
Microbe-Immune Crosstalk: Evidence That T Cells Influence the Development of the Brain Metabolome


Abstract
:
1. Introduction
2. Results
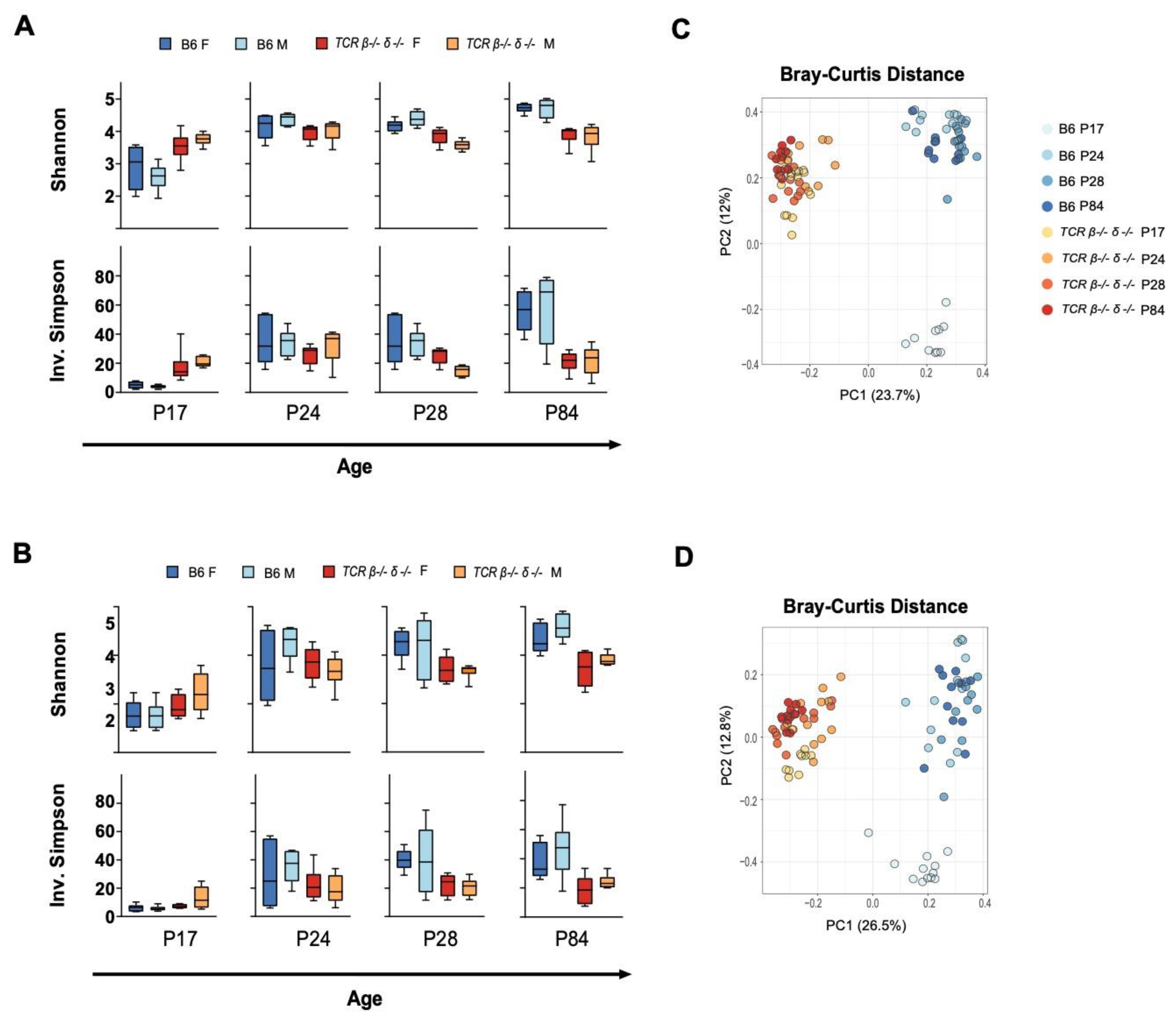
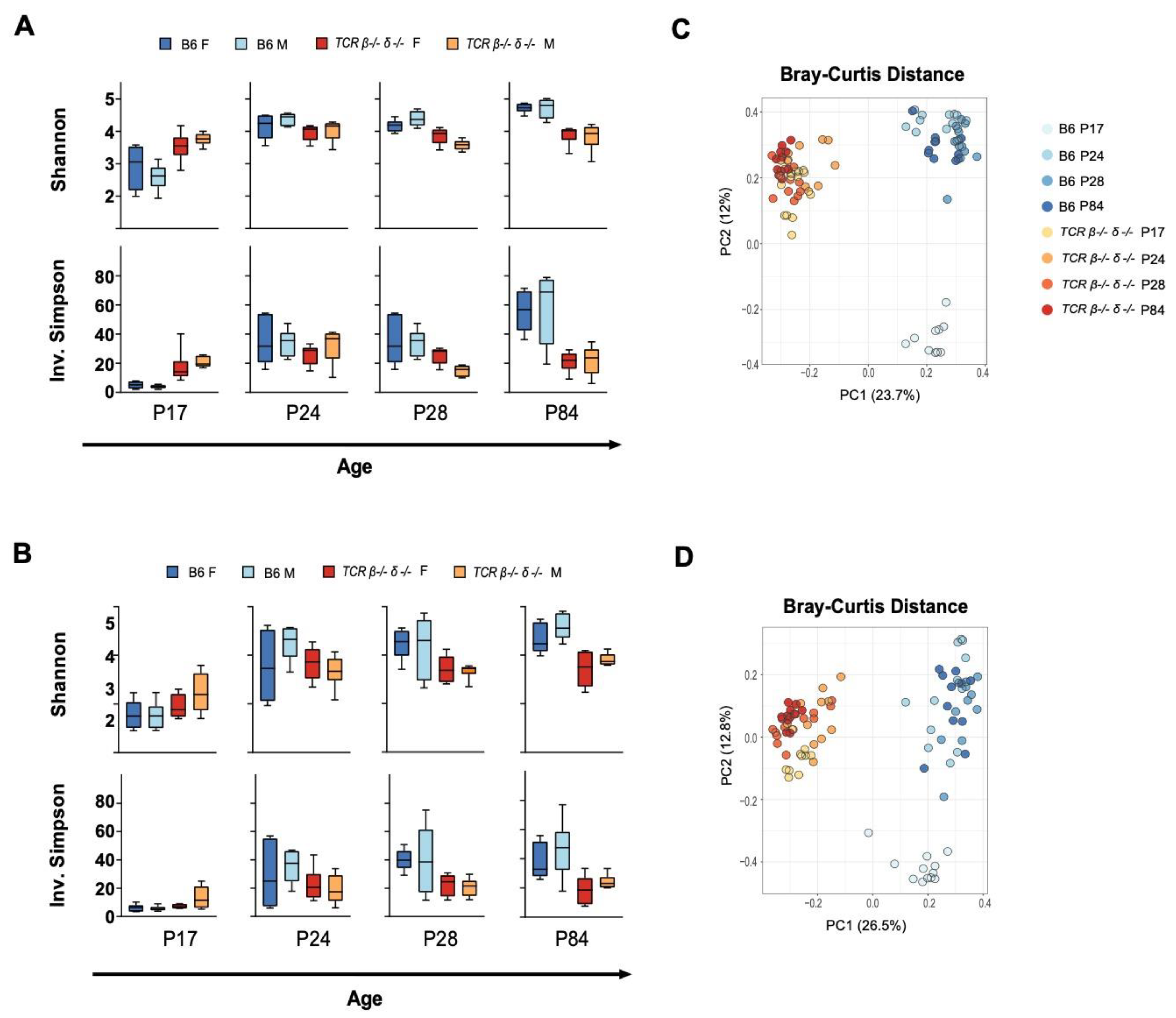
2.1. T-Cell Deficiency Altered the Developmental Trajectory of Alpha Diversity
2.2. Beta Diversity Analysis Revealed Significant Clustering by Genotype
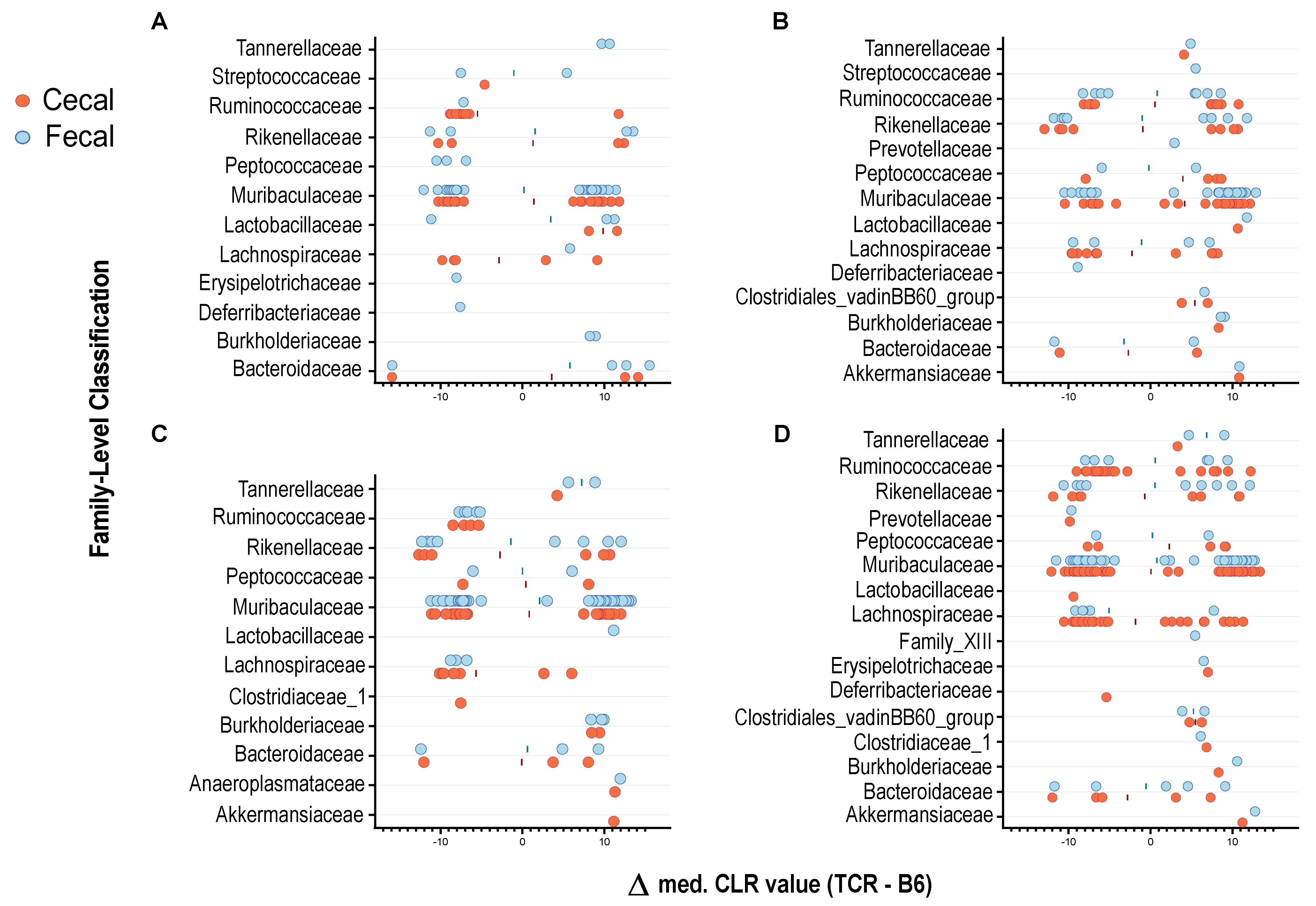
2.3. T-Cell Deficiency Affects the Abundance of Specific Bacterial Taxa
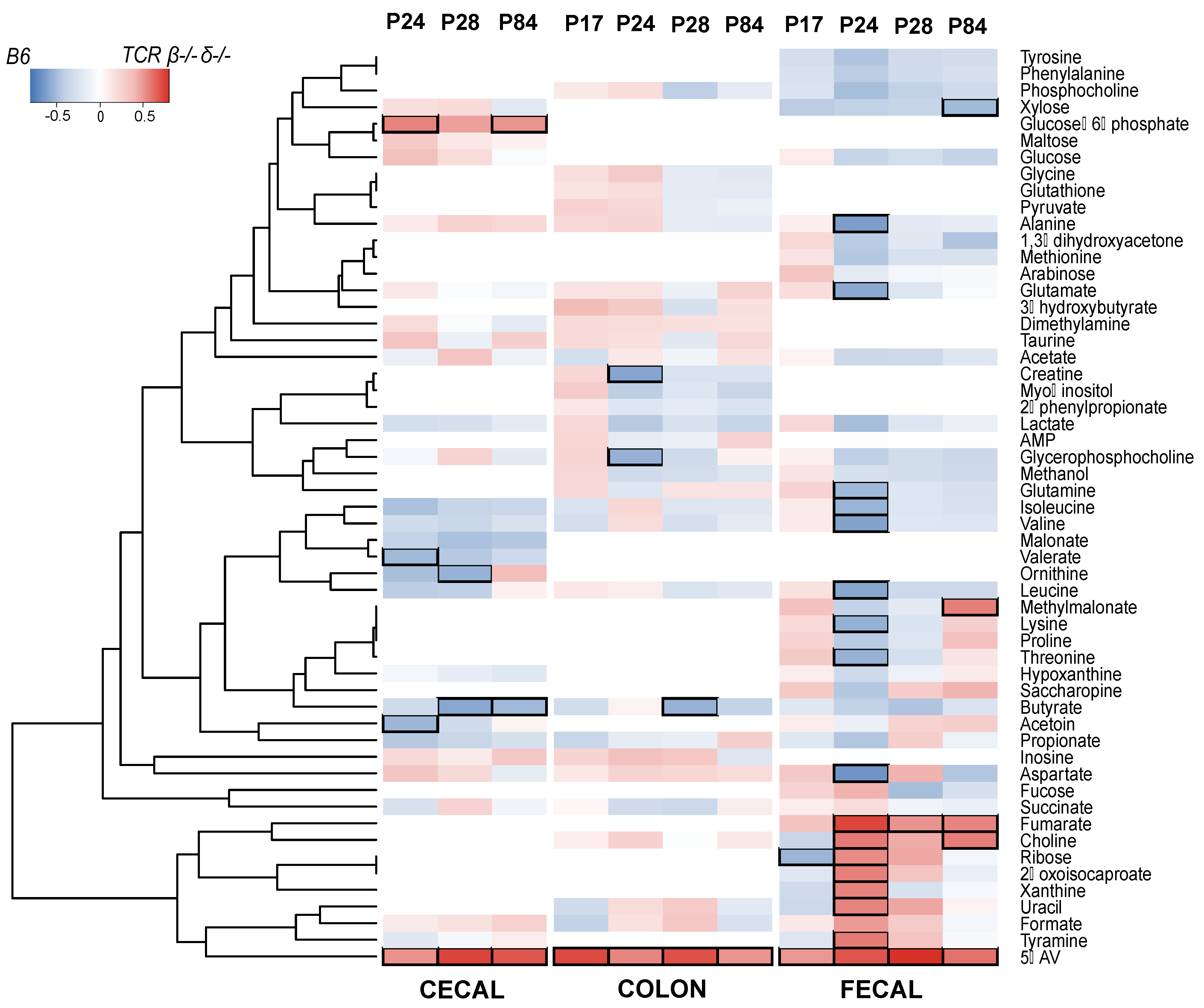
2.4. T-Cell-Deficient Mice Exhibit Altered Development of Colon, Cecal and Fecal Metabolomes
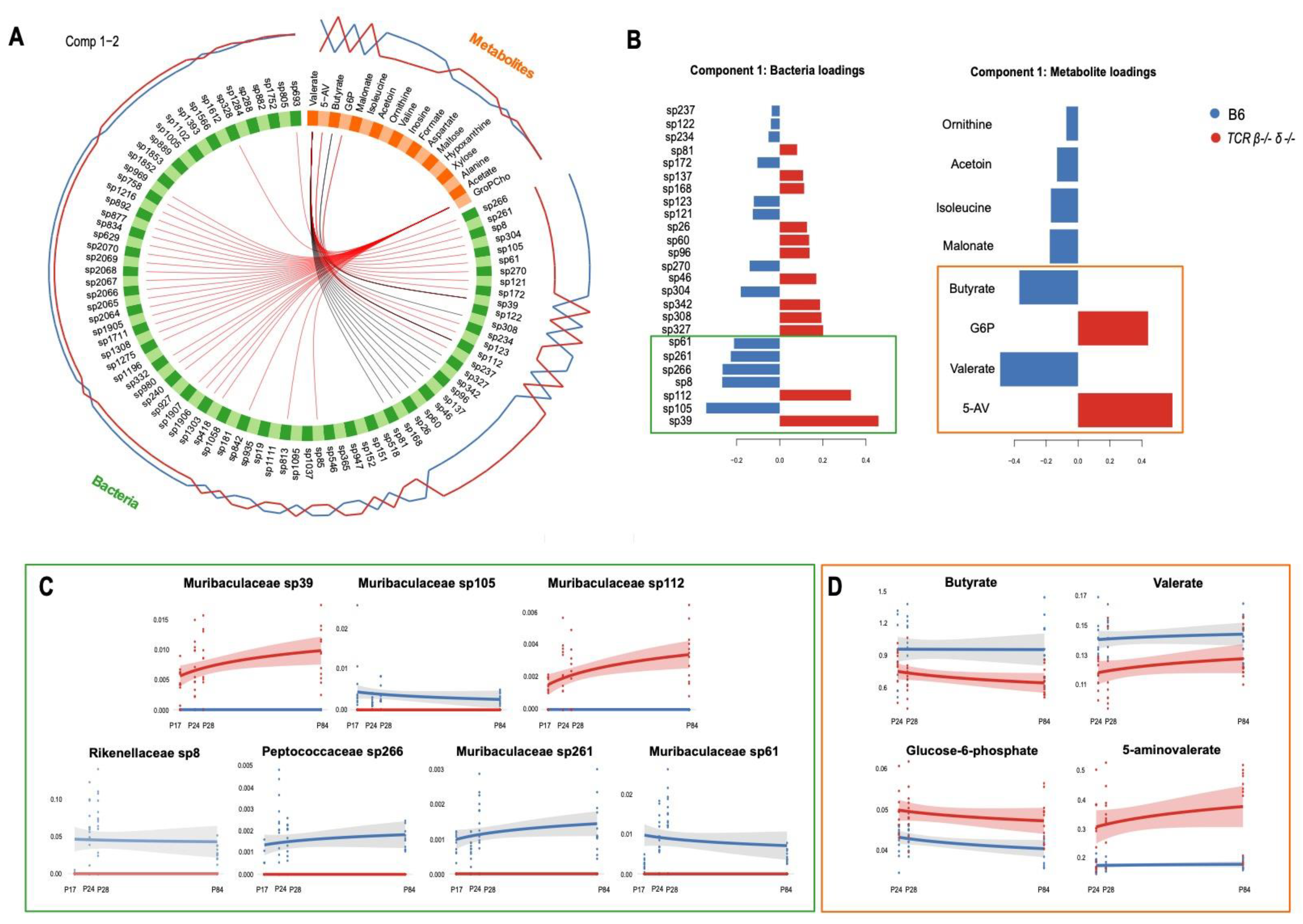
2.5. Abundance of Muribaculaceae Is Linked to Cecal Concentration of Microbial Metabolites
2.6. T-Cell-Deficient Mice Exhibited Altered Development of Cortical and Hippocampal, but Not Hypothalamic, Metabolomes
2.7. T-Cell Deficiency Affects the Expression of Neuroactive Metabolites
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. 16S rRNA Sequencing
4.3. Sample Preparation for Metabolomics
4.4. Metabolic Phenotyping
4.5. Data Analysis
4.5.1. 16S rRNA Analysis
4.5.2. Metabolomic Analysis
4.5.3. Multi-Block Discriminant Analysis with DIABLO
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pronovost, G.N.; Hsiao, E.Y. Perinatal Interactions between the Microbiome, Immunity, and Neurodevelopment. Immunity 2019, 50, 18–36. [Google Scholar] [CrossRef] [PubMed]
- de Lima, K.A.; Rustenhoven, J.; Da Mesquita, S.; Wall, M.; Salvador, A.F.; Smirnov, I.; Martelossi Cebinelli, G.; Mamuladze, T.; Baker, W.; Papadopoulos, Z.; et al. Meningeal gammadelta T cells regulate anxiety-like behavior via IL-17a signaling in neurons. Nat. Immunol. 2020, 21, 1421–1429. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Rescigno, M. The gut immune barrier and the blood-brain barrier: Are they so different? Immunity 2009, 31, 722–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belkaid, Y.; Hand, T.W. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Round, J.L.; Mazmanian, S.K. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 2009, 9, 313–323. [Google Scholar] [CrossRef]
