
Helicobacter pylori Urease: Potential Contributions to Alzheimer’s Disease

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. In Vitro Effects of HPU in CNS Derived Cell Lines
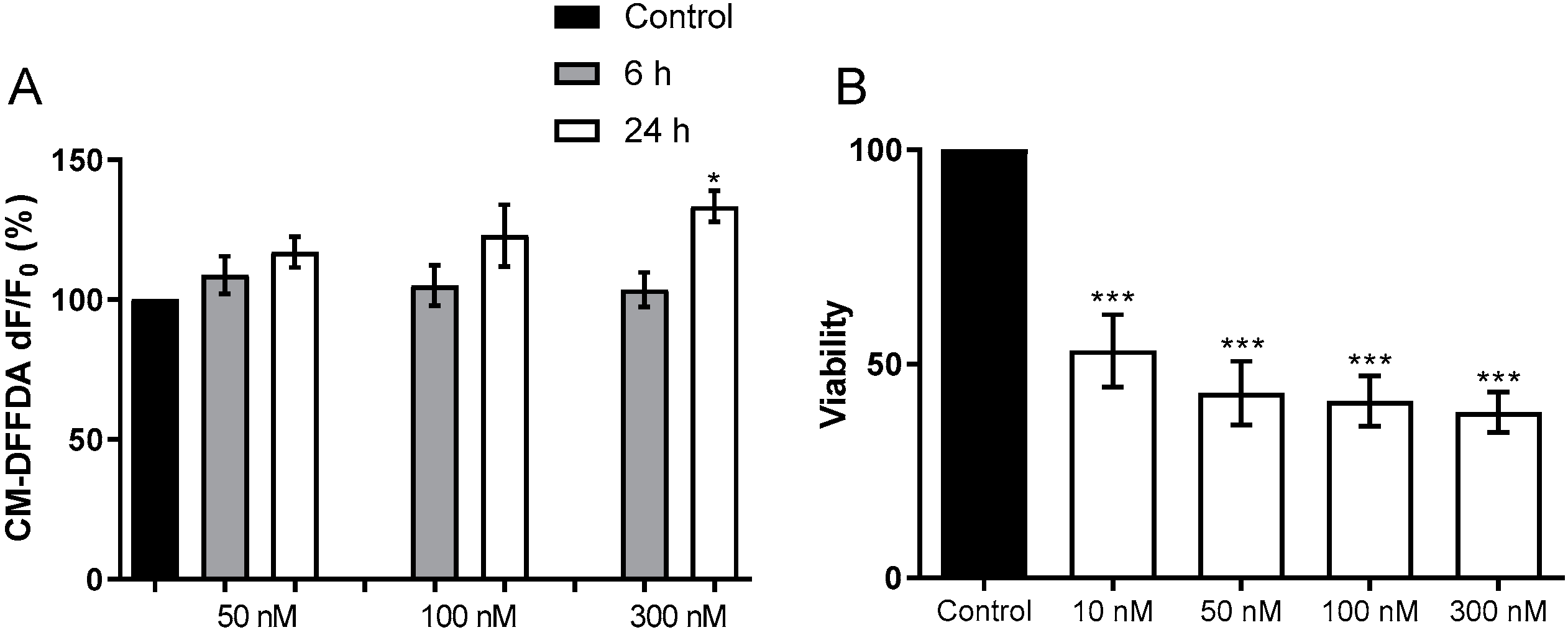
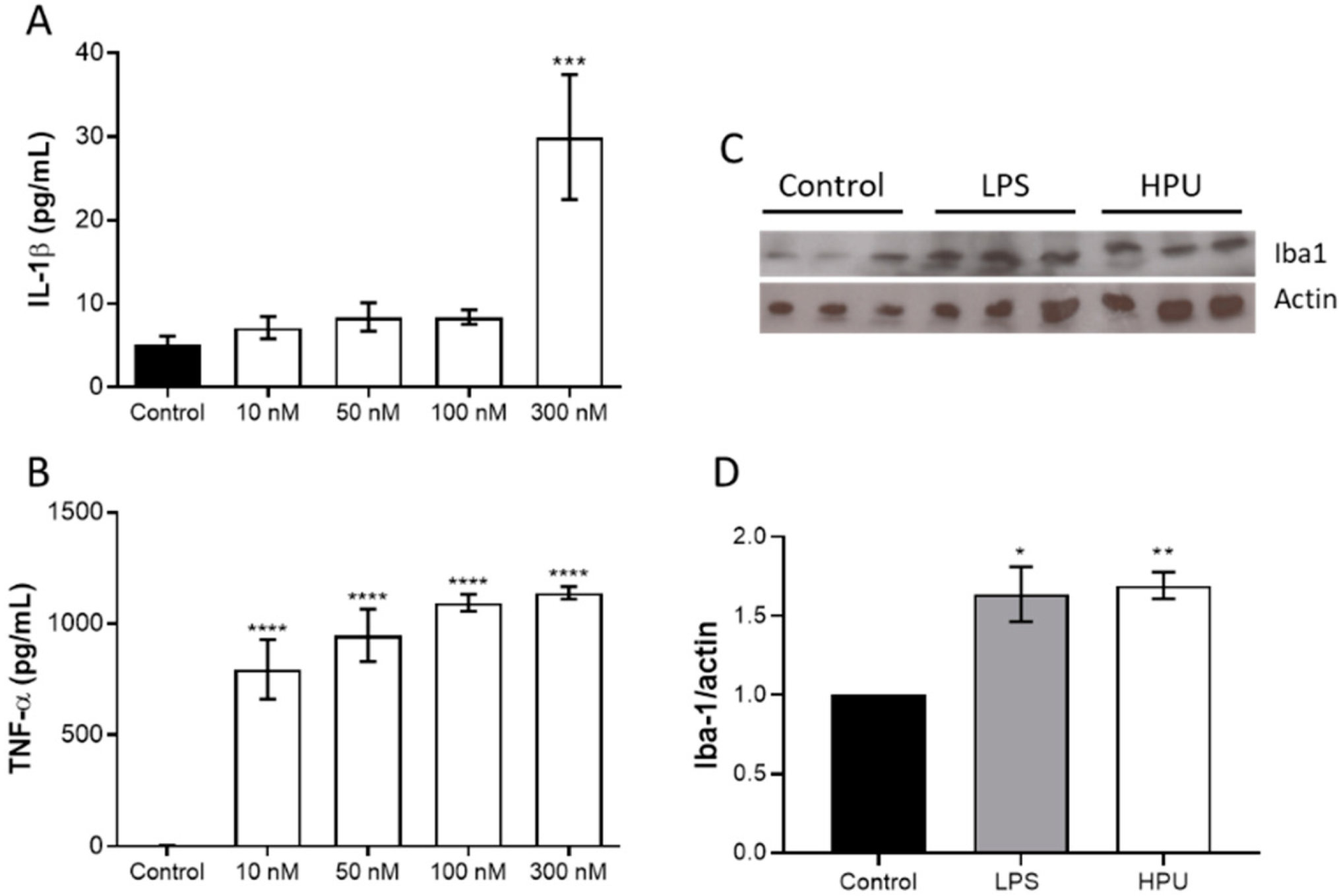
2.1.1. Toxicity to Human Neuroblastoma Cells
2.1.2. Activation of BV-2 Microglial Cells
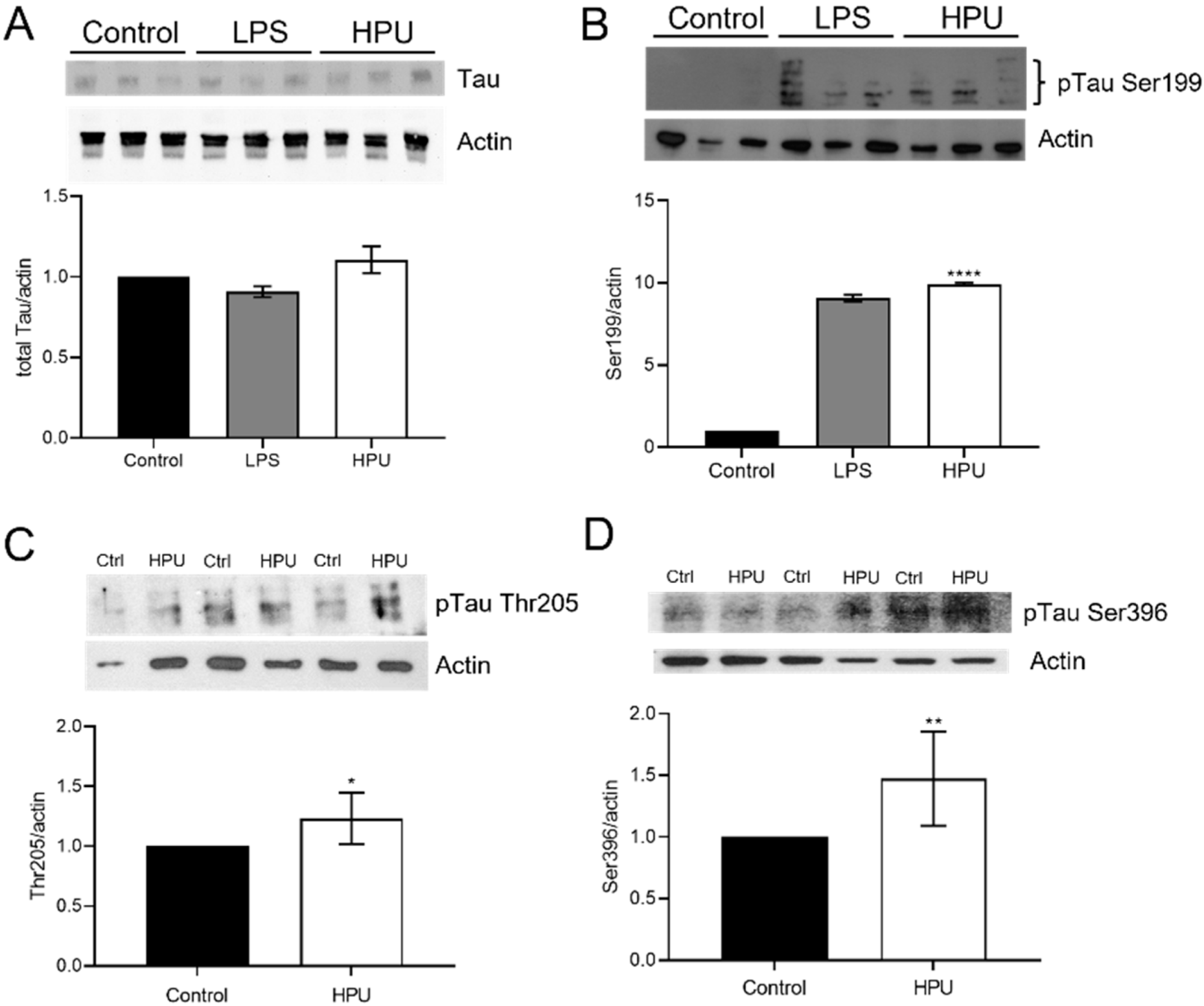
2.2. Evaluation of Total and Phosphorylated Hippocampal Tau Levels in Rats Treated with HPU
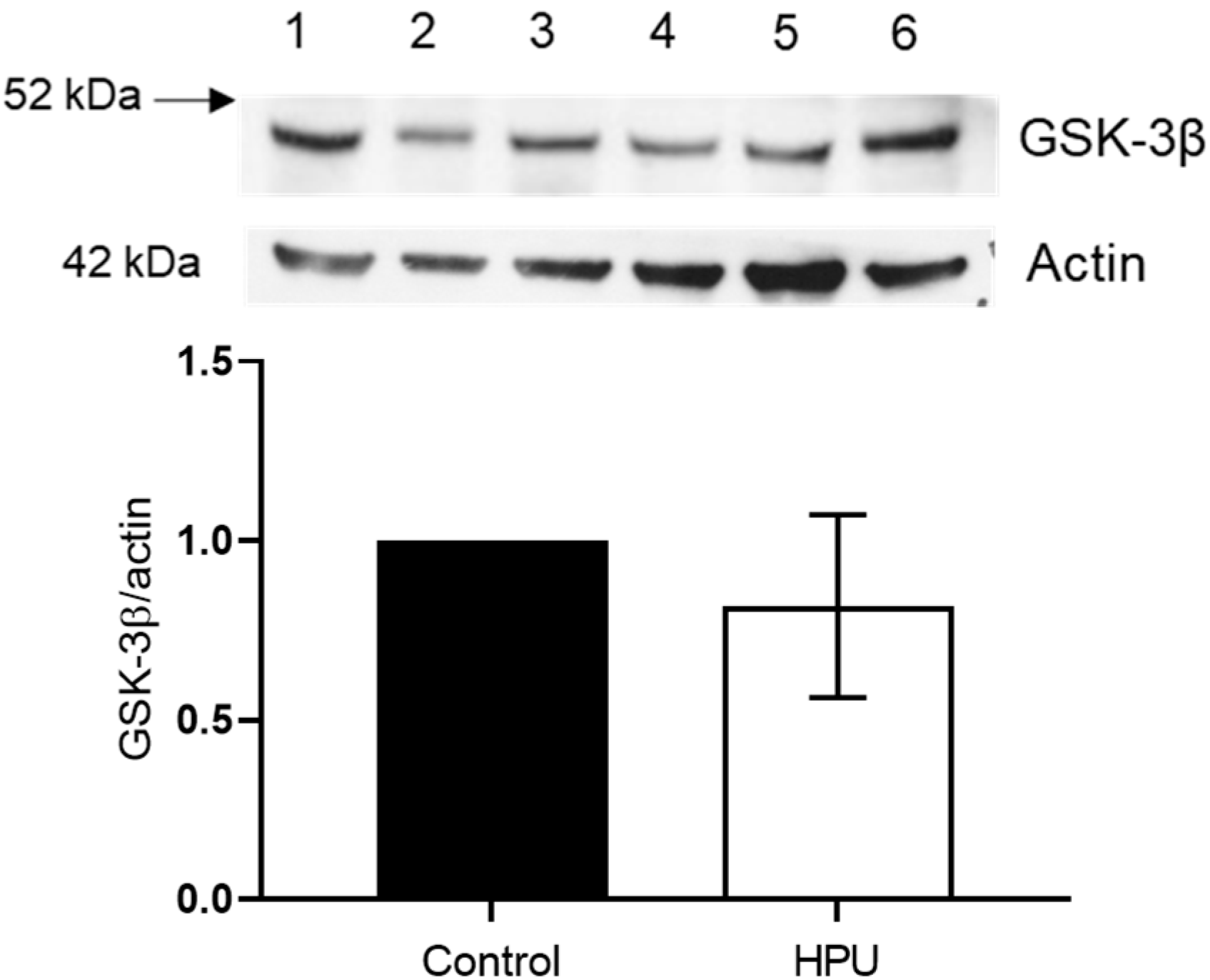
2.3. Evaluation of GSK-3β Levels
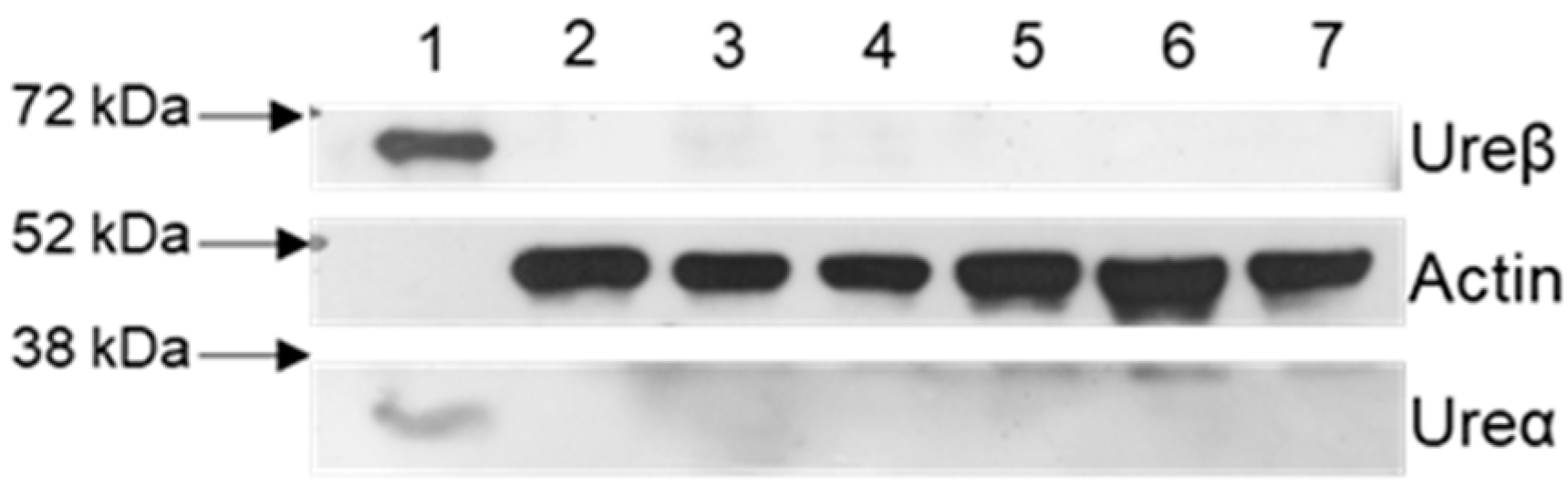
2.4. Blood–Brain Barrier Permeability of Rats Treated with HPU
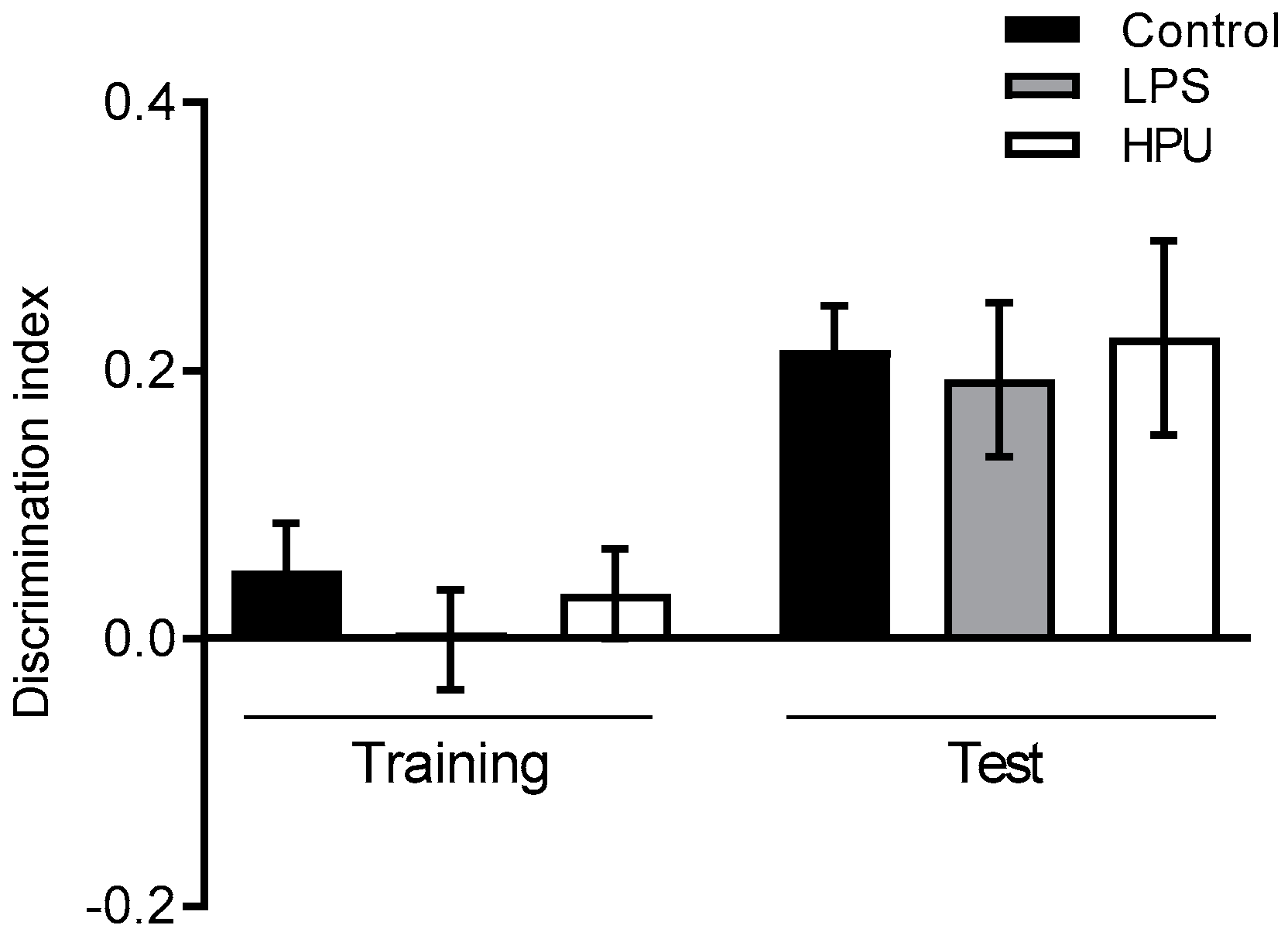
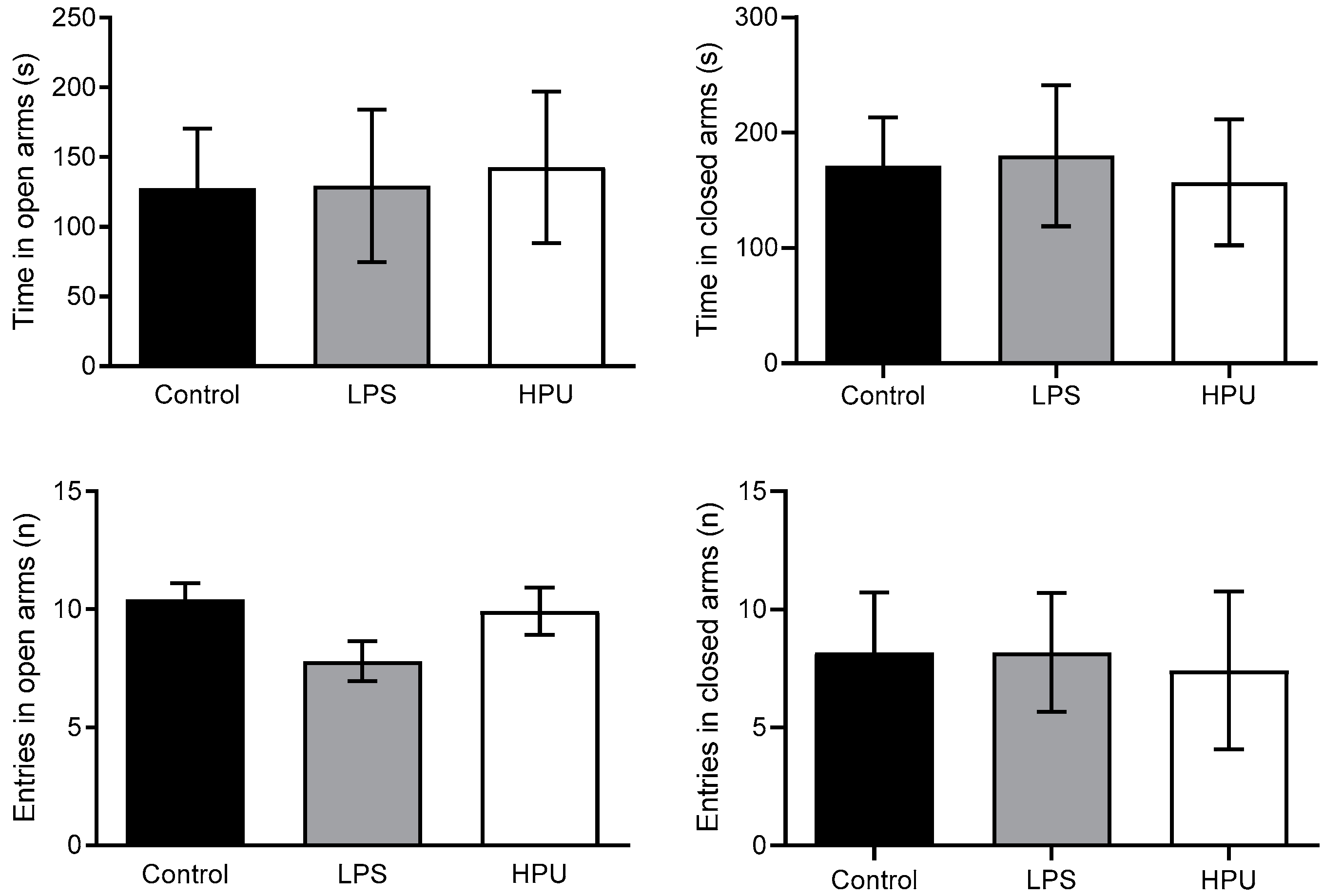
2.5. Behavioral Tests
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. HPU Purification
4.3. Protein Determination
4.4. Urease Activity
4.5. Rat In Vivo Assays
4.6. Preparation of Brain Homogenates
4.7. Western Blot Analysis
4.8. Reactive Oxygen Species (ROS) Measurement
4.9. Intracellular Calcium Measurement
4.10. Cytokine Measurement
4.11. Cell Viability
4.12. Behavioral Tests
4.12.1. Object Recognition Task
4.12.2. Elevated Plus Maze
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Doulberis, M.; Kotronis, G.; Thomann, R.; Polyzos, S.; Boziki, M.; Gialamprinou, D.; Deretzi, G.; Katsinelos, P.; Kountouras, J. Review: Impact of Helicobacter pylori on Alzheimer’s disease: What do we know so far? Helicobacter 2018, 23, e12454. [Google Scholar] [CrossRef] [PubMed]
- Hooi, J.K.; Lai, W.Y.; Ng, W.K.; Suen, M.M.; Underwood, F.E.; Tanyingoh, D.; Malfertheiner, P.; Graham, D.Y.; Wong, V.W.; Wu, J.C.; et al. Global Prevalence of Helicobacter pylori Infection: Systematic Review and Meta-Analysis. Gastroenterology 2017, 153, 420–429. [Google Scholar] [CrossRef]
- Wang, A.Y.; Peura, D.A. The prevalence and incidence of Helicobacter pylori-associated peptic ulcer disease and upper gastrointestinal bleeding throughout the world. Gastrointest. Endosc. Clin. 2011, 21, 613–635. [Google Scholar] [CrossRef] [PubMed]
- Ernst, P.B.; Gold, B.D. The disease spectrum of Helicobacter pylori: The immunopathogenesis of gastroduodenal ulcer and gastric cancer. Annu. Rev. Microbiol. 2000, 54, 615–640. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, F.; Covino, M.; Roubaud Baudron, C. Review: Helicobacter pylori and extragastric diseases. Helicobacter 2019, 24 (Suppl. S1), e12636. [Google Scholar] [CrossRef] [PubMed]
- Baj, J.; Forma, A.; Flieger, W.; Morawska, I.; Michalski, A.; Buszewicz, G.; Sitarz, E.; Portincasa, P.; Garruti, G.; Flieger, M.; et al. Helicobacter pylori Infection and Extragastric Diseases—A Focus on the Central Nervous System. Cells 2021, 10, 2191. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; McMahon, A.D.; Patel, H.; Packard, C.J.; Rathbone, B.J.; Samani, N.J. Prospective analysis of the association of infection with CagA bearing strains of Helicobacter pylori and coronary heart disease. Heart 2002, 88, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Kountouras, J.; Zavos, C.; Katsinelos, P.; Vardaka, E. Glaucoma and Helicobacter pylori: Eyes “wide open”! Dig. Liver Dis. 2012, 44, 962–963. [Google Scholar] [CrossRef] [PubMed]
