
Prediction of the Neurotoxic Potential of Chemicals Based on Modelling of Molecular Initiating Events Upstream of the Adverse Outcome Pathways of (Developmental) Neurotoxicity

 ,
, 
Abstract
:1. Introduction
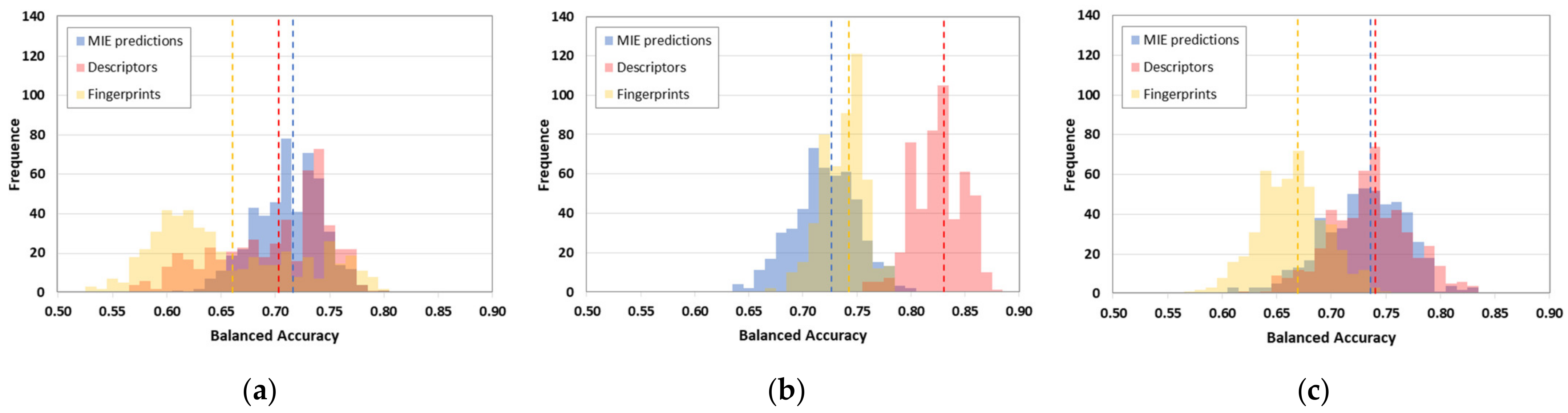
2. Results
3. Discussion
4. Materials and Methods
4.1. Data Selection for Molecular Initiating Events (MIEs)
- Glutamate ionotropic receptors, i.e., N-methyl-D-aspartate (NMDAR), alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPAR) and kainate (KAR) are responsible for excitatory synaptic transmission and synaptic plasticity, which are fundamental for learning and memory [46]. Sustained over-activation of these receptors (MIE A) can induce excitotoxicity due to increased Ca2+ influx, with consequent cell death, memory problems, and convulsions [4]. Analogously, the chronic blockage of NMDAR by chemicals during synaptogenesis (MIE B) disrupts neuronal network formation, resulting in the impairment of learning and memory processes [47] and increasing the risk of developing Alzheimer’s-type NDs in later life [2].
- Protein adduct formation is the covalent interaction between an electrophilic chemical and the nucleophilic part of a protein, and may lead to damage of the protein and the potential loss of its function. This may affect thiol- and seleno-containing proteins, which offer antioxidant protection [48]. The binding of xenobiotics (e.g., heavy metals and mercury) to these or other proteins during brain development (MIE D and H) may lead to several functional impairments, such as in learning and memory. Cytochrome P450 2E1 (CYP2E1) is relevant to this mechanism as well, as it is one of the enzymes responsible for the metabolism of small compounds. The induction of CYP2E1 (MIE E) leads to an increase in reactive metabolites, which can form protein adducts. For example, a high concentration of ethanol leads to an increased expression of CYP2E1 and consequent increased production of acetaldehyde metabolite, which can form protein adducts [49]. The consequences include oxidative stress, lipid peroxidation, unfolded protein responses and, ultimately the apoptosis of neuronal cells [50].
- The function of the Na+/I− symporter (NIS) is critical for the physiological production and maintenance of thyroid hormone levels in the serum, as it mediates the transport of iodide into thyroid cells. Its inhibition (MIE F) results in decreased thyroid hormone synthesis, with effects on neurocognitive function in children [51,52].
- Acetylcholinesterase (AChE) is an enzyme present in both central and peripheral nervous systems and in muscular motor plaques. It is responsible for the enzymatic cleavage of the neurotransmitter acetylcholine [53]. Inhibition of AChE (MIE I), e.g., by organophosphates and carbamates, leads to an increase in levels of acetylcholine and overstimulation of both muscarinic and nicotinic receptors, resulting in multiple adverse outcomes affecting a wide variety of functions [54].
- Ryanodine-sensitive Ca2+ channels (RyR) contribute to neurotransmission and synaptic plasticity. Polychlorinated biphenyl (PCB) exposure has been reported to alter intracellular Ca2+ levels and to interfere with normal neuronal dendritic growth and plasticity in a RyR-dependent manner (MIE L) [55].
