
Interactions between the Re-Emerging Pathogen Corynebacterium diphtheriae and Host Cells




Abstract
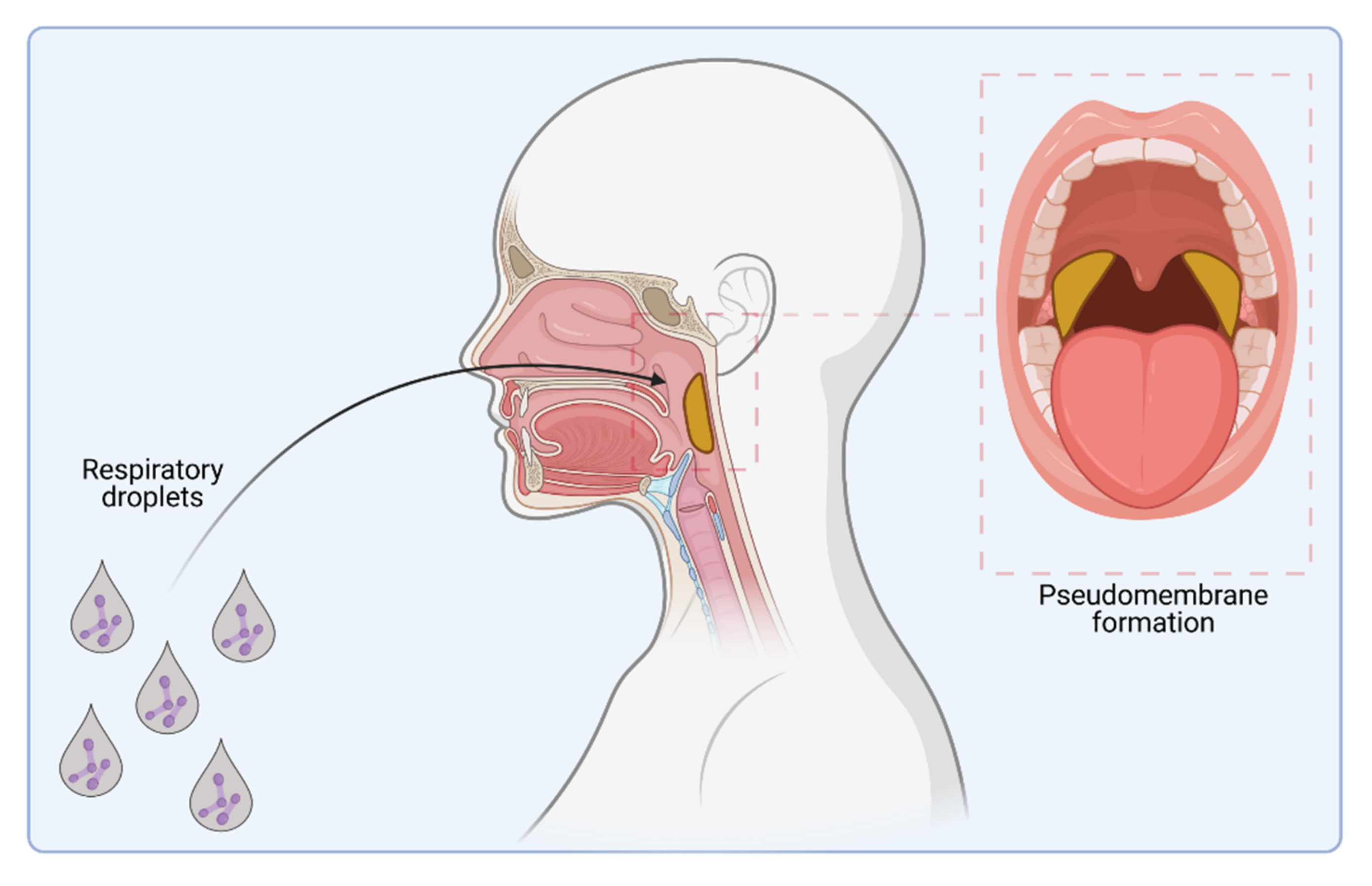
1. Introduction
2. Host Cell Binding Properties of C. diphtheriae
3. Invasion of C. diphtheriae—What We Know to Date
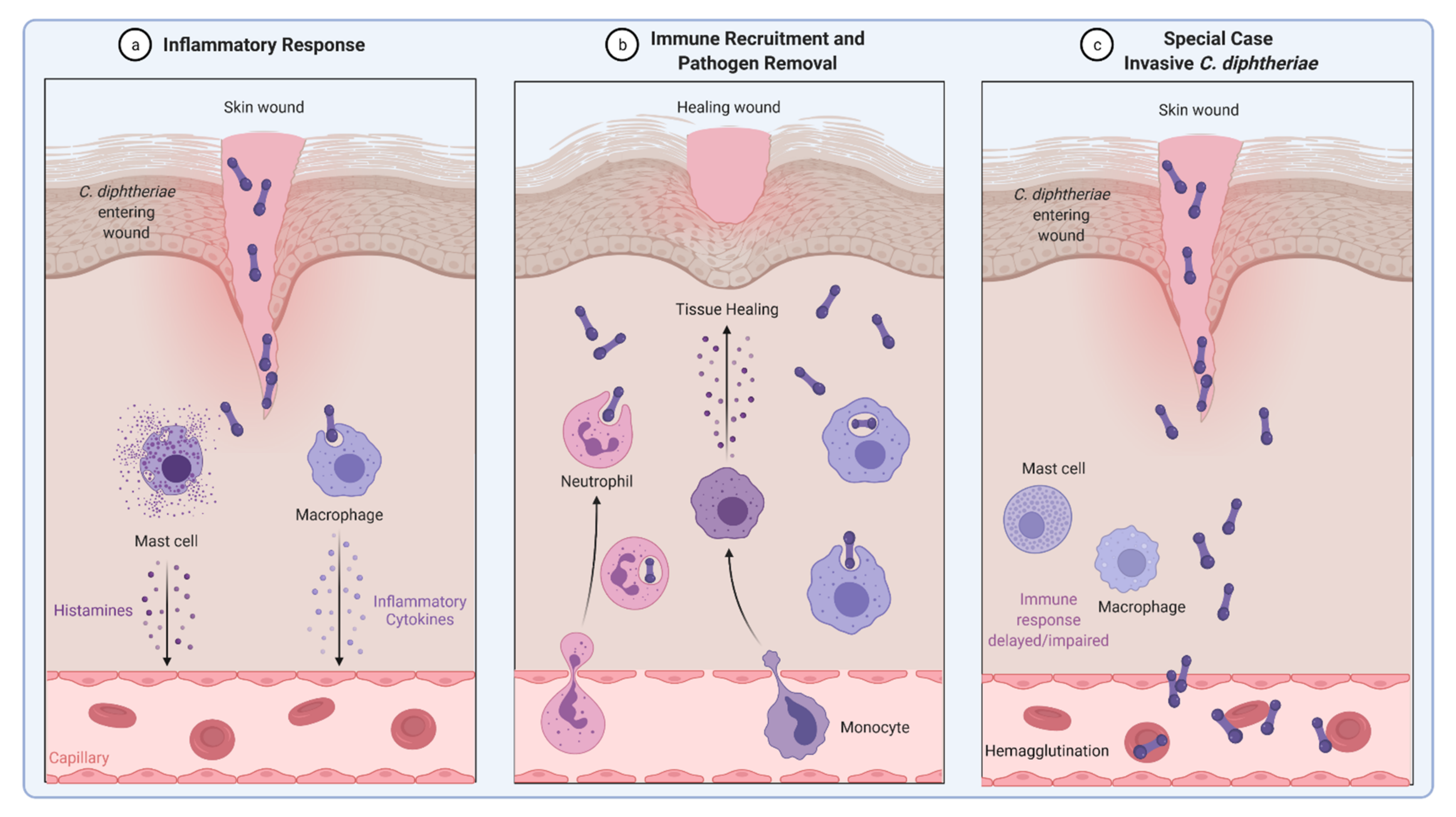
4. Inflammatory Signaling in Response to C. diphtheriae Infection
5. C. diphtheriae-Induced Apoptosis and Necrosis
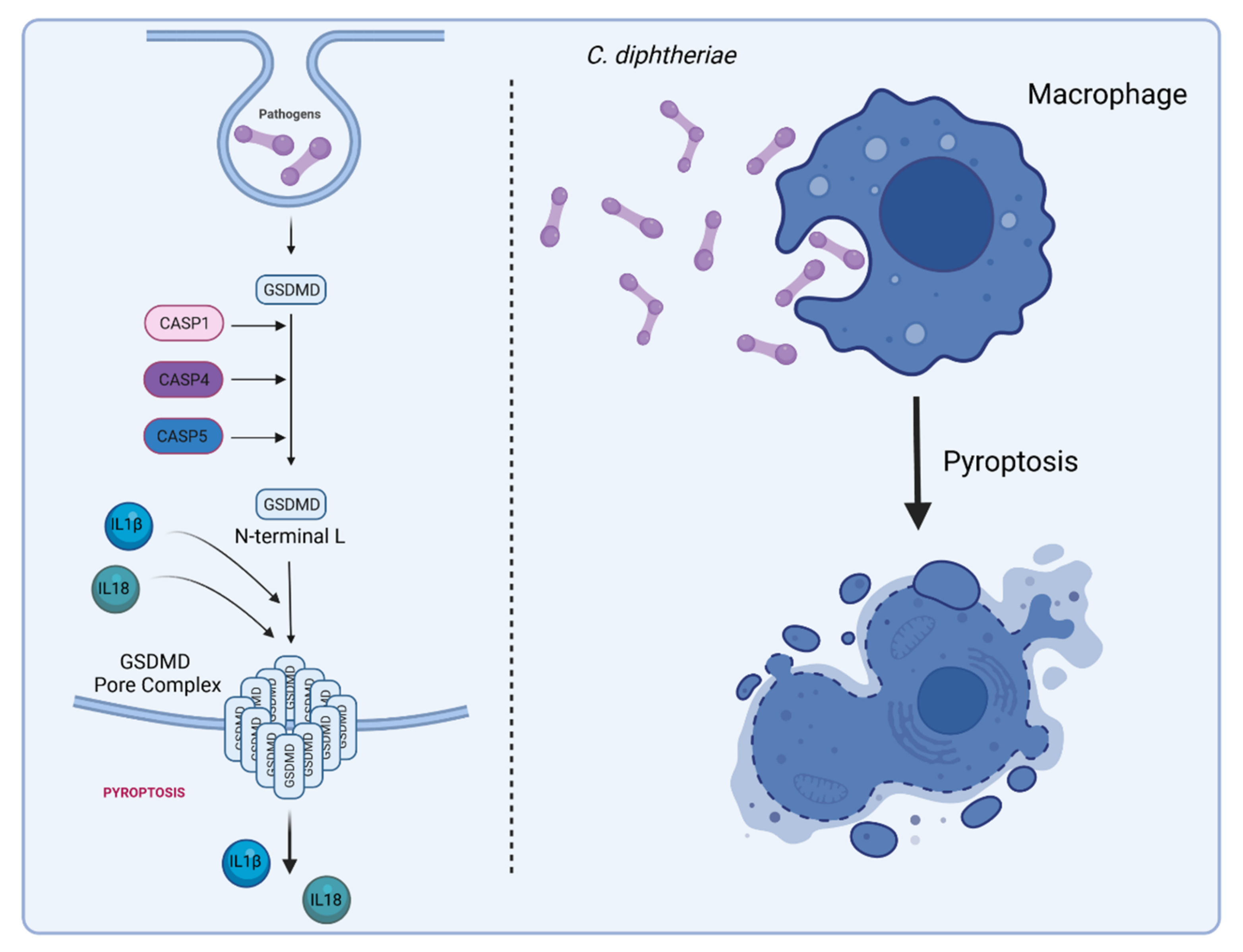
6. Inflammasome Activation and Pyroptosis
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Löffler, F. Untersuchungen über die Bedeutung der Mikroorganismen für die Entstehung der Diphtherie beim Menschen, bei der Taube und beim Kalbe. Mitt. Dem Kais. Gesundh. 1884, 2, 421–499. [Google Scholar]
- Sangal, V.; Hoskisson, P.A. Evolution, epidemiology and diversity of Corynebacterium diphtheriae: New perspectives on an old foe. Infect. Genet. Evol. 2016, 43, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Hoskisson, P.A. Microbe Profile: Corynebacterium diphtheriae—An old foe always ready to seize opportunity. Microbiology 2018, 164, 865–867. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.C.; Efstratiou, A.; Mokrousov, I.; Mutreja, A.; Das, B.; Ramamurthy, T. Diphtheria. Nat. Rev. Dis. Primers 2019, 5, 81. [Google Scholar] [CrossRef]
- Burkovski, A. Diphtheria and its etiological agents. In Corynebacterium diphtheriae and Related Toxigenic Species; Burkovski, A., Ed.; Springer: Dordrecht, The Netherlands, 2014; pp. 1–14. [Google Scholar]
- Zasada, A.A. Corynebacterium diphtheriae infections currently and in the past. Przegl. Epidemiol. 2015, 69, 439–444, 569–574, (In English and Polish). [Google Scholar]