- Grau, A.J.; Buggle, F.; Lichy, C.; Brandt, T.; Becher, H.; Rudi, J. Helicobacter pylori infection as an independent risk factor for cerebral ischemia of atherothrombotic origin. J. Neurol. Sci. 2001, 186, 1–5. [Google Scholar] [CrossRef]
- Kunovsky, L.; Dite, P.; Jabandziev, P.; Dolina, J.; Vaculova, J.; Blaho, M.; Bojkova, M.; Dvorackova, J.; Uvirova, M.; Kala, Z.; et al. Helicobacter pylori infection and other bacteria in pancreatic cancer and autoimmune pancreatitis. World J. Gastrointest. Oncol. 2021, 13, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Kountouras, J.; Boziki, M.; Gavalas, E.; Zavos, C.; Deretzi, G.; Grigoriadis, N.; Tsolaki, M.; Chatzopoulos, D.; Katsinelos, P.; Tzilves, D.; et al. Increased Cerebrospinal Fluid Helicobacter Pylori Antibody in Alzheimer’s Disease. Int. J. Neurosci. 2009, 119, 765–777. [Google Scholar] [CrossRef] [PubMed]
- Kountouras, J.; Tsolaki, M.; Gavalas, E.; Boziki, M.; Zavos, C.; Karatzoglou, P.; Chatzopoulos, D.; Venizelos, I. Relationship between Helicobacter pylori infection and Alzheimer disease. J. Neurol. 2006, 66, 938–940. [Google Scholar] [CrossRef] [PubMed]
- Kountouras, J.; Tsolaki, M.; Boziki, M.; Gavalas, E.; Zavos, C.; Stergiopoulos, C.; Kapetanakis, N.; Chatzopoulos, D.; Venizelos, I. Association between Helicobacter pylori infection and mild cognitive impairment. Eur. J. Neurol. 2007, 14, 976–982. [Google Scholar] [CrossRef] [PubMed]
- Lolekha, P.; Sriphanom, T.; Vilaichone, R.-K. Helicobacter pylori eradication improves motor fluctuations in advanced Parkinson’s disease patients: A prospective cohort study (HP-PD trial). PLoS ONE 2021, 16, e0251042. [Google Scholar] [CrossRef]
- Zhong, R.; Chen, Q.; Zhang, X.; Li, M.; Lin, W. Helicobacter pylori infection is associated with a poor response to levodopa in patients with Parkinson’s disease: A systematic review and meta-analysis. J. Neurol. 2021, 269, 703–711. [Google Scholar] [CrossRef]
- Roubaud Baudron, C.; Letenneur, L.; Langlais, A.; Buissonnière, A.; Mégraud, F.; Dartigues, J.F.; Salles, N.; Study, P.A.Q. Does Helicobacter pylori infection increase incidence of dementia? The Personnes Agées QUID Study. J. Am. Geriatr. Soc. 2013, 61, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, M.; Bella, R.; Alagona, G.; Ferri, R.; Carnemolla, A.; Pennisi, G. Helicobacter pylori and Alzheimer’s disease: A possible link. Eur. J. Intern. Med. 2004, 15, 381–386. [Google Scholar] [CrossRef]
- Wang, X.L.; Zeng, J.; Yang, Y.; Xiong, Y.; Zhang, Z.H.; Qiua, M.; Yan, X.; Sun, X.Y.; Tuo, Q.Z.; Liua, R.; et al. Helicobacter pylori Filtrate Induces Alzheimer-Like Tau Hyperphosphorylation by Activating Glycogen Synthase Kinase-3 beta. J. Alzheimer’s Dis. 2014, 43, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Marcus, E.A.; Scott, D.R. Cell lysis is responsible for the appearance of extracellular urease in Helicobacter pylori. Helicobacter 2001, 6, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Marshall, B.J.; Barrett, L.J.; Prakash, C.; McCallum, R.W.; Guerrant, R.L. Urea protects Helicobacter (Campylobacter) pylori from the bactericidal effect of acid. Gastroenterology 1990, 99, 697–702. [Google Scholar] [CrossRef]
