar] [CrossRef]
- Wu, J.; Prole, D.L.; Shen, Y.; Lin, Z.; Gnanasekaran, A.; Liu, Y.; Chen, L.; Zhou, H.; Chen, S.R.W.; Usachev, Y.M.; et al. Red Fluorescent Genetically Encoded Ca2+ Indicators for Use in Mitochondria and Endoplasmic Reticulum. Biochem. J. 2014, 464, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Santo-Domingo, J.; Demaurex, N. Perspectives on: SGP Symposium on Mitochondrial Physiology and Medicine: The Renaissance of Mitochondrial PH. J. Gen. Physiol. 2012, 139, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Porcelli, A.M.; Ghelli, A.; Zanna, C.; Pinton, P.; Rizzuto, R.; Rugolo, M. PH Difference across the Outer Mitochondrial Membrane Measured with a Green Fluorescent Protein Mutant. Biochem. Biophys. Res. Commun. 2005, 326, 799–804. [Google Scholar] [CrossRef]
- Gonzalez-Hunt, C.P.; Rooney, J.P.; Ryde, I.T.; Anbalagan, C.; Joglekar, R.; Meyer, J.N. PCR-Based Analysis of Mitochondrial DNA Copy Number, Mitochondrial DNA Damage, and Nuclear DNA Damage. Curr. Protoc. Toxicol. 2016, 67, 20.11.1–20.11.25. [Google Scholar] [CrossRef]
- Memon, A.A.; Zöller, B.; Hedelius, A.; Wang, X.; Stenman, E.; Sundquist, J.; Sundquist, K. Quantification of Mitochondrial DNA Copy Number in Suspected Cancer Patients by a Well Optimized DdPCR Method. Biomol. Detect. Quantif. 2017, 13, 32–39. [Google Scholar] [CrossRef]
- O’Hara, R.; Tedone, E.; Ludlow, A.; Huang, E.; Arosio, B.; Mari, D.; Shay, J.W. Quantitative Mitochondrial DNA Copy Number Determination Using Droplet Digital PCR with Single-Cell Resolution. Genome Res. 2019, 29, 1878–1888. [Google Scholar] [CrossRef] [PubMed]
- Refinetti, P.; Warren, D.; Morgenthaler, S.; Ekstrøm, P.O. Quantifying Mitochondrial DNA Copy Number Using Robust Regression to Interpret Real Time PCR Results. BMC Res. Notes 2017, 10, 593. [Google Scholar] [CrossRef]
- Chazotte, B. Labeling Mitochondria with MitoTracker Dyes. Cold Spring Harb. Protoc. 2011, 2011, 990–992. [Google Scholar] [CrossRef]
- Sasaki, S. Determination of Altered Mitochondria Ultrastructure by Electron Microscopy. Methods Mol. Biol. 2010, 648, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, R.J.; Tarloff, J.B. Evaluation of Hepatic Subcellular Fractions for Alamar Blue and MTT Reductase Activity. Toxicol. In Vitro Int. J. Publ. Assoc. BIBRA 2001, 15, 257–259. [Google Scholar] [CrossRef]
- Vistica, D.T.; Skehan, P.; Scudiero, D.; Monks, A.; Pittman, A.; Boyd, M.R. Tetrazolium-Based Assays for Cellular Viability: A Critical Examination of Selected Parameters Affecting Formazan Production. Cancer Res. 1991, 51, 2515–2520. [Google Scholar]
- Van Tonder, A.; Joubert, A.M.; Cromarty, A.D. Limitations of the 3-(4,5-Dimethylthiazol-2-Yl)-2,5-Diphenyl-2H-Tetrazolium Bromide (MTT) Assay When Compared to Three Commonly Used Cell Enumeration Assays. BMC Res. Notes 2015, 8, 47. [Google Scholar] [CrossRef] [PubMed]
- Rampersad, S.N. Multiple Applications of Alamar Blue as an Indicator of Metabolic Function and Cellular Health in Cell Viability Bioassays. Sensors 2012, 12, 12347–12360. [Google Scholar] [CrossRef] [PubMed]
- Riss, T.L.; Moravec, R.A.; Niles, A.L.; Duellman, S.; Benink, H.A.; Worzella, T.J.; Minor, L. Cell Viability Assays. In Assay Guidance Manual; Markossian, S., Grossman, A., Brimacombe, K., Arkin, M., Auld, D., Austin, C.P., Baell, J., Chung, T.D.Y., Coussens, N.P., Dahlin, J.L., et al., Eds.; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004. [Google Scholar]
- Hamid, R.; Rotshteyn, Y.; Rabadi, L.; Parikh, R.; Bullock, P. Comparison of Alamar Blue and MTT Assays for High Through-Put Screening. Toxicol. In Vitro Int. J. Publ. Assoc. BIBRA 2004, 18, 703–710. [Google Scholar] [CrossRef]
- Erikstein, B.S.; Hagland, H.R.; Nikolaisen, J.; Kulawiec, M.; Singh, K.K.; Gjertsen, B.T.; Tronstad, K.J. Cellular Stress Induced by Resazurin Leads to Autophagy and Cell Death via Production of Reactive Oxygen Species and Mitochondrial Impairment. J. Cell. Biochem. 2010, 111, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Morciano, G.; Naumova, N.; Koprowski, P.; Valente, S.; Sardão, V.A.; Potes, Y.; Rimessi, A.; Wieckowski, M.R.; Oliveira, P.J. The Mitochondrial Permeability Transition Pore: An Evolving Concept Critical for Cell Life and Death. Biol. Rev. 2021, 96, 2489–2521. [Google Scholar] [CrossRef] [PubMed]