- English, P.C. Diphtheria and theories of infectious disease: Centennial appreciation of the critical role of diphtheria in the history of medicine. Pediatrics 1985, 76, 1–9. [Google Scholar]
- Behring, E. Ueber ein neues Diphtherieschutzmittel. Dtsch. Med. Wochenschr. 1913, 19, 873–876. [Google Scholar] [CrossRef]
- Holmes, R.K. Biology and molecular epidemiology of diphtheria toxin and the tox gene. J. Infect. Dis. 2000, 181, S156–S167. [Google Scholar] [CrossRef]
- Relyveld, E.H. A history of toxoids. In A History of Vaccine Development; Plotkin, S.A., Ed.; Springer: New York, NY, USA, 2011; pp. 57–64. [Google Scholar]
- Tiwari, T.S.P.; Wharton, M. Diphtheria toxoid. In Plotkin’s Vaccines, 7th ed.; Plotkin, S.A., Offit, P.A., Orenstein, W.A., Edwards, K.M., Eds.; Elsevier: Philadelphia, PA, USA, 2012; pp. 261–275. [Google Scholar]
- Vitek, C.R.; Wharton, M. Diphtheria in the former Soviet Union: Reemergence of a pandemic disease. Emerg. Infect. Dis. 1998, 4, 539–550. [Google Scholar] [CrossRef]
- Dittmann, S.; Wharton, M.; Vitek, C.; Ciotti, M.; Galazka, A.; Guichard, S.; Hardy, I.; Kartoglu, U.; Koyama, S.; Kreysler, J.; et al. Successful control of epidemic diphtheria in the states of the former Union of Soviet Socialist Republics: Lessons learned. J. Infect. Dis. 2000, 181, S10–S22. [Google Scholar] [CrossRef]
- Markina, S.S.; Maksimova, N.M.; Vitek, C.R.; Bogatyreva, E.Y.; Monisov, A.A. Diphtheria in the Russian Federation in the 1990s. J. Infect. Dis. 2000, 181, S27–S34. [Google Scholar] [CrossRef]