- Ostman, S.; Rask, C.; Wold, A.E.; Hultkrantz, S.; Telemo, E. Impaired regulatory T cell function in germ-free mice. Eur. J. Immunol. 2006, 36, 2336–2346. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Zhao, T.; Yuan, S.; Yang, L.; Ding, J.; Cui, L.; Xu, M. Immunodeficiency Promotes Adaptive Alterations of Host Gut Microbiome: An Observational Metagenomic Study in Mice. Front. Microbiol. 2019, 10, 2415. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, S.; Maruya, M.; Kato, L.M.; Suda, W.; Atarashi, K.; Doi, Y.; Tsutsui, Y.; Qin, H.; Honda, K.; Okada, T.; et al. Foxp3(+) T cells regulate immunoglobulin a selection and facilitate diversification of bacterial species responsible for immune homeostasis. Immunity 2014, 41, 152–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, C.; Dikiy, S.; Bhattarai, S.K.; Chinen, T.; Matheis, F.; Calafiore, M.; Hoyos, B.; Hanash, A.; Mucida, D.; Bucci, V.; et al. Extrathymically Generated Regulatory T Cells Establish a Niche for Intestinal Border-Dwelling Bacteria and Affect Physiologic Metabolite Balance. Immunity 2018, 48, 1245–1257.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, K.; Baptista, A.P.; Tamoutounour, S.; Zhuang, L.; Bouladoux, N.; Martins, A.J.; Huang, Y.; Gerner, M.Y.; Belkaid, Y.; Germain, R.N. Innate and adaptive lymphocytes sequentially shape the gut microbiota and lipid metabolism. Nature 2018, 554, 255–259. [Google Scholar] [CrossRef]
- Cushman, J.; Lo, J.; Huang, Z.; Wasserfall, C.; Petitto, J.M. Neurobehavioral changes resulting from recombinase activation gene 1 deletion. Clin. Diagn. Lab. Immunol. 2003, 10, 13–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sankar, A.; Mackenzie, R.N.; Foster, J.A. Loss of class I MHC function alters behavior and stress reactivity. J. Neuroimmunol. 2012, 244, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Rilett, K.C.; Friedel, M.; Ellegood, J.; MacKenzie, R.N.; Lerch, J.P.; Foster, J.A. Loss of T cells influences sex differences in behavior and brain structure. Brain Behav. Immun. 2015, 46, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Rilett, K.C.; Luo, O.D.; McVey-Neufeld, K.A.; MacKenzie, R.N.; Foster, J.A. Loss of T cells influences sex differences in stress-related gene expression. J. Neuroimmunol. 2020, 343, 577213. [Google Scholar] [CrossRef] [PubMed]