- Levene, A.P.; Kudo, H.; Thursz, M.R.; Anstee, Q.M.; Goldin, R.D. Is Oil Red-O Staining and Digital Image Analysis the Gold Standard for Quantifying Steatosis in the Liver? Hepatology 2010, 51, 1859–1860, author reply. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, H.N.; Chaubal, K.A. Evaluation of Intracellular Lipids by Standardized Staining with a Sudan Black B Fraction. J. Biochem. Biophys. Methods 1990, 21, 9–16. [Google Scholar] [CrossRef]
- McMillian, M.K.; Grant, E.R.; Zhong, Z.; Parker, J.B.; Li, L.; Zivin, R.A.; Burczynski, M.E.; Johnson, M.D. Nile Red Binding to HepG2 Cells: An Improved Assay for in Vitro Studies of Hepatosteatosis. In Vitro Mol. Toxicol. 2001, 14, 177–190. [Google Scholar] [CrossRef]
- Qiu, B.; Simon, M.C. BODIPY 493/503 Staining of Neutral Lipid Droplets for Microscopy and Quantification by Flow Cytometry. Bio-Protocol 2016, 6, e1912. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A Simple Method for the Isolation and Purification of Total Lipides from Animal Tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Bligh, E.G.; Dyer, W.J. A Rapid Method of Total Lipid Extraction and Purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Matyash, V.; Liebisch, G.; Kurzchalia, T.V.; Shevchenko, A.; Schwudke, D. Lipid Extraction by Methyl-Tert-Butyl Ether for High-Throughput Lipidomics. J. Lipid Res. 2008, 49, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Tabernilla, A.; dos Santos Rodrigues, B.; Pieters, A.; Caufriez, A.; Leroy, K.; Van Campenhout, R.; Cooreman, A.; Gomes, A.R.; Arnesdotter, E.; Gijbels, E.; et al. In Vitro Liver Toxicity Testing of Chemicals: A Pragmatic Approach. Int. J. Mol. Sci. 2021, 22, 5038. [Google Scholar] [CrossRef]
- Simonnet, H.; Vigneron, A.; Pouysségur, J. Conventional Techniques to Monitor Mitochondrial Oxygen Consumption. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2014; Volume 542, pp. 151–161. ISBN 978-0-12-416618-9. [Google Scholar]
- Papkovsky, D.B.; Zhdanov, A.V. Phosphorescence Based O2 Sensors—Essential Tools for Monitoring Cell and Tissue Oxygenation and Its Impact on Metabolism. Free Radic. Biol. Med. 2016, 101, 202–210. [Google Scholar] [CrossRef]
- Takahashi, E.; Yamaoka, Y. Simple and Inexpensive Technique for Measuring Oxygen Consumption Rate in Adherent Cultured Cells. J. Physiol. Sci. 2017, 67, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Haraux, F.; Lombès, A. Kinetic Analysis of ATP Hydrolysis by Complex V in Four Murine Tissues: Towards an Assay Suitable for Clinical Diagnosis. PLoS ONE 2019, 14, e0221886. [Google Scholar] [CrossRef] [PubMed]
- Connolly, N.M.C.; Theurey, P.; Adam-Vizi, V.; Bazan, N.G.; Bernardi, P.; Bolaños, J.P.; Culmsee, C.; Dawson, V.L.; Deshmukh, M.; Duchen, M.R.; et al. Guidelines on Experimental Methods to Assess Mitochondrial Dysfunction in Cellular Models of Neurodegenerative Diseases. Cell Death Differ. 2018, 25, 542–572. [Google Scholar] [CrossRef]
- Lu, W.; Wang, L.; Chen, L.; Hui, S.; Rabinowitz, J.D. Extraction and Quantitation of Nicotinamide Adenine Dinucleotide Redox Cofactors. Antioxid. Redox Signal. 2018, 28, 167–179. [Google Scholar] [CrossRef]
- Zhao, Y.; Jin, J.; Hu, Q.; Zhou, H.-M.; Yi, J.; Yu, Z.; Xu, L.; Wang, X.; Yang, Y.; Loscalzo, J. Genetically Encoded Fluorescent Sensors for Intracellular NADH Detection. Cell Metab. 2011, 14, 555–566. [Google Scholar] [CrossRef]
- Figueira, T.R.; Melo, D.R.; Vercesi, A.E.; Castilho, R.F. Safranine as a Fluorescent Probe for the Evaluation of Mitochondrial Membrane Potential in Isolated Organelles and Permeabilized Cells. Methods Mol. Biol. 2012, 810, 103–117. [Google Scholar] [CrossRef]
- Moreno, A.J.; Santos, D.L.; Magalhães-Novais, S.; Oliveira, P.J. Measuring Mitochondrial Membrane Potential with a Tetraphenylphosphonium-Selective Electrode. Curr. Protoc. Toxicol. 2015, 65, 25.5.1–25.5.16. [Google Scholar] [CrossRef] [PubMed]
- Polster, B.M.; Nicholls, D.G.; Ge, S.X.; Roelofs, B.A. Use of Potentiometric Fluorophores in the Measurement of Mitochondrial Reactive Oxygen Species. Methods Enzymol. 2014, 547, 225–250. [Google Scholar] [CrossRef] [PubMed]