- Clarke, K.E.N.; MacNeil, A.; Hadler, S.; Scott, C.; Tiwari, T.S.P.; Cherian, T. Global epidemiology of diphtheria, 2000–2017. Emerg. Infect. Dis. 2019, 25, 1834–1842. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, R.; Akhmetzhanov, A.R.; Endo, A.; Lee, H.; Yamaguchi, T.; Tsuzuki, S.; Nishiura, H. Uncertainty and sensitivity analysis of the basic reproduction number of diphtheria: A case study of a Rohingya refugee camp in Bangladesh, November-December 2017. Peer J. 2018, 6, e4583. [Google Scholar] [CrossRef] [PubMed]
- Exavier, M.M.; Hanna, M.P.; Muscadin, E.; Freishstat, R.J.; Brisma, J.-P.; Canarie, M.F. Diphtheria in children in Northern Haiti. J. Trop. Pediatr. 2019, 65, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Mahomed, S.; Archary, M.; Mutevedzi, P.; Mahabeer, Y.; Govender, P.; Ntshoe, G.; Kuhn, W.; Thomas, J.; Olowolagba, A.; Blumberg, L.; et al. An isolated outbreak of diphtheria in South Africa, 2015. Epidemiol. Infect. 2017, 145, 2100–2108. [Google Scholar] [CrossRef]
- Strauss, R.A.; Herrera-Leon, L.; Guillén, A.C.; Castro, J.S.; Lorenz, E.; Carvajal, A.; Hernandez, E.; Navas, T.; Vielma, S.; Lopez, N.; et al. Molecular and epidemiologic characterization of the diphtheria outbreak in Venezuela. Sci. Rep. 2021, 11, 6378. [Google Scholar] [CrossRef]
- Dureab, F.; Al-Sakkaf, M.; Ismail, O.; Kuunibe, N.; Krisam, J.; Müller, O.; Jahn, A. Diphtheria outbreak in Yemen: The impact of conflict on a fragile health system. Confl. Health. 2019, 13, 19. [Google Scholar] [CrossRef]
- World Health Organization. Diphtheria Reported Cases. Available online: https://apps.who.int/immunization_monitoring/globalsummary/timeseries/tsincidencediphtheria.html (accessed on 22 February 2022).