- Montecucco, C.; Rappuoli, R. Living dangerously: How Helicobacter pylori survives in the human stomach. Nat. Rev. Mol. Cell Biol. 2001, 2, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Eaton, K.A.; Brooks, C.L.; Morgan, D.R.; Krakowka, S. Essential role of urease in pathogenesis of gastritis induced by Helicobacter pylori in gnotobiotic piglets. Infect. Immun. 1991, 59, 2470–2475. [Google Scholar] [CrossRef]
- Tsuda, M.; Karita, M.; Morshed, M.G.; Okita, K.; Nakazawa, T. A urease-negative mutant of Helicobacter pylori constructed by allelic exchange mutagenesis lacks the ability to colonize the nude mouse stomach. Infect. Immun. 1994, 62, 3586–3589. [Google Scholar] [CrossRef] [PubMed]
- Khalilpour, A.; Kazemzadeh-Narbat, M.; Tamayol, A.; Oklu, R.; Khademhosseini, A. Biomarkers and diagnostic tools for detection of Helicobacter pylori. Appl. Microbiol. Biotechnol. 2016, 100, 4723–4734. [Google Scholar] [CrossRef] [PubMed]
- Eslaminejad, A.; Marashian, S.M.; Aboutorabi, M.; Sadr, M.; Agah, S. Determination of optimal time for reading of rapid urease test diagnosis of Helicobacter pylori. Gastroenterol. Hepatol. Bed Bench 2020, 13, 232–237. [Google Scholar] [PubMed]
- Carlini, C.R.; Ligabue-Braun, R. Ureases as multifunctional toxic proteins: A review. Toxicon 2015, 110, 90–109. [Google Scholar] [CrossRef] [PubMed]
- Kappaun, K.; Piovesan, A.R.; Carlini, C.R.; Ligabue-Braun, R. Ureases: Historical aspects, catalytic, and non-catalytic properties—A review. J. Adv. Res. 2018, 13, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Wassermann, G.E.; Olivera-Severo, D.; Uberti, A.F.; Carlini, C.R. Helicobacter pylori urease activates blood platelets through a lipoxygenase-mediated pathway. J. Cell. Mol. Med. 2010, 14, 2025–2034. [Google Scholar] [CrossRef] [PubMed]
- Scopel-Guerra, A.; Olivera-Severo, D.; Staniscuaski, F.; Uberti, A.F.; Callai-Silva, N.; Jaeger, N.; Porto, B.N.; Carlini, C.R. The impact of Helicobacter pylori urease upon platelets and consequent contributions to inflammation. Front. Microbiol. 2017, 8, 2447. [Google Scholar] [CrossRef] [PubMed]
- Uberti, A.F.; Olivera-Severo, D.; Wassermann, G.E.; Scopel-Guerra, A.; Moraes, J.A.; Barcellos-de-Souza, P.; Barja-Fidalgo, C.; Carlini, C.R. Pro-inflammatory properties and neutrophil activation by Helicobacter pylori urease. Toxicon 2013, 69, 240–249. [Google Scholar] [CrossRef]
- Iba, T.; Levy, J.H. Inflammation and thrombosis: Roles of neutrophils, platelets and endothelial cells and their interactions in thrombus formation during sepsis. J. Thromb. Haemost. 2017, 16, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Olivera-Severo, D.; Uberti, A.F.; Marques, M.S.; Pinto, M.T.; Gomez-Lazaro, M.; Figueiredo, C.; Leite, M.; Carlini, C.R. A new role for Helicobacter pylori urease: Contributions to angiogenesis. Front. Microbiol. 2017, 8, 1883. [Google Scholar] [CrossRef] [PubMed]