- Gutscher, M.; Pauleau, A.-L.; Marty, L.; Brach, T.; Wabnitz, G.H.; Samstag, Y.; Meyer, A.J.; Dick, T.P. Real-Time Imaging of the Intracellular Glutathione Redox Potential. Nat. Methods 2008, 5, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Belousov, V.V.; Fradkov, A.F.; Lukyanov, K.A.; Staroverov, D.B.; Shakhbazov, K.S.; Terskikh, A.V.; Lukyanov, S. Genetically Encoded Fluorescent Indicator for Intracellular Hydrogen Peroxide. Nat. Methods 2006, 3, 281–286. [Google Scholar] [CrossRef]
- Imamura, H.; Nhat, K.P.H.; Togawa, H.; Saito, K.; Iino, R.; Kato-Yamada, Y.; Nagai, T.; Noji, H. Visualization of ATP Levels inside Single Living Cells with Fluorescence Resonance Energy Transfer-Based Genetically Encoded Indicators. Proc. Natl. Acad. Sci. USA 2009, 106, 15651–15656. [Google Scholar] [CrossRef]
- Nagai, T.; Yamada, S.; Tominaga, T.; Ichikawa, M.; Miyawaki, A. Expanded Dynamic Range of Fluorescent Indicators for Ca(2+) by Circularly Permuted Yellow Fluorescent Proteins. Proc. Natl. Acad. Sci. USA 2004, 101, 10554–10559. [Google Scholar] [CrossRef]
- Nagai, T.; Sawano, A.; Park, E.S.; Miyawaki, A. Circularly Permuted Green Fluorescent Proteins Engineered to Sense Ca2+. Proc. Natl. Acad. Sci. USA 2001, 98, 3197–3202. [Google Scholar] [CrossRef]
- Benčina, M. Illumination of the Spatial Order of Intracellular PH by Genetically Encoded PH-Sensitive Sensors. Sensors 2013, 13, 16736–16758. [Google Scholar] [CrossRef]
- Abad, M.F.C.; Di Benedetto, G.; Magalhães, P.J.; Filippin, L.; Pozzan, T. Mitochondrial PH Monitored by a New Engineered Green Fluorescent Protein Mutant. J. Biol. Chem. 2004, 279, 11521–11529. [Google Scholar] [CrossRef]
- Tantama, M.; Hung, Y.P.; Yellen, G. Imaging Intracellular PH in Live Cells with a Genetically Encoded Red Fluorescent Protein Sensor. J. Am. Chem. Soc. 2011, 133, 10034–10037. [Google Scholar] [CrossRef]
- Li, Y.; Tsien, R.W. PHTomato, a Red, Genetically Encoded Indicator That Enables Multiplex Interrogation of Synaptic Activity. Nat. Neurosci. 2012, 15, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Sims, D.; Sudbery, I.; Ilott, N.E.; Heger, A.; Ponting, C.P. Sequencing Depth and Coverage: Key Considerations in Genomic Analyses. Nat. Rev. Genet. 2014, 15, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Ashar, F.N.; Moes, A.; Moore, A.Z.; Grove, M.L.; Chaves, P.H.M.; Coresh, J.; Newman, A.B.; Matteini, A.M.; Bandeen-Roche, K.; Boerwinkle, E.; et al. Association of Mitochondrial DNA Levels with Frailty and All-Cause Mortality. J. Mol. Med. 2015, 93, 177–186. [Google Scholar] [CrossRef]
- Niles, A.L.; Moravec, R.A.; Riss, T.L. Update on in Vitro Cytotoxicity Assays for Drug Development. Expert Opin. Drug Discov. 2008, 3, 655–669. [Google Scholar] [CrossRef] [PubMed]
- Murayama, H.; Ikemoto, M.; Fukuda, Y.; Tsunekawa, S.; Nagata, A. Advantage of Serum Type-I Arginase and Ornithine Carbamoyltransferase in the Evaluation of Acute and Chronic Liver Damage Induced by Thioacetamide in Rats. Clin. Chim. Acta Int. J. Clin. Chem. 2007, 375, 63–68. [Google Scholar] [CrossRef]
- Ozawa, K.; Chance, B.; Tanaka, A.; Iwata, S.; Kitai, T.; Ikai, I. Linear Correlation between Acetoacetate/Beta-Hydroxybutyrate in Arterial Blood and Oxidized Flavoprotein/Reduced Pyridine Nucleotide in Freeze-Trapped Human Liver Tissue. Biochim. Biophys. Acta 1992, 1138, 350–352. [Google Scholar] [CrossRef]
- Rinaldo, P. Fatty Acid Transport and Mitochondrial Oxidation Disorders. Semin. Liver Dis. 2001, 21, 489–500. [Google Scholar] [CrossRef]
- Knapp, A.C.; Todesco, L.; Beier, K.; Terracciano, L.; Sägesser, H.; Reichen, J.; Krähenbühl, S. Toxicity of Valproic Acid in Mice with Decreased Plasma and Tissue Carnitine Stores. J. Pharmacol. Exp. Ther. 2008, 324, 568–575. [Google Scholar] [CrossRef]
- Takahashi, Y.; Fukusato, T. Histopathology of Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis. World J. Gastroenterol. 2014, 20, 15539–15548. [Google Scholar] [CrossRef]