- Clarke, G.; Grenham, S.; Scully, P.; Fitzgerald, P.; Moloney, R.D.; Shanahan, F.; Dinan, T.G.; Cryan, J.F. The microbiome-gut-brain axis during early life regulates the hippocampal serotonergic system in a sex-dependent manner. Mol. Psychiatry 2013, 18, 666–673. [Google Scholar] [CrossRef] [Green Version]
- Heijtz, R.D.; Wang, S.; Anuar, F.; Qian, Y.; Bjorkholm, B.; Samuelsson, A.; Hibberd, M.L.; Forssberg, H.; Pettersson, S. Normal gut microbiota modulates brain development and behavior. Proc. Natl. Acad. Sci. USA 2011, 108, 3047–3052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neufeld, K.M.; Kang, N.; Bienenstock, J.; Foster, J.A. Reduced anxiety-like behavior and central neurochemical change in germ-free mice. Neurogastroenterol. Motil. 2011, 23, 255–264.e119. [Google Scholar] [CrossRef] [PubMed]
- Mombaerts, P.; Mizoguchi, E.; Ljunggren, H.G.; Iacomini, J.; Ishikawa, H.; Wang, L.; Grusby, M.J.; Glimcher, L.H.; Winn, H.J.; Bhan, A.K.; et al. Peripheral lymphoid development and function in TCR mutant mice. Int. Immunol. 1994, 6, 1061–1070. [Google Scholar] [CrossRef]
- Callery, P.S.; Geelhaar, L.A. 1-Piperideine as an in vivo precursor of the gamma-aminobutyric acid homologue 5-aminopentanoic acid. J. Neurochem. 1985, 45, 946–948. [Google Scholar] [CrossRef]
- Fothergill, J.C.; Guest, J.R. Catabolism of L-lysine by Pseudomonas aeruginosa. J. Gen. Microbiol. 1977, 99, 139–155. [Google Scholar] [CrossRef] [Green Version]
- Vital, M.; Howe, A.C.; Tiedje, J.M. Revealing the bacterial butyrate synthesis pathways by analyzing (meta)genomic data. mBio 2014, 5, e00889. [Google Scholar] [CrossRef] [Green Version]
- Dominguez-Gerpe, L.; Rey-Mendez, M. Evolution of the thymus size in response to physiological and random events throughout life. Microsc. Res. Tech. 2003, 62, 464–476. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.M.; Garrett, W.S. The gut microbiota and mucosal T cells. Front. Microbiol. 2011, 2, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swann, J.R.; Spitzer, S.O.; Diaz Heijtz, R. Developmental Signatures of Microbiota-Derived Metabolites in the Mouse Brain. Metabolites 2020, 10, 172. [Google Scholar] [CrossRef] [PubMed]