- Wroblewski, L.E.; Shen, L.; Ogden, S.; Romero-Gallo, J.; Lapierre, L.A.; Israel, D.A.; Turner, J.R.; Peek, R.M., Jr. Helicobacter pylori dysregulation of gastric epithelial tight junctions by urease-mediated myosin II activation. Gastroenterology 2009, 136, 236–246. [Google Scholar] [CrossRef] [PubMed]
- de Jesus Souza, M.; de Moraes, J.A.; Da Silva, V.N.; Helal-Neto, E.; Uberti, A.F.; Scopel-Guerra, A.; Olivera-Severo, D.; Carlini, C.R.; Barja-Fidalgo, C. Helicobacter pylori urease induces pro-inflammatory effects and differentiation of human endothelial cells: Cellular and molecular mechanism. Helicobacter 2019, 24, e12573. [Google Scholar] [CrossRef] [PubMed]
- Hanzel, C.E.; Iulita, M.F.; Eyjolfsdottir, H.; Hjorth, E.; Schultzberg, M.; Eriksdotter, M.; Cuello, A.C. Analysis of matrix metallo-proteases and the plasminogen system in mild cognitive impairment and Alzheimer’s disease cerebrospinal fluid. J. Alzheimer’s Dis. 2014, 40, 667–678. [Google Scholar] [CrossRef]
- Drake, C.; Boutin, H.; Jones, M.S.; Denes, A.; McColl, B.W.; Selvarajah, J.R.; Hulme, S.; Georgiou, R.F.; Hinz, R.; Gerhard, A.; et al. Brain inflammation is induced by co-morbidities and risk factors for stroke. Brain Behav. Immun. 2011, 25, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Barrientos, R.M.; Kitt, M.M.; Watkins, L.R.; Maier, S.F. Neuroinflammation in the normal aging hippocampus. Neuroscience 2015, 309, 84–99. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, A.; Vallström, A.; Petzold, K.; Tegtmeyer, N.; Schleucher, J.; Carlsson, S.; Haas, R.; Backert, S.; Wai, S.N.; Gröbner, G.; et al. Biochemical and functional characterization of Helicobacter pylori vesicles. Mol. Microbiol. 2010, 77, 1539–1555. [Google Scholar] [CrossRef] [PubMed]
- Bruno, L.; Karagil, S.; Mahmood, A.; Elbediwy, A.; Stolinski, M.; Mackenzie, F.E. Mechanosensing and the Hippo Pathway in Microglia: A Potential Link to Alzheimer’s Disease Pathogenesis? Cells 2021, 10, 3144. [Google Scholar] [CrossRef]
- Loeffler, D.A.; Smith, L.M.; Klaver, A.C.; Martić, S. Effects of antibodies to phosphorylated and non-phosphorylated tau on in vitro tau phosphorylation at Serine-199: Preliminary report. Exp. Gerontol. 2015, 67, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Mondragonrodriguez, S.; Perry, G.; Lunamunoz, J.; Acevedo-Aquino, M.C.; Williams, S.E. Phosphorylation of tau protein at sites Ser(396-404) is one of the earliest events in Alzheimer’s disease and Down syndrome. Neuropathol. Appl. Neurobiol. 2014, 40, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Avila, J.; Leon-Espinosa, G.; Garcia, E.; Garcia-Escudero, V.; Hernandez, F.; Defelipe, J. Tau Phosphorylation by GSK3 in Different Conditions. Int. J. Alzheimer’s Dis. 2012, 2012, 578373. [Google Scholar] [CrossRef]
- Schaffer, B.A.J.; Bertram, L.; Miller, B.L.; Mullin, K.; Weintraub, S.; Johnson, N.; Bigio, E.H.; Mesulam, M.; Wiedau-Pazos, M.; Jackson, G.R.; et al. Association of GSK3B with Alzheimer disease and frontotemporal dementia. Arch. Neurol. 2008, 65, 1368–1374. [Google Scholar] [CrossRef]
- Hernandez, F.; Lucas, J.J.; Avila, J. GSK3 and tau: Two convergence points in Alzheimer’s disease. J. Alzheimer’s Dis. 2013, 33 (Suppl. S1), S141–S144. [Google Scholar] [CrossRef] [PubMed]