- Begriche, K.; Igoudjil, A.; Pessayre, D.; Fromenty, B. Mitochondrial Dysfunction in NASH: Causes, Consequences and Possible Means to Prevent It. Mitochondrion 2006, 6, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Kaplowitz, N. Biochemical and Cellular Mechanisms of Toxic Liver Injury. Semin. Liver Dis. 2002, 22, 137–144. [Google Scholar] [CrossRef]
- Nadanaciva, S.; Will, Y. Investigating Mitochondrial Dysfunction to Increase Drug Safety in the Pharmaceutical Industry. Curr. Drug Targets 2011, 12, 774–782. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Checa, J.C.; Bagnaninchi, P.; Ye, H.; Sancho-Bru, P.; Falcon-Perez, J.M.; Royo, F.; Garcia-Ruiz, C.; Konu, O.; Miranda, J.; Lunov, O.; et al. Advanced Preclinical Models for Evaluation of Drug-Induced Liver Injury—Consensus Statement by the European Drug-Induced Liver Injury Network [PRO-EURO-DILI-NET]. J. Hepatol. 2021, 75, 935–959. [Google Scholar] [CrossRef] [PubMed]
- Andersson, T.B. Evolution of Novel 3D Culture Systems for Studies of Human Liver Function and Assessments of the Hepatotoxicity of Drugs and Drug Candidates. Basic Clin. Pharmacol. Toxicol. 2017, 121, 234–238. [Google Scholar] [CrossRef]
- Nadanaciva, S.; Bernal, A.; Aggeler, R.; Capaldi, R.; Will, Y. Target Identification of Drug Induced Mitochondrial Toxicity Using Immunocapture Based OXPHOS Activity Assays. Toxicol. In Vitro Int. J. Publ. Assoc. BIBRA 2007, 21, 902–911. [Google Scholar] [CrossRef] [PubMed]
- Hynes, J.; Marroquin, L.D.; Ogurtsov, V.I.; Christiansen, K.N.; Stevens, G.J.; Papkovsky, D.B.; Will, Y. Investigation of Drug-Induced Mitochondrial Toxicity Using Fluorescence-Based Oxygen-Sensitive Probes. Toxicol. Sci. Off. J. Soc. Toxicol. 2006, 92, 186–200. [Google Scholar] [CrossRef]
- Bell, C.C.; Hendriks, D.F.G.; Moro, S.M.L.; Ellis, E.; Walsh, J.; Renblom, A.; Fredriksson Puigvert, L.; Dankers, A.C.A.; Jacobs, F.; Snoeys, J.; et al. Characterization of Primary Human Hepatocyte Spheroids as a Model System for Drug-Induced Liver Injury, Liver Function and Disease. Sci. Rep. 2016, 6, 25187. [Google Scholar] [CrossRef]
- Gaskell, H.; Sharma, P.; Colley, H.E.; Murdoch, C.; Williams, D.P.; Webb, S.D. Characterization of a Functional C3A Liver Spheroid Model. Toxicol. Res. 2016, 5, 1053–1065. [Google Scholar] [CrossRef]
- Vilas-Boas, V.; Gijbels, E.; Leroy, K.; Pieters, A.; Baze, A.; Parmentier, C.; Vinken, M. Primary Human Hepatocyte Spheroids as Tools to Study the Hepatotoxic Potential of Non-Pharmaceutical Chemicals. Int. J. Mol. Sci. 2021, 22, 11005. [Google Scholar] [CrossRef]
- Bouwmeester, M.C.; Bernal, P.N.; Oosterhoff, L.A.; van Wolferen, M.E.; Lehmann, V.; Vermaas, M.; Buchholz, M.-B.; Peiffer, Q.C.; Malda, J.; van der Laan, L.J.W.; et al. Bioprinting of Human Liver-Derived Epithelial Organoids for Toxicity Studies. Macromol. Biosci. 2021, 21, e2100327. [Google Scholar] [CrossRef] [PubMed]
- Godoy, P.; Hewitt, N.J.; Albrecht, U.; Andersen, M.E.; Ansari, N.; Bhattacharya, S.; Bode, J.G.; Bolleyn, J.; Borner, C.; Böttger, J.; et al. Recent Advances in 2D and 3D in Vitro Systems Using Primary Hepatocytes, Alternative Hepatocyte Sources and Non-Parenchymal Liver Cells and Their Use in Investigating Mechanisms of Hepatotoxicity, Cell Signaling and ADME. Arch. Toxicol. 2013, 87, 1315–1530. [Google Scholar] [CrossRef]
- Beckwitt, C.H.; Clark, A.M.; Wheeler, S.; Taylor, D.L.; Stolz, D.B.; Griffith, L.; Wells, A. Liver “Organ on a Chip”. Exp. Cell Res. 2018, 363, 15–25. [Google Scholar] [CrossRef]
- Kuna, L.; Bozic, I.; Kizivat, T.; Bojanic, K.; Mrso, M.; Kralj, E.; Smolic, R.; Wu, G.Y.; Smolic, M. Models of Drug Induced Liver Injury (DILI)—Current Issues and Future Perspectives. Curr. Drug Metab. 2018, 19, 830–838. [Google Scholar] [CrossRef] [PubMed]
- Schwenger, K.J.; Clermont-Dejean, N.; Allard, J.P. The Role of the Gut Microbiome in Chronic Liver Disease: The Clinical Evidence Revised. JHEP Rep. Innov. Hepatol. 2019, 1, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Philips, C.A.; Augustine, P.; Yerol, P.K.; Ramesh, G.N.; Ahamed, R.; Rajesh, S.; George, T.; Kumbar, S. Modulating the Intestinal Microbiota: Therapeutic Opportunities in Liver Disease. J. Clin. Transl. Hepatol. 2020, 8, 87–99. [Google Scholar] [CrossRef]
- Zheng, Z.; Wang, B. The Gut-Liver Axis in Health and Disease: The Role of Gut Microbiota-Derived Signals in Liver Injury and Regeneration. Front. Immunol. 2021, 12, 775526. [Google Scholar] [CrossRef]