- World Health Organization. Third Dose of Diphtheria Toxoid, Tetanus Toxoid and Pertussis Vaccine—Reported Estimates of DTP3 Coverage. Available online: https://apps.who.int/immunization_monitoring/globalsummary/timeseries/tscoveragedtp3.html (accessed on 22 February 2022).
- Burkovski, A. Pathogenesis of Corynebacterium diphtheriae and Corynebacterium ulcerans. In Human Emerging and Re-Emerging Infections; Singh, S.K., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2016; Volume 2, pp. 697–708. [Google Scholar]
- Lai, Y.; Purnima, P.; Ho, M.; Ang, M.; Deepak, R.N.; Chew, K.L.; Vasoo, S.; Capulong, D.F.; Lee, V. Fatal case of diphtheria and risk for reemergence, Singapore. Emerg. Infect. Dis. 2018, 24, 2084–2086. [Google Scholar] [CrossRef]
- Scheifer, C.; Rolland-Debord, C.; Badell, E.; Reibel, F.; Aubry, A.; Perignon, A.; Patey, O.; Brisse, S.; Caumes, E. Re-emergence of Corynebacterium diphtheriae. Med. Mal. Infect. 2019, 49, 463–466. [Google Scholar] [CrossRef]
- Hessling, M.; Feiertag, J.; Hoenes, K. Pathogens provoking most deaths worldwide: A review. Heal. Sci. Commun. Biosci. Bio. Res. Comm. 2017, 10, 1–7. [Google Scholar]
- World Health Organisation. Diphtheria vaccine: WHO position paper. Wkly. Epidemiol. Rec. 2017, 31, 417–436. [Google Scholar]
- Hennart, M.; Panunzi, L.G.; Rodrigues, C.; Gaday, Q.; Baines, S.L.; Barros-Pinkelnig, M.; Carmi-Leroy, A.; Dazas, M.; Wehenkel, A.M.; Didelot, X.; et al. Population genomics and antimicrobial resistance in Corynebacterium diphtheriae. Genome Med. 2020, 12, 107. [Google Scholar] [CrossRef] [PubMed]
- Forde, B.M.; Henderson, A.; Playford, E.G.; Looke, D.; Henderson, B.C.; Watson, C.; Steen, J.A.; Sidjabat, H.E.; Laurie, G.; Muttaiyah, S.; et al. Fatal respiratory diphtheria caused by ß-lactam-resistant Corynebacterium diphtheriae. Clin. Infect. Dis. 2021, 73, e4531–e4538. [Google Scholar] [CrossRef] [PubMed]
- Bisht, D.; Meena, L.S. Adhesion molecules facilitate host-pathogen interaction & mediate Mycobacterium tuberculosis pathogenesis. Indian J. Med. Res. 2019, 150, 23–32. [Google Scholar]
- Ott, L. Adhesion properties of toxigenic corynebacteria. AIMS Microbiol. 2018, 4, 85–103. [Google Scholar] [CrossRef]
- Gaspar, A.H.; Ton-That, H. Assembly of distinct pilus structures on the surface of Corynebacterium diphtheriae. J. Bacteriol. 2006, 188, 1526–1533. [Google Scholar] [CrossRef]
- Swierczynski, A.; Ton-That, H. Type III pilus of corynebacteria: Pilus length is determined by the level of its major pilin subunit. J. Bacteriol. 2006, 188, 6318–6325. [Google Scholar] [CrossRef]
- Mandlik, A.; Swierczynski, A.; Das, A.; Ton-That, H. Corynebacterium diphtheriae employs specific minor pilins to target human pharyngeal epithelial cells. Mol. Microbiol. 2007, 64, 111–124. [Google Scholar] [CrossRef]
- Sangal, V.; Blom, J.; Sutcliffe, I.C.; von Hunolstein, C.; Burkovski, A.; Hoskisson, P.A. Adherence and invasive properties of Corynebacterium diphtheriae strains correlates with the predicted membrane-associated and secreted proteome. BMC Genom. 2015, 16, 765. [Google Scholar] [CrossRef]
- Mandlik, A.; Das, A.; Ton-That, H. The molecular switch that activates the cell wall anchoring step of pilus assembly in gram-positive bacteria. Proc. Natl. Acad. Sci. USA 2008, 105, 14147–14152. [Google Scholar] [CrossRef]
- Broadway, M.M.; Rogers, E.A.; Chang, C.; Huang, I.H.; Dwivedi, P.; Yildirim, S.; Schmitt, M.P.; Das, A.; Ton-That, H. Pilus gene pool variation and the virulence of Corynebacterium diphtheriae clinical isolates during infection of a nematode. J. Bacteriol. 2013, 195, 3774–3783. [Google Scholar] [CrossRef] [PubMed]
- Ott, L.; Höller, M.; Rheinlaender, J.; Schaeffer, T.E.; Hensel, M.; Burkovski, A. Strain-specific differences in pili formation and the interaction of Corynebacterium diphtheriae with host cells. BMC Microbiol. 2010, 10, 257. [Google Scholar] [CrossRef] [PubMed]
- Ghasemian, A.; Najar Peerayeh, S.; Bakhshi, B.; Mirzaee, M. The microbial surface components recognizing adhesive matrix molecules (MSCRAMMs) genes among clinical isolates of Staphylococcus aureus from hospitalized children. Iranian J. Pathol. 2015, 10, 258–264. [Google Scholar]