- Salzman, N.H.; de Jong, H.; Paterson, Y.; Harmsen, H.J.M.; Welling, G.W.; Bos, N.A. Analysis of 16S libraries of mouse gastrointestinal microflora reveals a large new group of mouse intestinal bacteria. Microbiology 2002, 148 Pt 11, 3651–3660. [Google Scholar] [CrossRef] [Green Version]
- Ormerod, K.L.; Wood, D.L.; Lachner, N.; Gellatly, S.L.; Daly, J.N.; Parsons, J.D.; Dal’Molin, C.G.; Palfreyman, R.W.; Nielsen, L.K.; Cooper, M.A.; et al. Genomic characterization of the uncultured Bacteroidales family S24-7 inhabiting the guts of homeothermic animals. Microbiome 2016, 4, 36. [Google Scholar] [CrossRef] [Green Version]
- Lagkouvardos, I.; Lesker, T.R.; Hitch, T.C.A.; Galvez, E.J.C.; Smit, N.; Neuhaus, K.; Wang, J.; Baines, J.F.; Abt, B.; Stecher, B.; et al. Sequence and cultivation study of Muribaculaceae reveals novel species, host preference, and functional potential of this yet undescribed family. Microbiome 2019, 7, 28. [Google Scholar] [CrossRef] [Green Version]
- Lagkouvardos, I.; Pukall, R.; Abt, B.; Foesel, B.U.; Meier-Kolthoff, J.P.; Kumar, N.; Bresciani, A.; Martinez, I.; Just, S.; Ziegler, C.; et al. The Mouse Intestinal Bacterial Collection (miBC) provides host-specific insight into cultured diversity and functional potential of the gut microbiota. Nat. Microbiol. 2016, 1, 16131. [Google Scholar] [CrossRef] [Green Version]
- Smith, B.J.; Miller, R.A.; Schmidt, T.M. Muribaculaceae genomes assembled from metagenomes suggest genetic drivers of differential response to acarbose treatment in mice. mSphere 2020, 6, e00851-21. [Google Scholar] [CrossRef]
- Mortensen, P.B.; Holtug, K.; Bonnen, H.; Clausen, M.R. The degradation of amino acids, proteins, and blood to short-chain fatty acids in colon is prevented by lactulose. Gastroenterology 1990, 98, 353–360. [Google Scholar] [CrossRef]
- Yoon, S.H.; Ha, S.M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Barker, H.A.; D’Ari, L.; Kahn, J. Enzymatic reactions in the degradation of 5-aminovalerate by Clostridium aminovalericum. J. Biol. Chem. 1987, 262, 8994–9003. [Google Scholar] [CrossRef]