- Lee, S.Y.; Sung, J.H. Gut-Liver on a Chip toward an in Vitro Model of Hepatic Steatosis. Biotechnol. Bioeng. 2018, 115, 2817–2827. [Google Scholar] [CrossRef]
- Jeon, J.-W.; Lee, S.H.; Kim, D.; Sung, J.H. In Vitro Hepatic Steatosis Model Based on Gut-Liver-on-a-Chip. Biotechnol. Prog. 2021, 37, e3121. [Google Scholar] [CrossRef]
- Tang, L. In Vitro Intestine Model for Gut Microbiome. Nat. Methods 2019, 16, 578. [Google Scholar] [CrossRef] [PubMed]
- Biagini, F.; Calvigioni, M.; Lapomarda, A.; Vecchione, A.; Magliaro, C.; De Maria, C.; Montemurro, F.; Celandroni, F.; Mazzantini, D.; Mattioli-Belmonte, M.; et al. A Novel 3D in Vitro Model of the Human Gut Microbiota. Sci. Rep. 2020, 10, 21499. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Peng, Y.; Xu, X.; Wang, Z.; Wu, Z.; Li, W.; Tang, Y.; Liu, G. In Silico Prediction of Mitochondrial Toxicity of Chemicals Using Machine Learning Methods. J. Appl. Toxicol. 2021, 41, 1518–1526. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-T.; Lin, K.-H.; Huang, C.-J.; Wei, A.-C. MitoTox: A Comprehensive Mitochondrial Toxicity Database. BMC Bioinform. 2021, 22, 369. [Google Scholar] [CrossRef] [PubMed]
- Luechtefeld, T.; Hartung, T. Computational Approaches to Chemical Hazard Assessment. ALTEX 2017, 34, 459–478. [Google Scholar] [CrossRef]
- Vall, A.; Sabnis, Y.; Shi, J.; Class, R.; Hochreiter, S.; Klambauer, G. The Promise of AI for DILI Prediction. Front. Artif. Intell. 2021, 4, 638410. [Google Scholar] [CrossRef] [PubMed]
- Ankley, G.T.; Bennett, R.S.; Erickson, R.J.; Hoff, D.J.; Hornung, M.W.; Johnson, R.D.; Mount, D.R.; Nichols, J.W.; Russom, C.L.; Schmieder, P.K.; et al. Adverse Outcome Pathways: A Conceptual Framework to Support Ecotoxicology Research and Risk Assessment. Environ. Toxicol. Chem. 2010, 29, 730–741. [Google Scholar] [CrossRef]
- Vinken, M. The Adverse Outcome Pathway Concept: A Pragmatic Tool in Toxicology. Toxicology 2013, 312, 158–165. [Google Scholar] [CrossRef]
- Vinken, M.; Knapen, D.; Vergauwen, L.; Hengstler, J.G.; Angrish, M.; Whelan, M. Adverse Outcome Pathways: A Concise Introduction for Toxicologists. Arch. Toxicol. 2017, 91, 3697–3707. [Google Scholar] [CrossRef]
- Vinken, M. Adverse Outcome Pathways and Drug-Induced Liver Injury Testing. Chem. Res. Toxicol. 2015, 28, 1391–1397. [Google Scholar] [CrossRef]
- Gijbels, E.; Vilas-Boas, V.; Annaert, P.; Vanhaecke, T.; Devisscher, L.; Vinken, M. Robustness Testing and Optimization of an Adverse Outcome Pathway on Cholestatic Liver Injury. Arch. Toxicol. 2020, 94, 1151–1172. [Google Scholar] [CrossRef]
- Horvat, T.; Landesmann, B.; Lostia, A.; Vinken, M.; Munn, S.; Whelan, M. Adverse Outcome Pathway Development from Protein Alkylation to Liver Fibrosis. Arch. Toxicol. 2017, 91, 1523–1543. [Google Scholar] [CrossRef]
- Burden, N.; Sewell, F.; Andersen, M.E.; Boobis, A.; Chipman, J.K.; Cronin, M.T.D.; Hutchinson, T.H.; Kimber, I.; Whelan, M. Adverse Outcome Pathways Can Drive Non-Animal Approaches for Safety Assessment. J. Appl. Toxicol. JAT 2015, 35, 971–975. [Google Scholar] [CrossRef] [PubMed]
- Vinken, M.; Kramer, N.; Allen, T.E.H.; Hoffmans, Y.; Thatcher, N.; Levorato, S.; Traussnig, H.; Schulte, S.; Boobis, A.; Thiel, A.; et al. The Use of Adverse Outcome Pathways in the Safety Evaluation of Food Additives. Arch. Toxicol. 2020, 94, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Fontana, R.J. Pathogenesis of Idiosyncratic Drug-Induced Liver Injury and Clinical Perspectives. Gastroenterology 2014, 146, 914–928. [Google Scholar] [CrossRef] [PubMed]
- Tolosa, L.; Jiménez, N.; Pérez, G.; Castell, J.V.; Gómez-Lechón, M.J.; Donato, M.T. Customised in Vitro Model to Detect Human Metabolism-Dependent Idiosyncratic Drug-Induced Liver Injury. Arch. Toxicol. 2018, 92, 383–399. [Google Scholar] [CrossRef] [PubMed]
- Uetrecht, J. Mechanistic Studies of Idiosyncratic DILI: Clinical Implications. Front. Pharmacol. 2019, 10, 837. [Google Scholar] [CrossRef]
- Ballotin, V.R.; Bigarella, L.G.; de Mello Brandão, A.B.; Balbinot, R.A.; Balbinot, S.S.; Soldera, J. Herb-Induced Liver Injury: Systematic Review and Meta-Analysis. World J. Clin. Cases 2021, 9, 5490–5513. [Google Scholar] [CrossRef]