- Antunes, C.A.; dos Santos, L.S.; Hacker, E.; Köhler, S.; Bösl, K.; Ott, L.; das Graças de Luna, M.; Hirata, R., Jr.; de Carvalho Azevedo, V.A.; Mattos-Guaraldi, A.L.; et al. Characterization of DIP0733, a multi-functional virulence factor of Corynebacterium diphtheriae. Microbiology 2015, 161, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, R.S.; Antunes, C.A.; Lourêdo, L.S.; Viana, V.G.; Santos, C.S.D.; Fuentes Ribeiro da Silva, J.; Hirata, R., Jr.; Hacker, E.; Mattos-Guaraldi, A.L.; Burkovski, A. Functional characterization of the collagen-binding protein DIP2093 and its influence on host-pathogen interaction and arthritogenic potential of Corynebacterium diphtheriae. Microbiology 2017, 163, 692–701. [Google Scholar] [CrossRef]
- Ott, L.; Höller, M.; Gerlach, R.G.; Hensel, M.; Rheinlaender, J.; Schaeffer, T.E.; Burkovski, A. Corynebacterium diphtheriae invasion-associated protein (DIP1281) is involved in cell surface organization, adhesion and internalization in epithelial cells. BMC Microbiol. 2010, 10, 2. [Google Scholar] [CrossRef]
- Ott, L.; McKenzie, A.; Baltazar, M.T.; Britting, S.; Bischof, A.; Burkovski, A.; Hoskisson, P. Evaluation of invertebrate infection models for pathogenic corynebacteria. FEMS Immunol. Med. Microbiol. 2012, 65, 413–421. [Google Scholar] [CrossRef]
- Kolodkina, V.; Denisevich, T.; Titov, L. Identification of Corynebacterium diphtheriae gene involved in adherence to epithelial cells. Infect. Genet. Evol. 2011, 11, 518–521. [Google Scholar] [CrossRef]
- Moreira, L.O.; Mattos-Guaraldi, A.L.; Andrade, A.F. Novel lipoarabinomannan-like lipoglycan (CdiLAM) contributes to the adherence of Corynebacterium diphtheriae to epithelial cells. Arch. Microbiol. 2008, 190, 521–530. [Google Scholar] [CrossRef]
- Patey, O.; Bimet, F.; Riegel, P.; Halioua, B.; Emond, J.P.; Estrangin, E.; Dellion, S.; Alonso, J.M.; Kiredjian, M.; Dublanchet, A.; et al. Clinical and molecular study of Corynebacterium diphtheriae systemic infections in France. Coryne Study Group. J. Clin. Microbiol. 1997, 35, 441–445. [Google Scholar] [CrossRef]
- Mattos-Guaraldi, A.L.; Formiga, L.C. Bacteriological properties of a sucrose-fermenting Corynebacterium diphtheriae strain isolated from a case of endocarditis. Curr. Microbiol. 1998, 37, 156–158. [Google Scholar] [CrossRef] [PubMed]
- Fricchione, M.J.; Deyro, H.J.; Jensen, C.Y.; Hoffman, J.F.; Singh, K.; Logan, L.K. Non-toxigenic penicillin and cephalosporin-resistant Corynebacterium diphtheriae endocarditis in a child: A case report and review of the literature. J. Pediat. Infect. Dis. Soc. 2014, 3, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, R.S.; Hacker, E.; Antunes, C.A.; Weerasekera, D.; Dias, A.A.; Martins, C.A.; Hirata, R., Jr.; Santos, K.R.N.D.; Burkovski, A.; Mattos-Guaraldi, A.L. Pathogenic properties of a Corynebacterium diphtheriae strain isolated from a case of osteomyelitis. J. Med. Microbiol. 2016, 65, 1311–1321. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, L.; Priyadarshi, K.; Kumaresan, M.; Sivaradjy, M.; Upadhyay, P.; Elamurugan, T.P.; Sastry, A.S. A rare case report of non-toxigenic Corynebacterium diphtheriae bloodstream infection in an uncontrolled diabetic with peripheral vascular disease. Cureus 2021, 13, e14947. [Google Scholar] [CrossRef]
- Sabbadini, P.S.; Assis, M.C.; Trost, E.; Gomes, D.L.; Moreira, L.O.; Dos Santos, C.S.; Pereira, G.A.; Nagao, P.E.; Azevedo, V.A.; Hirata, R., Jr.; et al. Corynebacterium diphtheriae 67–72p hemagglutinin, characterized as the protein DIP0733, contributes to invasion and induction of apoptosis in HEp-2 cells. Microbial Path. 2012, 52, 165–176. [Google Scholar] [CrossRef]
- Santos, L.S.; Antunes, C.A.; Santos, C.S.; Pereira, J.A.; Sabbadini, P.S.; Luna, M.; Azevedo, V.; Hirata, R., Jr.; Burkovski, A.; Asad, L.M.; et al. Corynebacterium diphtheriae putative tellurite-resistance protein (CDCE8392_0813) contributes to the intracellular survival in human epithelial cells and lethality of Caenorhabditis elegans. Mem. Inst. Oswaldo Cruz. 2015, 110, 662–668. [Google Scholar] [CrossRef]
- dos Santos, L.S.; Antunes, C.A.; de Oliveira, D.M.; Sant’ Anna, L.d.O.; Pereira, J.A.A.; Hirata, R., Jr.; Burkovski, A.; Mattos-Guaraldi, A.L. Tellurite resistance: A putative pitfall in Corynebacterium diphtheriae diagnosis? Antonie Van Leeuwenhoek 2015, 108, 1275–1279. [Google Scholar] [CrossRef]
- Pei, B.; Wang, Y.; Katzianer, D.S.; Wang, H.; Wu, H.; Zhong, Z.; Zhu, J. Role of a TehA homolog in Vibrio cholerae C6706 antibiotic resistance and intestinal colonization. Can. J. Microbiol. 2013, 59, 136–139. [Google Scholar] [CrossRef]