- Wang, H.B.; Wang, P.Y.; Wang, X.; Wan, Y.L.; Liu, Y.C. Butyrate enhances intestinal epithelial barrier function via up-regulation of tight junction protein Claudin-1 transcription. Dig. Dis. Sci. 2012, 57, 3126–3135. [Google Scholar] [CrossRef] [PubMed]
- Ploger, S.; Stumpff, F.; Penner, G.B.; Schulzke, J.D.; Gabel, G.; Martens, H.; Shen, Z.; Gunzel, D.; Aschenbach, J.R. Microbial butyrate and its role for barrier function in the gastrointestinal tract. Ann. N. Y. Acad. Sci. 2012, 1258, 52–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burger-van Paassen, N.; Vincent, A.; Puiman, P.J.; van der Sluis, M.; Bouma, J.; Boehm, G.; van Goudoever, J.B.; van Seuningen, I.; Renes, I.B. The regulation of intestinal mucin MUC2 expression by short-chain fatty acids: Implications for epithelial protection. Biochem. J. 2009, 420, 211–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, C.J.; Zheng, L.; Campbell, E.L.; Saeedi, B.; Scholz, C.C.; Bayless, A.J.; Wilson, K.E.; Glover, L.E.; Kominsky, D.J.; Magnuson, A.; et al. Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe 2015, 17, 662–671. [Google Scholar] [CrossRef] [Green Version]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; van der Veeken, J.; deRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef]
- Atarashi, K.; Honda, K. Microbiota in autoimmunity and tolerance. Curr. Opin. Immunol. 2011, 23, 761–768. [Google Scholar] [CrossRef]
- Becattini, S.; Sorbara, M.T.; Kim, S.G.; Littmann, E.L.; Dong, Q.; Walsh, G.; Wright, R.; Amoretti, L.; Fontana, E.; Hohl, T.M.; et al. Rapid transcriptional and metabolic adaptation of intestinal microbes to host immune activation. Cell Host Microbe 2021, 29, 378–393.e5. [Google Scholar] [CrossRef] [PubMed]
- Schjoldager, K.T.; Maltesen, H.R.; Balmer, S.; Lund, L.R.; Claesson, M.H.; Sjostrom, H.; Troelsen, J.T.; Olsen, J. Cellular cross talk in the small intestinal mucosa: Postnatal lymphocytic immigration elicits a specific epithelial transcriptional response. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G1335–G1343. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Tome, S.; Takei, Y. Intraventricular IL-17A administration activates microglia and alters their localization in the mouse embryo cerebral cortex. Mol. Brain 2020, 13, 93. [Google Scholar] [CrossRef] [PubMed]