- Hanger, D.P.; Anderton, B.H.; Noble, W. Tau phosphorylation: The therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 2009, 15, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Berlyne, D.E. Novelty and curiosity as determinants of exploratory behaviour. Br. J. Psychol. Gen. Sect. 1950, 41, 68–80. [Google Scholar] [CrossRef]
- Rodgers, R.J.; Dalvi, A. Anxiety, defence and the elevated plus-maze. Neurosci. Biobehav. Rev. 1997, 21, 801–810. [Google Scholar] [CrossRef]
- Lo, Y.C.; Shih, Y.T.; Wu, D.C.; Lee, Y.C. In vitro effects of Helicobacter pylori-induced infection in gastric epithelial AGS cells on microglia-mediated toxicity in neuroblastoma SH-SY5Y cells. Inflamm. Res. 2009, 58, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Alom-Ruiz, S.P.; Anilkumar, N.; Shah, A.M. Reactive oxygen species and endothelial activation. Antioxidants Redox Signal. 2008, 10, 1089–1100. [Google Scholar] [CrossRef] [PubMed]
- Frey, R.S.; Ushio-Fukai, M.; Malik, A.B. NADPH oxidase-dependent signaling in endothelial cells: Role in physiology and pathophysiology. Antioxidants Redox Signal. 2009, 11, 791–810. [Google Scholar] [CrossRef] [PubMed]
- Puri, B.K. Calcium Signaling and Gene Expression. In Calcium Signaling. Advances in Experimental Medicine and Biology; Islam, M., Ed.; Springer: Cham, Switzerland, 2020. [Google Scholar] [CrossRef]
- Sattler, R.; Tymianski, M. Molecular mechanisms of calcium-dependent excitotoxicity. J. Mol. Med. 2000, 78, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Franklin, J.L.; Johnson, E.M. Block of neuronal apoptosis by a sustained increase of steady-state free Ca2+ concentration. Philos. Trans. R. Soc. B Biol. Sci. 1994, 345, 251–256. [Google Scholar] [CrossRef][Green Version]
- Franklin, J.L.; Johnson, E.M. Suppression of programmed neuronal death by sustained elevation of cytoplasmic calcium. Trends Neurosci. 1992, 15, 501–508. [Google Scholar] [CrossRef]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Wei, H. Anesthetic neurotoxicity: Apoptosis and autophagic cell death mediated by calcium dysregulation. Neurotoxicology Teratol. 2017, 60, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Bocchini, V.; Mazzolla, R.; Barluzzi, R.; Blasi, E.; Sick, P.; Kettenmann, H. An immortalized cell line expresses properties of activated microglial cells. J. Neurosci. Res. 1992, 31, 616–621. [Google Scholar] [CrossRef] [PubMed]
- Grahl, M.V.C.; Uberti, A.F.; Broll, V.; Bacaicoa-Caruso, P.; Meirelles, E.F.; Carlini, C.R. Proteus mirabilis Urease: Unsuspected Non-Enzymatic Properties Relevant to Pathogenicity. Int. J. Mol. Sci. 2021, 22, 7205. [Google Scholar] [CrossRef] [PubMed]
- Boutajangout, A.; Wisniewski, T. The Innate Immune System in Alzheimer’s Disease. Int. J. Cell Biol. 2013, 2013, 576383. [Google Scholar] [CrossRef]