- Frenzel, C.; Teschke, R. Herbal Hepatotoxicity: Clinical Characteristics and Listing Compilation. Int. J. Mol. Sci. 2016, 17, 588. [Google Scholar] [CrossRef]
- Faure, J.-E.; Dyląg, T.; Norstedt, I.; Matthiessen, L. The European Innovative Medicines Initiative: Progress to Date. Pharm. Med. 2018, 32, 243–249. [Google Scholar] [CrossRef]
- Goldman, M. The Innovative Medicines Initiative: A European Response to the Innovation Challenge. Clin. Pharmacol. Ther. 2012, 91, 418–425. [Google Scholar] [CrossRef]





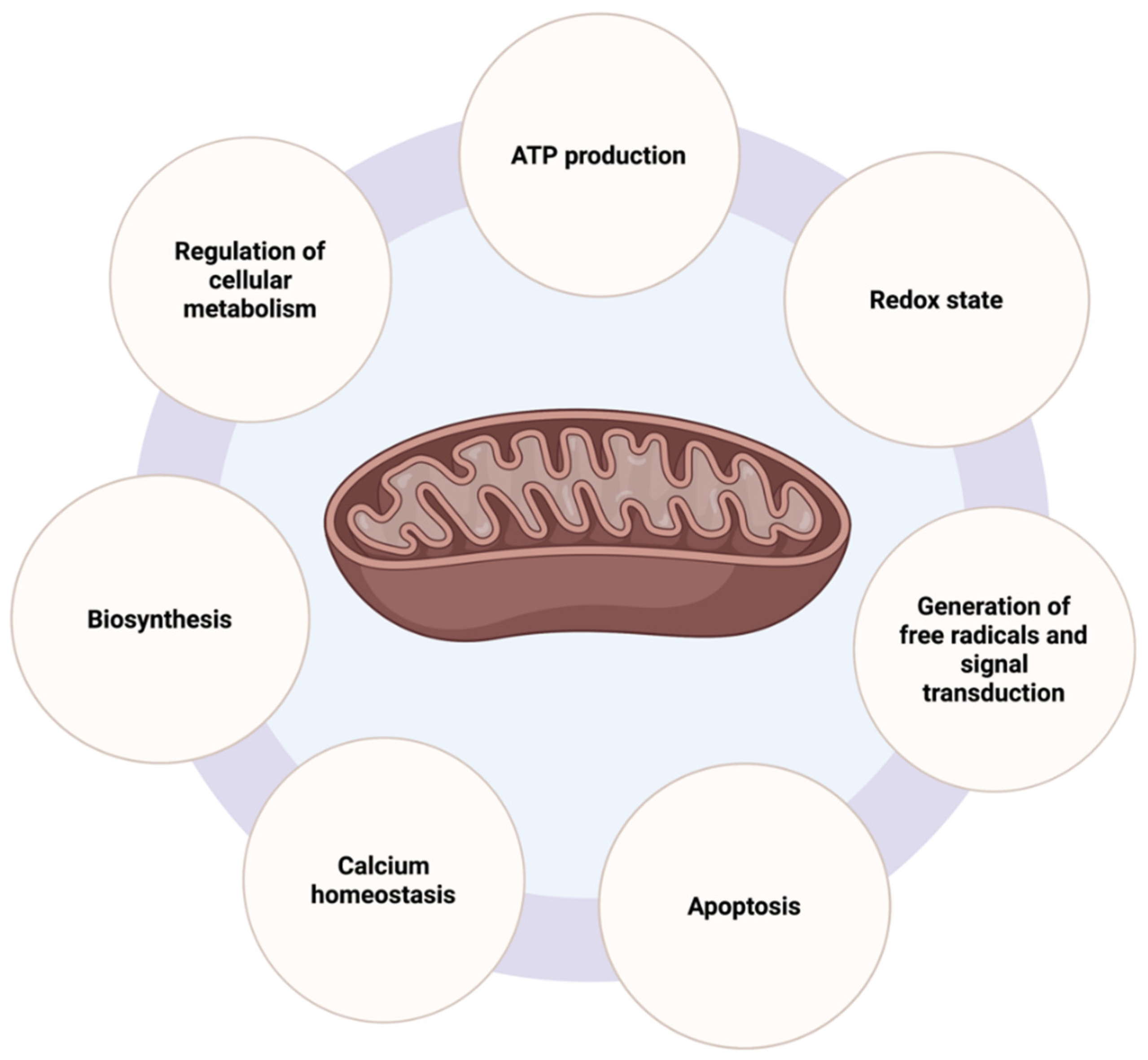{kind=link}
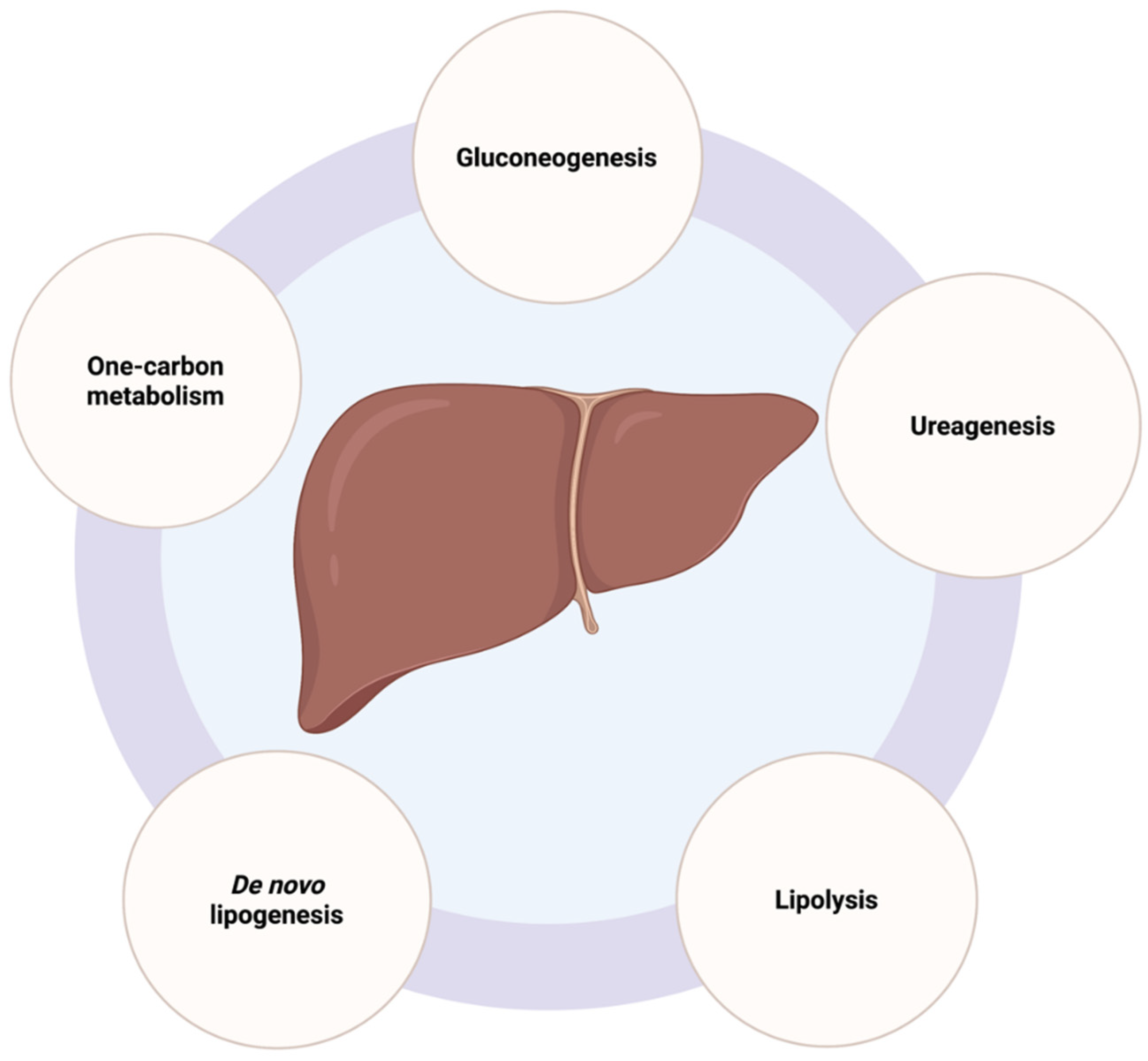{kind=link}
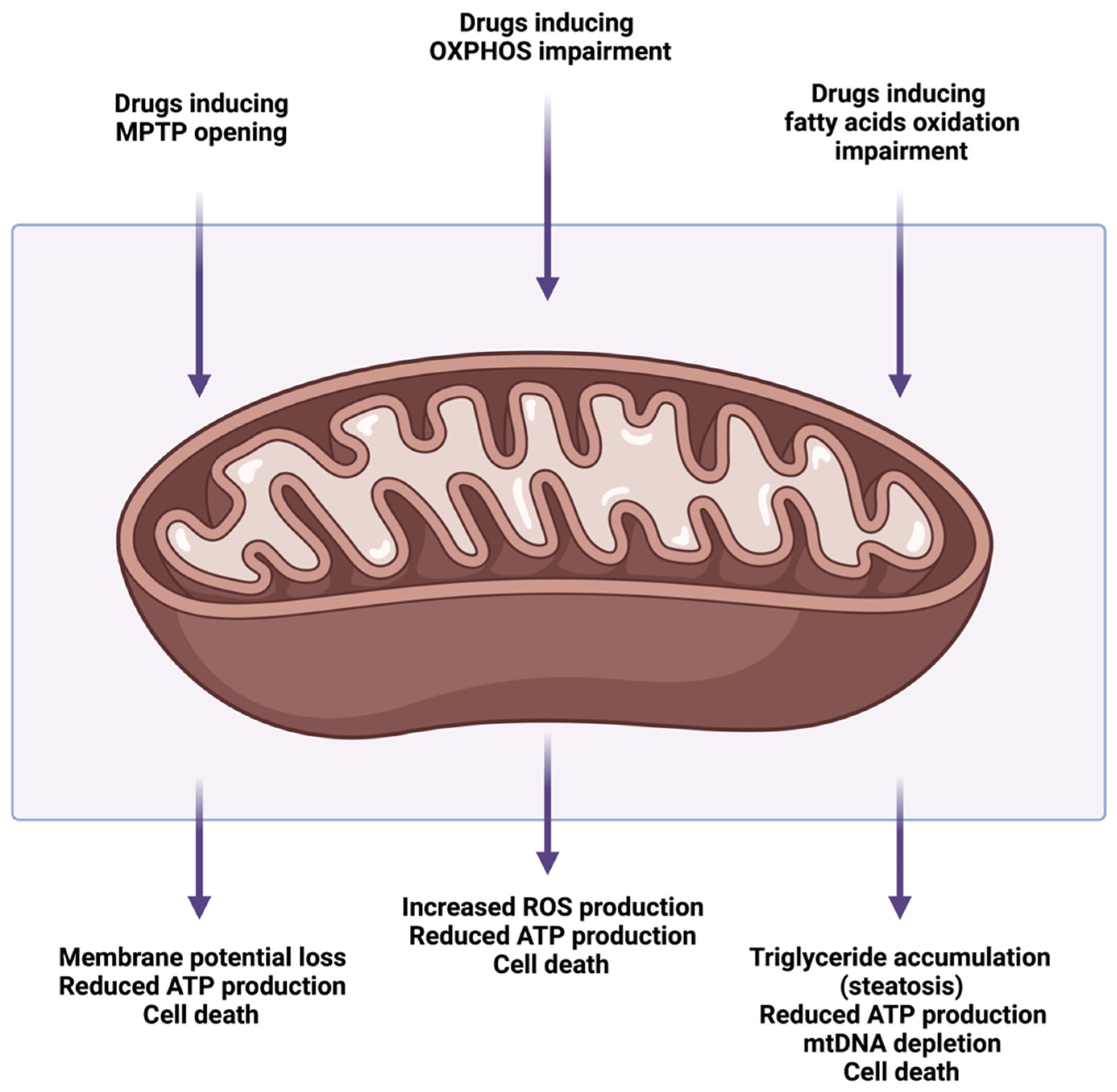{kind=link}
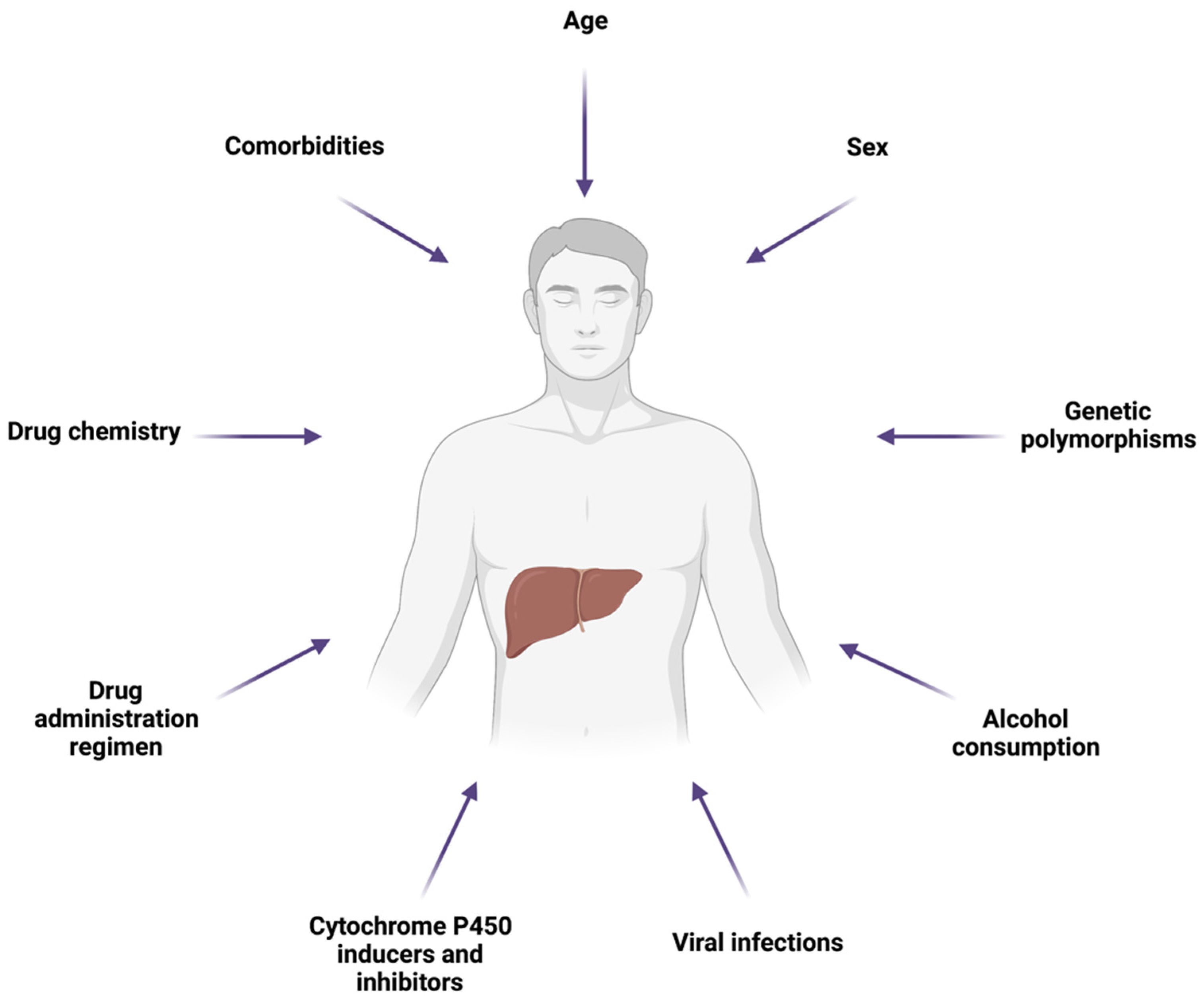{kind=link}
| Drug | Therapeutic Class | Mechanism Leading to MPTP Opening | References |
|---|---|---|---|
| Acetaminophen | Analgesic | JNK activation, intracellular Fe2+ increase, translocation into mitochondria | [76,77,78] |
| Alpidem | Anxiolytic | Ligand | [73] |
| Amiodarone | Antiarrhythmic | Oxidative stress | [79] |
| Diclofenac | NSAID | Oxidative stress, intracellular Ca2+ increase | [74] |
| Disulfiram | Aversion therapy for alcoholism | Oxidative stress | [80] |
| Nimesulide | NSAID | Oxidative stress, intracellular Ca2+ increase | [81] |
| Salicylic acid | NSAID | Oxidative stress, intracellular Ca2+ increase | [82,83] |
| Troglitazone | Antidiabetic | JNK activation, oxidative stress, intracellular Ca2+ increase | [84,85,86] |
| Valproic acid | Antiepileptic | Oxidative stress | [69] |
| Drug | Therapeutic Class | Mechanism Leading to Impaired OXPHOS | References |
|---|---|---|---|
| Acetaminophen | Analgesic | Direct inhibition of ETC activity (inhibition of complexes I and II) | [93,94] |
| Alpidem | Anxiolytic | OXPHOS uncoupling, direct inhibition of ETC activity | [73] |
| Amiodarone | Antiarrhythmic | OXPHOS uncoupling, direct inhibition of ETC activity (inhibition of complexes I, II, and III) | [90] |
| Benzarone | Thrombolytic | OXPHOS uncoupling | [95] |
| Benzbromarone | Uricosuric | OXPHOS uncoupling | [95] |
| Buprenophrine | Therapy for opioid dependence | OXPHOS uncoupling, direct inhibition of ETC activity | [91] |
| Diclofenac | NSAID | OXPHOS uncoupling | [96] |
| Disulfiram | Aversion therapy for alcoholism | Direct inhibition of ETC activity | [80] |
| Ibuprofen | NSAID | OXPHOS uncoupling | [97] |
| Nilutamide | Antineoplastic | Direct inhibition of ETC activity (inhibition of complex I) | [92] |
| Nimesulide | NSAID | OXPHOS uncoupling | [81] |
| Perhexiline | Antianginal | OXPHOS uncoupling, direct inhibition of ETC activity (inhibition of complexes I and II) | [98] |
| Salicylic acid | NSAID | OXPHOS uncoupling | [96] |
| Tacrine | Anti-dementia | OXPHOS uncoupling | [99] |
| Tamoxifen | Antineoplastic | OXPHOS uncoupling, direct inhibition of ETC activity (inhibition of complexes III and IV) | [100,101] |
| Tetracyclines | Antibiotic | Direct inhibition of ETC activity (inhibition of complexes I and IV) | [102] |
| Troglitazone | Antidiabetic | Direct inhibition of ETC activity (inhibition of complex II, III, IV, and V) | [103] |
| Drug | Therapeutic Class | Type of Steatosis Induced | Mechanism Leading to Impaired Fatty Acid Oxidation | References |
|---|---|---|---|---|
| Acetaminophen | Analgesic | Microvesicular | Inhibition of fatty acid oxidation enzymes and inhibition of ETC activity | [89,93,126] |
| Amineptine | Antidepressant | Microvesicular | Inhibition of fatty acid oxidation enzymes and sequestration of fatty acid oxidation cofactors | [127] |
| Amiodarone | Antiarrhythmic | Microvesicular, macrovesicular | Inhibition of fatty acid oxidation enzymes and inhibition of ETC activity | [90,116] |
| Buprenophrine | Therapy for opioid dependence | Microvesicular | Inhibition of ETC activity | [91] |
| Didanosine | Antiretroviral | Microvesicular, macrovesicular | mtDNA depletion and inhibition of mtDNA polymerase γ | [119,128] |
| Fialuridine | Antiviral | Microvesicular | mtDNA depletion and inhibition of mtDNA polymerase γ | [117,119] |