- Pasciuto, E.; Burton, O.T.; Roca, C.P.; Lagou, V.; Rajan, W.D.; Theys, T.; Mancuso, R.; Tito, R.Y.; Kouser, L.; Callaerts-Vegh, Z.; et al. Microglia Require CD4 T Cells to Complete the Fetal-to-Adult Transition. Cell 2020, 182, 625–640.e24. [Google Scholar] [CrossRef]
- Erny, D.; Hrabe de Angelis, A.L.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef]
- Ziv, Y.; Ron, N.; Butovsky, O.; Landa, G.; Sudai, E.; Greenberg, N.; Cohen, H.; Kipnis, J.; Schwartz, M. Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat. Neurosci. 2006, 9, 268–275. [Google Scholar] [CrossRef]
- Lee, Y.K.; Menezes, J.S.; Umesaki, Y.; Mazmanian, S.K. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4615–4622. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Kim, H.; Yim, Y.S.; Ha, S.; Atarashi, K.; Tan, T.G.; Longman, R.S.; Honda, K.; Littman, D.R.; Choi, G.B.; et al. Maternal gut bacteria promote neurodevelopmental abnormalities in mouse offspring. Nature 2017, 549, 528–532. [Google Scholar] [CrossRef] [Green Version]
- Shin Yim, Y.; Park, A.; Berrios, J.; Lafourcade, M.; Pascual, L.M.; Soares, N.; Yeon Kim, J.; Kim, S.; Kim, H.; Waisman, A.; et al. Reversing behavioural abnormalities in mice exposed to maternal inflammation. Nature 2017, 549, 482–487. [Google Scholar] [CrossRef] [Green Version]
- Choi, G.B.; Yim, Y.S.; Wong, H.; Kim, S.; Kim, H.; Kim, S.V.; Hoeffer, C.A.; Littman, D.R.; Huh, J.R. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 2016, 351, 933–939. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.H.; Bai, J.; Wu, D.; Yu, S.F.; Qiang, X.L.; Bai, H.; Wang, H.N.; Peng, Z.W. Association between fecal microbiota and generalized anxiety disorder: Severity and early treatment response. J. Affect. Disord. 2019, 259, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Sprockett, D.D.; Martin, M.; Costello, E.K.; Burns, A.R.; Holmes, S.P.; Gurven, M.D.; Relman, D.A. Microbiota assembly, structure, and dynamics among Tsimane horticulturalists of the Bolivian Amazon. Nat. Commun. 2020, 11, 3772. [Google Scholar] [CrossRef] [PubMed]