- Hunter, J.M.; Kwan, J.; Malek-Ahmadi, M.; Maarouf, C.L.; Kokjohn, T.A.; Belden, C.; Sabbagh, M.N.; Beach, T.G.; Roher, A.E. Morphological and pathological evolution of the brain microcirculation in aging and Alzheimer’s disease. PLoS ONE 2012, 7, e36893. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.G.; Zhu, S.G.; Jones, R.A.; Griffin, W.S.; Mrak, R.E. Interleukin-1 promotes expression and phosphorylation of neurofilament and tau proteins in vivo. Exp. Neurol. 2000, 163, 388–391. [Google Scholar] [CrossRef] [PubMed]
- Koch, K.N.; Hartung, M.L.; Urban, S.; Kyburz, A.; Bahlmann, A.S.; Lind, J.; Backert, S.; Taube, C.; Muller, A. Helicobacter urease-induced activation of the TLR2/NLRP3/IL-18 axis protects against asthma. J. Clin. Investig. 2015, 125, 3297–3302. [Google Scholar] [CrossRef]
- Kacimi, R.; Giffard, R.G.; Yenari, M.A. Endotoxin-activated microglia injure brain derived endothelial cells via NF-κB, JAK-STAT and JNK stress kinase pathways. J. Inflamm. 2011, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Baik, S.C.; Kang, H.L.; Seo, J.-H.; Park, E.S.; Rhee, K.; Cho, M.J. Helicobacter pylori urease induces mouse death. J. Bacteriol. Virol. 2005, 35, 175–181. [Google Scholar]
- Carlini, C.R.; Gomes, C.; Guimaraes, J.A.; Markus, R.P.; Sato, H.; Trolin, G. Central nervous effects of the convulsant protein canatoxin. Acta Pharmacol. Toxicol. 1984, 54, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Almeida, C.G.M.; Costa-Higuchi, K.; Piovesan, A.R.; Moro, C.F.; Venturin, G.T.; Greggio, S.; Costa-Ferro, Z.S.; Salamoni, S.D.; Peigneur, S.; Tytgat, J.; et al. Neurotoxic and convulsant effects induced by jack bean ureases on the mammalian nervous system. Toxicology 2021, 454, 152737. [Google Scholar] [CrossRef]
- Adlimoghaddam, A.; Sabbir, M.G.; Albensi, B.C. Ammonia as a Potential Neurotoxic Factor in Alzheimer’s Disease. Front. Mol. Neurosci. 2016, 9, 57. [Google Scholar] [CrossRef] [PubMed]
- Skowronska, M.; Albrecht, J. Alterations of blood brain barrier function in hyperammonemia: An overview. Neurotox. Res. 2012, 21, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Skowrońska, M.; Zielińska, M.; Wójcik-Stanaszek, L.; Ruszkiewicz, J.; Milatovic, D.; Aschner, M.; Albrecht, J. Ammonia increases paracellular permeability of rat brain endothelial cells by a mechanism encompassing oxidative/nitrosative stress and activation of matrix metalloproteinases. J. Neurochem. 2012, 121, 125–134. [Google Scholar] [CrossRef]
- Wilhelm, I.; Nyul-Toth, A.; Suciu, M.; Hermenean, A.; Krizbai, I.A. Heterogeneity of the blood-brain barrier. Tissue Barriers 2016, 4, e1143544. [Google Scholar] [CrossRef] [PubMed]
- Bentivoglio, M.; Kristensson, K.; Rottenberg, M.E. Circumventricular Organs and Parasite Neurotropism: Neglected Gates to the Brain? Front. Immunol. 2018, 9, 2877. [Google Scholar] [CrossRef] [PubMed]
- Shaimardanova, A.A.; Solovyeva, V.V.; Chulpanova, D.S.; James, V.; Kitaeva, K.V.; Rizvanov, A.A. Extracellular vesicles in the diagnosis and treatment of central nervous system diseases. Neural Regen. Res. 2020, 15, 586–596. [Google Scholar] [CrossRef] [PubMed]
- Turkina, M.V.; Olofsson, A.; Magnusson, K.E.; Arnqvist, A.; Vikstrom, E. Helicobacter pylori vesicles carrying CagA localize in the vicinity of cell-cell contacts and induce histone H1 binding to ATP in epithelial cells. FEMS Microbiol. Lett. 2015, 362, fnv076. [Google Scholar] [CrossRef] [PubMed]