- Franks, S.E.; Ebrahimi, C.; Hollands, A.; Okumura, C.Y.; Aroian, R.V.; Nizet, V.; McGillivray, S.M. Novel role for the yceGH tellurite resistance genes in the pathogenesis of Bacillus anthracis. Infect. Immun. 2014, 82, 1132–1140. [Google Scholar] [CrossRef]
- Cappelli, E.A.; do Espírito Santos Cucinelli, A.; Simpson-Louredo, L.; Freire Canellas, M.E.; Azevedo Antunes, C.; Burkovski, A.; Fuentes Ribeiro da Silva, J.; Sanches dos Santos, L.; Mattos Saliba, A.; Mattos-Guaraldi, A.L. Effects of OxyR as a negative regulator on NO production and mechanisms of host-pathogen interaction of Corynebacterium diphtheriae CDC-E8392 with human epithelial cell lines, Caenorhabditis elegans and murine infection models. Braz. J. Microbiol. 2022, in press. [Google Scholar]
- Schick, J.; Etschel, P.; Bailo, R.; Ott, L.; Bhatt, A.; Lepenies, B.; Kirschning, C.; Burkovski, A.; Lang, R. Toll-like receptor 2 and Mincle cooperatively sense corynebacterial cell wall glycolipids. Infect. Immun. 2017, 85, e00075-17. [Google Scholar] [CrossRef] [PubMed]
- Weerasekera, D.; Fastner, T.; Lang, R.; Burkovski, A.; Ott, L. Of mice and men: Interaction of Corynebacterium diphtheriae strains with murine and human phagocytes. Virulence 2019, 10, 414–428. [Google Scholar] [CrossRef] [PubMed]
- Ton-That, H.; Marraffini, L.A.; Schneewind, O. Sortases and pilin elements involved in pilus assembly of Corynebacterium diphtheriae. Mol. Microbiol. 2004, 53, 251–261. [Google Scholar] [CrossRef]
- Hirata, R., Jr.; Souza, S.M.; Rocha-de-Souza, C.M.; Andrade, A.F.; Monteiro-Leal, L.H.; Formiga, L.C.; Mattos-Guaraldi, A.L. Patterns of adherence to HEp-2 cells and actin polymerisation by toxigenic Corynebacterium diphtheriae strains. Microbial Path. 2004, 36, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Weerasekera, D.; Möller, J.; Kraner, M.E.; Azevedo Antunes, C.; Mattos-Guaraldi, A.L.; Burkovski, A. Beyond diphtheria toxin: Cytotoxic proteins of Corynebacterium ulcerans and Corynebacterium diphtheriae. Microbiology 2019, 165, 876–890. [Google Scholar] [CrossRef] [PubMed]
- Shishido, Y.; Sharma, K.D.; Higashiyama, S.; Klagsbrun, M.; Mekada, E. Heparin-like molecules on the cell surface potentiate binding of diphtheria toxin to the diphtheria toxin receptor/membrane-anchored heparin-binding epidermal growth factor-like growth factor. J. Biol. Chem. 1995, 270, 29578–29585. [Google Scholar] [CrossRef]
- Collier, R.J. Diphtheria toxin: Mode of action and structure. Bacteriol. Rev. 1975, 39, 54–85. [Google Scholar] [CrossRef]
- Pappenheimer, A.M. Diphtheria toxin. Ann. Rev. Biochem. 1977, 46, 69–94. [Google Scholar] [CrossRef]
- Roux, E.; Yersin, A. Contribution à l’étude de la diphtérie. Ann. Inst. Pasteur. 1888, 2, 629–661. [Google Scholar]
- Ott, L.; Scholz, B.; Höller, M.; Hasselt, K.; Ensser, A.; Burkovski, A. Induction of the NFκ-B signal transduction pathway in response to Corynebacterium diphtheriae infection. Microbiology 2013, 159, 126–135. [Google Scholar] [CrossRef]
- Niederweis, M.; Danilchanka, O.; Huff, J.; Hoffmann, C.; Engelhardt, H. Mycobacterial outer membranes: In search of proteins. Trends Microbiol. 2010, 18, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Burkovski, A. Cell envelope of corynebacteria: Structure and influence on pathogenicity. ISRN Microbiol. 2013, 2013, 935736. [Google Scholar] [CrossRef] [PubMed]
- Schoenen, H.; Bodendorfer, B.; Hitchens, K.; Manzanero, S.; Werninghaus, K.; Nimmerjahn, F.; Agger, E.M.; Stenger, S.; Andersen, P.; Ruland, J.; et al. Cutting edge: Mincle is essential for recognition and adjuvanticity of the mycobacterial cord factor and its synthetic analog trehalose-dibehenate. J. Immunol. 2010, 184, 2756–2760. [Google Scholar] [CrossRef]
- Murphy, J.R. Mechanism of diphtheria toxin catalytic domain delivery to the eukaryotic cell cytosol and the cellular factors that directly participate in the process. Toxins 2011, 3, 294–308. [Google Scholar] [CrossRef] [PubMed]
- Sangal, V.; Burkovski, A.; Hunt, A.C.; Edwards, B.; Blom, J.; Hoskisson, P.A. A lack of genetic basis for biovar differentiation in clinically important Corynebacterium diphtheriae from whole genome sequencing. Infect. Genet. Evol. 2014, 21, 54–57. [Google Scholar] [CrossRef]
- Muthuirulandi Sethuvel, D.P.; Subramanian, N.; Pragasam, A.K.; Inbanathan, F.Y.; Gupta, P.; Johnson, J.; Sharma, N.C.; Hemvani, N.; Veeraraghavan, B.; Anandan, S.; et al. Insights to the diphtheria toxin encoding prophages amongst clinical isolates of Corynebacterium diphtheriae from India. Indian J. Med. Microbiol. 2019, 37, 423–425. [Google Scholar] [CrossRef]