- Stearns, J.C.; Davidson, C.J.; McKeon, S.; Whelan, F.J.; Fontes, M.E.; Schryvers, A.B.; Bowdish, D.M.; Kellner, J.D.; Surette, M.G. Culture and molecular-based profiles show shifts in bacterial communities of the upper respiratory tract that occur with age. ISME J. 2015, 9, 1246–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whelan, F.J.; Surette, M.G. A comprehensive evaluation of the sl1p pipeline for 16S rRNA gene sequencing analysis. Microbiome 2017, 5, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whelan, F.J.; Verschoor, C.P.; Stearns, J.C.; Rossi, L.; Luinstra, K.; Loeb, M.; Smieja, M.; Johnstone, J.; Surette, M.G.; Bowdish, D.M. The loss of topography in the microbial communities of the upper respiratory tract in the elderly. Ann. Am. Thorac. Soc. 2014, 11, 513–521. [Google Scholar] [CrossRef]
- Veselkov, K.A.; Lindon, J.C.; Ebbels, T.M.; Crockford, D.; Volynkin, V.V.; Holmes, E.; Davies, D.B.; Nicholson, J.K. Recursive segment-wise peak alignment of biological (1)h NMR spectra for improved metabolic biomarker recovery. Anal. Chem. 2009, 81, 56–66. [Google Scholar] [CrossRef]
- Cloarec, O.; Dumas, M.E.; Craig, A.; Barton, R.H.; Trygg, J.; Hudson, J.; Blancher, C.; Gauguier, D.; Lindon, J.C.; Holmes, E.; et al. Statistical total correlation spectroscopy: An exploratory approach for latent biomarker identification from metabolic 1H NMR data sets. Anal. Chem. 2005, 77, 1282–1289. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Glockner, F.O.; Yilmaz, P.; Quast, C.; Gerken, J.; Beccati, A.; Ciuprina, A.; Bruns, G.; Yarza, P.; Peplies, J.; Westram, R.; et al. 25 years of serving the community with ribosomal RNA gene reference databases and tools. J. Biotechnol. 2017, 261, 169–176. [Google Scholar] [CrossRef]
- Oksanen, J.; Guillaume Blanchet, F.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Mihchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. Available online: https://CRAN.R-project.org/package=vegan (accessed on 30 September 2020).