- Pachathundikandi, S.K.; Tegtmeyer, N.; Backert, S. Signal transduction of Helicobacter pylori during interaction with host cell protein receptors of epithelial and immune cells. Gut Microbes 2013, 4, 454–474. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, H.; Tachikawa, M.; Yagi, Y.; Umetsu, M.; Nurdin, A.; Miyauchi, E.; Watanabe, M.; Uchida, Y.; Terasaki, T. Cluster of Differentiation 46 Is the Major Receptor in Human Blood-Brain Barrier Endothelial Cells for Uptake of Exosomes Derived from Brain-Metastatic Melanoma Cells (SK-Mel-28). Mol. Pharm. 2019, 16, 292–304. [Google Scholar] [CrossRef] [PubMed]
- González-Domínguez, R.; García-Barrera, T.; Vitorica, J.; Gómez-Ariza, J.L. Metabolomic investigation of systemic manifestations associated with Alzheimer’s disease in the APP/PS1 transgenic mouse model. Mol. BioSyst. 2015, 11, 2429–2440. [Google Scholar] [CrossRef] [PubMed]
- Ghazaleh, F.A.; Francischetti, I.M.; Gombarovits, M.E.; Carlini, C.R. Stimulation of calcium influx and platelet activation by canatoxin: Methoxyverapamil inhibition and downregulation by cGMP. Arch. Biochem. Biophys. 1997, 339, 362–367. [Google Scholar] [CrossRef]
- Sawikr, Y.; Yarla, N.S.; Peluso, I.; Kamal, M.A.; Aliev, G.; Bishayee, A. Neuroinflammation in Alzheimer’s Disease: The Preventive and Therapeutic Potential of Polyphenolic Nutraceuticals. Adv. Protein Chem. Struct. Biol. 2017, 108, 33–57. [Google Scholar] [CrossRef]
- Valcarcel-Ares, M.N.; Tucsek, Z.; Kiss, T.; Giles, C.B.; Tarantini, S.; Yabluchanskiy, A.; Balasubramanian, P.; Gautam, T.; Galvan, V.; Ballabh, P.; et al. Obesity in Aging Exacerbates Neuroinflammation, Dysregulating Synaptic Function-Related Genes and Altering Eicosanoid Synthesis in the Mouse Hippocampus: Potential Role in Impaired Synaptic Plasticity and Cognitive Decline. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2019, 74, 290–298. [Google Scholar] [CrossRef]
- Ghosh, P.; Singh, R.; Ganeshpurkar, A.; Pokle, A.V.; Singh, R.B.; Singh, S.K.; Kumar, A. Cellular and molecular influencers of neuroinflammation in Alzheimer’s disease: Recent concepts & roles. Neurochem. Int. 2021, 151, 105212. [Google Scholar] [CrossRef] [PubMed]
- Biringer, R.G. The Role of Eicosanoids in Alzheimer’s Disease. Int. J. Environ. Res. Public Health 2019, 16, 2560. [Google Scholar] [CrossRef] [PubMed]
- Aisen, P.S.; Cummings, J.; Jack, C.R., Jr.; Morris, J.C.; Sperling, R.; Frölich, L.; Jones, R.W.; Dowsett, S.A.; Matthews, B.R.; Raskin, J.; et al. On the path to 2025: Understanding the Alzheimer’s disease continuum. Alzheimer’s Res. Ther. 2017, 9, 60. [Google Scholar] [CrossRef] [PubMed]
- Lehnardt, S.; Lachance, C.; Patrizi, S.; Lefebvre, S.; Follett, P.L.; Jensen, F.E.; Rosenberg, P.A.; Volpe, J.J.; Vartanian, T. The toll-like receptor TLR4 is necessary for lipopolysaccharide-induced oligodendrocyte injury in the CNS. J. Neurosci. 2002, 22, 2478–2486. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Choi, D.Y.; Yun, Y.P.; Han, S.B.; Oh, K.W.; Hong, J.T. Epigallocatechin-3-gallate prevents systemic inflammation-induced memory deficiency and amyloidogenesis via its anti-neuroinflammatory properties. J. Nutr. Biochem. 2013, 24, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Zakaria, R.; Wan Yaacob, W.M.; Othman, Z.; Long, I.; Ahmad, A.H.; Al-Rahbi, B. Lipopolysaccharide-induced memory impairment in rats: A model of Alzheimer’s disease. Physiol. Res. 2017, 66, 553–565. [Google Scholar] [CrossRef] [PubMed]