| Ibuprofen | NSAID | Microvesicular | Inhibition of fatty acid oxidation enzymes and sequestration of fatty acid oxidation cofactors | [113] |
| Panadiplon | Anxiolytic | Microvesicular | Sequestration of fatty acid oxidation cofactors | [129,130] |
| Perhexiline | Antianginal | Microvesicular, macrovesicular | Inhibition of fatty acid oxidation enzymes and inhibition of ETC activity | [98,111] |
| Salicylic acid | NSAID | Microvesicular | Sequestration of fatty acid oxidation cofactors | [114] |
| Stavudine | Antiretroviral | Microvesicular, macrovesicular | mtDNA depletion and inhibition of mtDNA polymerase γ | [128] |
| Tamoxifen | Antineoplastic | Macrovesicular | Inhibition of fatty acid oxidation enzymes, inhibition of ETC activity, and mtDNA depletion | [100,107,131,132] |
| Tetracyclines | Antibiotic | Microvesicular | Inhibition of fatty acid oxidation enzymes | [133,134,135] |
| Tianeptine | Antidepressant | Microvesicular | Inhibition of fatty acid oxidation enzymes | [136] |
| Troglitazone | Antidiabetic | Microvesicular | Inhibition of fatty acid oxidation enzymes | [137,138] |
| Valproic acid | Antiepileptic | Microvesicular, macrovesicular | Inhibition of fatty acid oxidation enzymes and sequestration of fatty acid oxidation cofactors | [70,71] |
| Zidovudine | Antiretroviral | Microvesicular | mtDNA depletion and inhibition of mtDNA polymerase γ | [119,139] |
| Method/Assay | Advantages | Disadvantages | References |
|---|---|---|---|
| Swelling assay for MPTP opening—absorbance | Allows the multiplex assessment of mitochondrial Ca2+ uptake and mitochondrial swelling due to loss of the inner mitochondrial membrane integrity | Only possible in isolated mitochondria; mitochondria isolation procedure can affect shape and morphology of mitochondria, reducing reliability of obtained data | [213,214,215,216] |
| Swelling assay for MPTP opening—microscopy | Intact cells and fixed cells/tissue samples can be used | Low resolution; diffraction limits; artifacts due to sample preparation and fixation; difficult to estimate the actual volume of mitochondria | [216,217,218] |
| Fatty acid oxidation—14C labeled palmitate | Direct measurement of mitochondrial fatty acid oxidation efficiency | Low 14CO2 recovery rate; large inter-assay variability; use of radiolabeled compounds | [219,220] |
| Steatosis—staining procedures (Oil Red O, Sudan Black B, Nile Red, BODIPYTM 493/503) | Simple and reproducible; allows determination of cellular localization and distribution of lipid droplets; compatible with other assays; compatible with various detection methods (microscopy, flow cytometry, plate readers) | Lower specificity; stability or the background of the signal | [221,222,223,224,225,226,227] |
| Steatosis—absolute lipid quantification | Specificity and sensibility; commercially available kits | Laborious procedure; does not provide information about cellular localization of lipids | [228,229,230] |
| OCR—Clark electrode | Simple; inexpensive | Potential artifacts due to oxygen consumption by the electrode; required cell detachment by trypsinization can affect OCR | [231,232] |
| OCR—Seahorse XF Flux Analyzer | Simultaneous measurement of OCR and extracellular acidification rate; reduced sample volume; high throughput | Expensive; limited to non-perfused cell population measurements | [233,234,235] |
| OCR—Oroboros Oxygraph-2k | Simultaneous measurement of ORC and Δψm and ADP-ATP exchange rate in suspension | Labor-intensive; low throughput | [233,236] |
| Mitochondrial NADPH and NADH—autofluorescence | Non-invasive; informative | Excessive exposure highly phototoxic; Exposure optimization required to improve signal-to-noise levels | [237,238,239] |
| Mitochondrial NADPH and NADH—fluorescent reporters | Improved sensitivity; low phototoxicity | pH sensitivity; transfection efficiency | [240,241,242,243] |
| Mitochondrial membrane potential variation—fluorescent dyes | Reliable and informative; compatible with various detection methods (microscopy, flow cytometry, plate readers) | Most probes are substrates of MDR transporters and mitochondrial loading can be affected; need for pharmacological inhibitors such as cyclosporin A; phototoxicity and photobleaching in confocal microscopy; possible binding to mitochondrial membrane and affecting mitochondrial respiration; some probes present high toxicity; low sensitivity | [244,245,246,247,248] |