- Fernandes, A.D.; Macklaim, J.M.; Linn, T.G.; Reid, G.; Gloor, G.B. ANOVA-like differential expression (ALDEx) analysis for mixed population RNA-Seq. PLoS ONE 2013, 8, e67019. [Google Scholar] [CrossRef] [PubMed]
- Gloor, G.B.; Macklaim, J.M.; Pawlowsky-Glahn, V.; Egozcue, J.J. Microbiome Datasets Are Compositional: And This Is Not Optional. Front. Microbiol. 2017, 8, 2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cloarec, O.; Dumas, M.E.; Trygg, J.; Craig, A.; Barton, R.H.; Lindon, J.C.; Nicholson, J.K.; Holmes, E. Evaluation of the orthogonal projection on latent structure model limitations caused by chemical shift variability and improved visualization of biomarker changes in 1H NMR spectroscopic metabonomic studies. Anal. Chem. 2005, 77, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Shannon, C.P.; Gautier, B.; Rohart, F.; Vacher, M.; Tebbutt, S.J.; Le Cao, K.A. DIABLO: An integrative approach for identifying key molecular drivers from multi-omics assays. Bioinformatics 2019, 35, 3055–3062. [Google Scholar] [CrossRef] [PubMed]
- Rohart, F.; Gautier, B.; Singh, A.; Le Cao, K.A. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herve, M. RVAideMemoire: Testing and Plotting Procedures for Biostatistics. Available online: https://CRAN.R-project.org/package=RVAideMemoire (accessed on 30 April 2021).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cortex | Hippocampus | Hypothalamus | ||||
|---|---|---|---|---|---|---|
| Genotype | Genotype/Age | Genotype | Genotype/Age | Genotype | Genotype/Age | |
| Aspartate | 0.003 | - | <0.001 | - | 0.002 | - |
| Glycerophosphocholine | 0.001 | - | 0.008 | - | <0.001 | - |
| Acetate | - | 0.049 | 0.035 | - | 0.030 | - |
| GABA | - | - | 0.044 | - | 0.023 | - |
| Glycine | - | 0.008 | ||||
| Alanine | - | - | 0.004 | - | - | - |
| Phosphocholine | - | - | 0.037 | - | 0.004 | - |
| IMP | - | - | - | <0.001 | - | - |
| Taurine | 0.011 | - | - | - | 0.007 | - |
| Glutamate | - | - | - | - | 0.042 | - |
| Myo-inositol | - | - | - | - | 0.013 | - |
| 3-hydroxyisovalerate | - | - | - | - | 0.039 | - |
| Carnitine | - | - | - | - | 0.035 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caspani, G.; Green, M.; Swann, J.R.; Foster, J.A. Microbe-Immune Crosstalk: Evidence That T Cells Influence the Development of the Brain Metabolome. Int. J. Mol. Sci. 2022, 23, 3259. https://doi.org/10.3390/ijms23063259
Caspani G, Green M, Swann JR, Foster JA. Microbe-Immune Crosstalk: Evidence That T Cells Influence the Development of the Brain Metabolome. International Journal of Molecular Sciences. 2022; 23(6):3259. https://doi.org/10.3390/ijms23063259
Chicago/Turabian StyleCaspani, Giorgia, Miranda Green, Jonathan R. Swann, and Jane A. Foster. 2022. "Microbe-Immune Crosstalk: Evidence That T Cells Influence the Development of the Brain Metabolome" International Journal of Molecular Sciences 23, no. 6: 3259. https://doi.org/10.3390/ijms23063259
APA StyleCaspani, G., Green, M., Swann, J. R., & Foster, J. A. (2022). Microbe-Immune Crosstalk: Evidence That T Cells Influence the Development of the Brain Metabolome. International Journal of Molecular Sciences, 23(6), 3259. https://doi.org/10.3390/ijms23063259