- Schmitt, M. Iron acquisition and iron-dependent gene expression in Corynebacterium diphtheriae. In Corynebacterium diphtheriae and Related Toxigenic Species; Burkovski, A., Ed.; Springer: Dordrecht, The Netherlands, 2014; pp. 95–121. [Google Scholar]
- Choe, S.; Bennett, M.J.; Fujii, G.; Curmi, P.M.; Kantardjieff, K.; Collier, R.J.; Eisenberg, D. The crystal structure of diphtheria toxin. Nature 1992, 357, 216–222. [Google Scholar] [CrossRef]
- Varol, B.; Bektaş, M.; Nurten, R.; Bermek, E. The cytotoxic effect of diphtheria toxin on the actin cytoskeleton. Cell. Mol. Biol. Lett. 2012, 17, 49–61. [Google Scholar] [CrossRef]
- Iwamoto, R.; Higashiyama, S. Heparin-binding EGF-like growth factor, which acts as the diphtheria toxin receptor, forms a complex with membrane protein DRAP27/CD9, which up-regulates functional receptors and diphtheria toxin sensitivity. EMBO J. 1994, 13, 2322–2330. [Google Scholar] [CrossRef]
- Donovan, J.J.; Simon, M.I.; Draper, R.K.; Montal, M. Diphtheria toxin forms transmembrane channels in planar lipid bilayers. Proc. Natl. Acad. Sci. USA 1981, 78, 172–176. [Google Scholar] [CrossRef]
- Kagan, B.L.; Finkelstein, A.; Colombini, M. Diphtheria toxin fragment forms large pores in phospholipid bilayer membranes. Proc. Natl. Acad. Sci. USA 1981, 78, 4950–4954. [Google Scholar] [CrossRef]
- Morimoto, H.; Bonavida, B. Diphtheria toxin- and Pseudomonas A toxin-mediated apoptosis. ADP ribosylation of elongation factor-2 is required for DNA fragmentation and cell lysis and synergy with tumor necrosis factor-alpha. J. Immunol. 1992, 149, 2089–2094. [Google Scholar] [PubMed]
- Hirata, R., Jr.; Pereira, G.A.; Filardy, A.A.; Gomes, D.L.; Damasco, P.V.; Rosa, A.C.; Nagao, P.E.; Pimenta, F.P.; Mattos-Guaraldi, A.L. Potential pathogenic role of aggregative-adhering Corynebacterium diphtheriae of different clonal groups in endocarditis. Brazilian J. Med. Biol. Res. 2008, 41, 986–991. [Google Scholar] [CrossRef] [PubMed]
- Trost, E.; Blom, J.; de Soares, S.C.; Huang, I.H.; Al-Dilaimi, A.; Schroeder, J.; Jaenicke, S.; Dorella, F.A.; Rocha, F.S.; Miyoshi, A.; et al. Pangenomic study of Corynebacterium diphtheriae that provides insights into the genomic diversity of pathogenic isolates from cases of classical diphtheria, endocarditis, and pneumonia. J. Bacteriol. 2012, 194, 3199–3215. [Google Scholar] [CrossRef] [PubMed]
- Weerasekera, D.; Hahn, J.; Herrmann, M.; Burkovski, A. Induction of necrosis in human macrophage cell lines by Corynebacterium diphtheriae and Corynebacterium ulcerans strains isolated from fatal cases of systemic infections. Int. J. Mol. Sci. 2019, 20, 4109. [Google Scholar] [CrossRef] [PubMed]
- Weerasekera, D.; Hahn, J.; Herrmann, M.; Burkovski, A. Live cell imaging of macrophage/bacterium interaction demonstrates cell lysis induced by Corynebacterium diphtheriae and Corynebacterium ulcerans. BMC Res. Notes 2019, 12, 695. [Google Scholar] [CrossRef]
- Rello, S.; Stockert, J.C.; Moreno, V.; Gámez, A.; Pacheco, M.; Juarranz, A.; Cañete, M.; Villanueva, A. Morphological criteria to distinguish cell death induced by apoptotic and necrotic treatments. Apoptosis 2005, 10, 201–208. [Google Scholar] [CrossRef]
- Galluzzi, L.; Maiuri, M.C.; Vitale, I.; Zischka, H.; Castedo, M.; Zitvogel, L.; Kroemer, G. Cell death modalities: Classification and pathophysiological implications. Cell Death Diff. 2007, 14, 1237–1243. [Google Scholar] [CrossRef]
- Holler, N.; Zaru, R.; Micheau, O.; Thome, M.; Attinger, A.; Valitutti, S.; Bodmer, J.L.; Schneider, P.; Seed, B.; Tschopp, J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nature Immunol. 2000, 1, 489–495. [Google Scholar] [CrossRef]
- Hopkins-Donaldson, S.; Bodmer, J.L.; Bourloud, K.B.; Brognara, C.B.; Tschopp, J.; Gross, N. Loss of caspase-8 expression in highly malignant human neuroblastoma cells correlates with resistance to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis. Cancer Res. 2000, 60, 4315–4319. [Google Scholar]
- Sawai, H. Characterization of TNF-induced caspase-independent necroptosis. Leuk. Res. 2014, 38, 706–713. [Google Scholar] [CrossRef]
- MacFarlane, M.; Williams, A.C. Apoptosis and disease: A life-or-death decision. EMBO Rep. 2004, 5, 674–678. [Google Scholar] [CrossRef] [PubMed]