| Respiratory chain complexes activity | Very informative when combined with other measurements such as OCR; useful for detecting molecular origin of mitochondrial defects | Not necessarily reflecting mitochondrial dysfunction (presence of compensatory mechanisms) | [232,249,250,251] |
| Mitochondrial ROS—redox-sensitive fluorophores | Relatively easy to perform and measure; compatible with live microscopy, flow cytometry, and plate readers | Not reliably attributable to mitochondrial ROS; requires some form of correction; optimization required to avoid dilution and saturation of the signal; non-linear fluorescence response; photosensitivity and pH sensitivity; auto-oxidation | [252,253,254,255,256,257] |
| Mitochondrial ROS—redox-sensitive fluorescent proteins | Suitable for monitoring ROS production over longer times | Lack of specificity; dye-specific pH sensitivity | [258,259,260] |
| Mitochondrial ROS—redox-sensitive enzymatic assays | Allows determination of enzymatic ROS origin | Provides only fixed time-point readouts | [261,262] |
| Mitochondrial ATP | Possible to multiplex with other fluorescent probes; reproducible; signal stability | Potential phototoxicity; potential pH and temperature sensitivity; weak signal in bioluminescence assays; expensive | [263,264,265,266,267,268] |
| Mitochondrial Ca2+ | Allows specific mitochondrial targeting with genetically encoded fluorescent reporters; can be combined with Δψm measurement | Incomplete intramitochondrial accumulation of traditional fluorescent probes; possibly can cause alterations of Ca2+ dynamics; mostly free and not total Ca2+ is measured | [269,270,271,272,273,274,275,276] |
| Mitochondrial pH | Allows reliable calculation of the proton-motive force; good indicator of energy metabolism fluctuations | Many pH sensor probes are not specific for mitochondria; requires the use of additional mitochondrial markers | [246,277,278] |
| mtDNA copy number | Easy to measure and accessible | High variability in experimental procedures related to DNA extraction, quality, cross-contaminations, accuracy | [279,280,281,282] |
| Microscopy methods for mitochondrial morphology, size, and number | Provide more detail and insight when combined with other methods of mitochondrial dysfunction | Some probes not compatible with paraformaldehyde fixation; low transfection efficiency for targeted reporter proteins; laborious optimization of experimental protocols | [283,284] |
| Tetrazolium salt assay | Easy to perform; reproducible; low cost | Not reliable for mitochondrial activity assessment; endpoint assay; dependent on cell type and cell culturing | [285,286,287] |
| Resazurin reduction assay | Easy to perform; compatible with other assays; high sensitivity | Not reliable for mitochondrial activity assessment; requires optimization of incubation times, possibly causing cellular alterations | [288,289,290,291] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mihajlovic, M.; Vinken, M. Mitochondria as the Target of Hepatotoxicity and Drug-Induced Liver Injury: Molecular Mechanisms and Detection Methods. Int. J. Mol. Sci. 2022, 23, 3315. https://doi.org/10.3390/ijms23063315
Mihajlovic M, Vinken M. Mitochondria as the Target of Hepatotoxicity and Drug-Induced Liver Injury: Molecular Mechanisms and Detection Methods. International Journal of Molecular Sciences. 2022; 23(6):3315. https://doi.org/10.3390/ijms23063315
Chicago/Turabian StyleMihajlovic, Milos, and Mathieu Vinken. 2022. "Mitochondria as the Target of Hepatotoxicity and Drug-Induced Liver Injury: Molecular Mechanisms and Detection Methods" International Journal of Molecular Sciences 23, no. 6: 3315. https://doi.org/10.3390/ijms23063315
APA StyleMihajlovic, M., & Vinken, M. (2022). Mitochondria as the Target of Hepatotoxicity and Drug-Induced Liver Injury: Molecular Mechanisms and Detection Methods. International Journal of Molecular Sciences, 23(6), 3315. https://doi.org/10.3390/ijms23063315