- MacRae, M.; Macrina, T.; Khoury, A.; Migliore, M.M.; Kentner, A.C. Tracing the trajectory of behavioral impairments and oxidative stress in an animal model of neonatal inflammation. Neuroscience 2015, 298, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Zubareva, O.E.; Postnikova, T.Y.; Grifluk, A.V.; Schwarz, A.P.; Smolensky, I.V.; Karepanov, A.A.; Vasilev, D.S.; Veniaminova, E.A.; Rotov, A.Y.; Kalemenev, S.V.; et al. Exposure to bacterial lipopolysaccharidein early life affects the expression of ionotropic glutamate receptor genes and is accompanied by disturbances in long-term potentiation and cognitive functions in young rats. Brain Behav. Immun. 2020, 90, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Zeng, J.; Feng, J.; Tian, Y.T.; Liu, Y.J.; Qiu, M.; Yan, X.; Yang, Y.; Xiong, Y.; Zhang, Z.H.; et al. Helicobacter pylori filtrate impairs spatial learning and memory in rats and increases beta-amyloid by enhancing expression of presenilin-2. Front. Aging Neurosci. 2014, 6, 66. [Google Scholar] [CrossRef] [PubMed]
- Ennaceur, A.; Delacour, J. A new one-trial test for neurobiological studies of memory in rats. 1: Behavioral data. Behav. Brain Res. 1988, 31, 47–59. [Google Scholar] [CrossRef]
- Furini, C.R.; Rossato, J.I.; Bitencourt, L.L.; Medina, J.H.; Izquierdo, I.; Cammarota, M. Beta-adrenergic receptors link NO/sGC/PKG signaling to BDNF expression during the consolidation of object recognition long-term memory. Hippocampus 2010, 20, 672–683. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uberti, A.F.; Callai-Silva, N.; Grahl, M.V.C.; Piovesan, A.R.; Nachtigall, E.G.; Furini, C.R.G.; Carlini, C.R. Helicobacter pylori Urease: Potential Contributions to Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 3091. https://doi.org/10.3390/ijms23063091
Uberti AF, Callai-Silva N, Grahl MVC, Piovesan AR, Nachtigall EG, Furini CRG, Carlini CR. Helicobacter pylori Urease: Potential Contributions to Alzheimer’s Disease. International Journal of Molecular Sciences. 2022; 23(6):3091. https://doi.org/10.3390/ijms23063091
Chicago/Turabian StyleUberti, Augusto F., Natalia Callai-Silva, Matheus V. C. Grahl, Angela R. Piovesan, Eduarda G. Nachtigall, Cristiane R. G. Furini, and Celia Regina Carlini. 2022. "Helicobacter pylori Urease: Potential Contributions to Alzheimer’s Disease" International Journal of Molecular Sciences 23, no. 6: 3091. https://doi.org/10.3390/ijms23063091
APA StyleUberti, A. F., Callai-Silva, N., Grahl, M. V. C., Piovesan, A. R., Nachtigall, E. G., Furini, C. R. G., & Carlini, C. R. (2022). Helicobacter pylori Urease: Potential Contributions to Alzheimer’s Disease. International Journal of Molecular Sciences, 23(6), 3091. https://doi.org/10.3390/ijms23063091