- De Miguel, C.; Pelegrín, P.; Baroja-Mazo, A.; Cuevas, S. Emerging role of the inflammasome and pyroptosis in hypertension. Int. J. Mol. Sci. 2021, 22, 1064. [Google Scholar] [CrossRef] [PubMed]
- Labbé, K. Saleh, M. Pyroptosis: A caspase-1-dependent programmed cell death and a barrier to infection. In The Inflammasomes; Couillin, I., Pétrilli, V., Martinon, F., Eds.; Springer: Basel, Switzerland, 2011; pp. 17–36. [Google Scholar]
- Ackers, I.; Malgor, R. Interrelationship of canonical and non-canonical Wnt signaling pathways in chronic metabolic diseases. Diabetes Vascular Dis. Res. 2018, 15, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, H.; Duan, X.; Dai, P.; Li, J. Comprehensive analysis of the canonical and non-canonical Wnt signaling pathways in gastric cancer. Digest. Dis. Sci. 2019, 64, 2830–2842. [Google Scholar] [CrossRef] [PubMed]
- Miao, E.A.; Rajan, J.V.; Aderem, A. Caspase-1-induced pyroptotic cell death. Immunol. Rev. 2011, 243, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Dixit, V. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signaling. Nature 2015, 526, 666–671. [Google Scholar]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef]
- Man, S.M.; Kanneganti, T.D. Regulation of inflammasome activation. Immunol. Rev. 2015, 265, 6–21. [Google Scholar] [CrossRef]
- Chan, A.H.; Schroder, K. Inflammasome signaling and regulation of interleukin-1 family cytokines. J. Exp. Med. 2020, 217, e20190314. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Number of Diphtheria Cases | Third Dose DTP Vaccination Coverage (%) |
|---|---|---|
| 2009 | 4349 | 89.06 |
| 2010 | 4603 | 89.48 |
| 2011 | 5626 | 89.90 |
| 2012 | 4490 | 90.33 |
| 2013 | 4680 | 89.54 |
| 2014 | 7774 | 89.84 |
| 2015 | 4535 | 89.03 |
| 2016 | 7102 | 89.16 |
| 2017 | 8819 | 88.77 |
| 2018 | 16,611 | 89.22 |
| 2019 | 22,986 | 89.70 |
| C. diphtheriae Component | Interacting Host Cell Receptor | Function and Experimental System | Reference |
|---|---|---|---|
| CdiLAM and other glycolipids | C-type lectin receptor Mincle, | adhesion to human epithelial cells agglutination of human erythrocytes | [45,57,58] |
| TLR2 | Mincle activation in primary mouse macrophages | ||
| Pili | laminin | adherence to human epithelial cells colonization of C. elegans | [34,36,38,59] |
| CpG methylated DNA | TLR9 | activation of TLR9 in human macrophages | [58] |
| DIP0733 | fibrinogen fibronectin collagen | agglutination of human erythrocytes adherence to human epithelial cells invasion of human epithelial cells collagen and fibrinogen-binding induction of apoptosis colonization of C. elegans lethal to Galleria.mellonella | [40,51,60] |
| DIP1281 | unknown | adherence to human epithelial cells | [42] |
| DIP1546 | unknown | adherence to human epithelial cells colonization of C. elegans | [43] |
| DIP1621 | unknown | adherence to human epithelial cells | [44] |
| DIP2093 | fibrinogen fibronectin collagen | collagen binding adherence to human epithelial cells invasion into human epithelial cells colonization of C. elegans arthritis in mice | [41] |
| Rbp | unknown | cytotoxic effect to Vero cells (green monkey kidney cells) apoptosis and necrosis in human macrophages and epithelial cells detrimental effects to C. elegans and G. mellonella | [61] |
| Diphtheria toxin | HB-EGF EF-2 | receptor-mediated endocytosis of the toxin in human cells ADP ribosylation and stop of protein synthesis lethal to guinea pigs | [62,63,64,65] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ott, L.; Möller, J.; Burkovski, A. Interactions between the Re-Emerging Pathogen Corynebacterium diphtheriae and Host Cells. Int. J. Mol. Sci. 2022, 23, 3298. https://doi.org/10.3390/ijms23063298
Ott L, Möller J, Burkovski A. Interactions between the Re-Emerging Pathogen Corynebacterium diphtheriae and Host Cells. International Journal of Molecular Sciences. 2022; 23(6):3298. https://doi.org/10.3390/ijms23063298
Chicago/Turabian StyleOtt, Lisa, Jens Möller, and Andreas Burkovski. 2022. "Interactions between the Re-Emerging Pathogen Corynebacterium diphtheriae and Host Cells" International Journal of Molecular Sciences 23, no. 6: 3298. https://doi.org/10.3390/ijms23063298
APA StyleOtt, L., Möller, J., & Burkovski, A. (2022). Interactions between the Re-Emerging Pathogen Corynebacterium diphtheriae and Host Cells. International Journal of Molecular Sciences, 23(6), 3298. https://doi.org/10.3390/ijms23063298