- Thyroid hormone receptors α and β (THRα and THRβ) mediate the effects of thyroid hormones, while thyroperoxidase (TPO) and deionidase are involved in the biosynthesis/catabolism of thyroid hormones. Transtyretrin serum binding protein (TTR), monocarboxylate transporters 8 and 10, and the solute carrier organic anion transporter family member 1C14 (OATP1C1) are involved in the transportation of thyroid hormones at various levels [56]. Interference at any of these levels (MIEs G and Q-T) may lead to decreased thyroxine (T4) and thyroid hormones in the brain, and ultimately alter neurodevelopmental processes such as neuronal proliferation, apoptosis, migration, neurite outgrowth, and neuronal network connectivity [57,58], culminating in irreversible mental retardation and motor deficits [59]. It has been reported that PCBs induce activation of xenobiotic nuclear receptors, e.g., the constitutive androstane receptor (CAR) and the pregnane X receptor (PXR), which represent MIE P, leading to thyroid hormone disruption during cochlear development and potentially resulting in permanent auditory loss [60].
- The complexes of the respiratory chain play a pivotal role in neuronal and glial cell survival and cell death, as they regulate both energy metabolism and apoptotic/necrotic pathways. The interaction of xenobiotics with these enzymes can interfere in various ways with their normal functionality, e.g., inhibiting the production of ATP (MIE M) or interfering with the redox cycle (MIE N and O), with consequent increased production of ROS and oxidative stress. Oxidative stress contributes to a loss of function of hippocampal neural progenitor cells and a decline in learning and memory performance [4]. Moreover, the inhibition of NADH-quinone oxidoreductase (NADHOX) (MIE C) by pesticides or toxins (e.g., neurotoxin 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine, MPTP) has been reported to cause mitochondrial dysfunction and degeneration of dopaminergic neurons of the nigro-striatal area, with consequent motor deficits typical of Parkinson’s disease [61].
- Voltage-gated sodium channels (VGSC) are the primary molecules responsible for the control of the electrophysiological potentials of electrically excitable cells. Various isoforms exist, with isoforms 1, 2, 3, and 6 reported to be mainly expressed in the central nervous system [62]. Neurotoxic effects in mammals have been associated with the ability of some neurotoxicants (e.g., p,p’-DDT and pyrethroids) to bind to and disrupt VGSC (MIE U), with consequent behavioural effects [4,63].
- Ionotropic GABA receptors (GABAR) are ligand-gated ion channels which play important roles in inhibitory neurotransmission [64]. Interference with GABA signalling (MIE V) during development and after brain maturation is likely to cause such varied adverse outcomes as autism, mental retardation, epilepsy, and schizophrenia [59]. Chemically-induced epileptic seizures can be caused by the binding of neurotoxicants (e.g., barbiturates, benzodiazepines, and picrotoxin) to the active sites of the GABA receptor [65].
4.2. QSARs for Molecular Initiating Events
4.3. Thyroperoxidase (TPO) Modelling
4.4. Reactivity SMARTS
4.5. Neurotoxicity Data
4.6. Neurotoxicity Modelling
4.7. Evaluation of MIE Relative Importance
- (1)
- MIEs were iteratively removed, then QSARs for neurotoxicity were developed with the remaining features, as described in Section 4.6. BAs were averaged among the various iterations and compared to the reference values of models developed using all of the variables. A reduction in performance after the removal of a specific MIE flags a strong relationship between the excluded MIE and neurotoxicity. On the contrary, MIEs are considered less relevant if their exclusion does not vary or improve baseline performance.
- (2)
- Variable importance was calculated for each MIE within RF models. A score was calculated based on the attribute usage statistics in the RF for each descriptor by counting how many times it was selected for a split (#split) and at which rank (“level”; the first two levels were considered) among all available attributes (#candidates) in the trees of the ensemble:
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AMPAR | alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptor |
| AChE | acetylcholinesterase |
| AO | adverse outcome |
| AOP | adverse outcome pathway |
| AUC | area under the ROC curve |
| AUR-TPO | Amplex UltraRed-thyroperoxidase |
| BA | balanced accuracy |
| BRF | balanced random forest |
| CAR | constitutive androstane receptor |
| CYP2E1 | cytochrome P450 2E1 |
| DNT | developmental neurotoxicity |
| FN | false negative |
| FP | false positive |
| GABAR | GABA receptor |
| IATA | integrated approaches to testing and assessment |
| KA | kainite receptor |
| KE | key event |
| k-NN | k-nearest neighbors |
| MCC | Matthew’s correlation coefficient |
| MIE | molecular initiating event |
| NADHOX | NADH-quinone oxidoreductase |
| NIS | Na+/I− symporter |
| ND | neurodegenerative disease |
| NNET | neural networks |
| NMDAR | N-methyl-D-aspartate receptor |
| OATP1C1 | solute carrier organic anion transporter family member 1C14 |
| PCB | polychlorinated biphenyls |
| PXR | pregnane X receptor |
| QSAR | quantitative structure-activity relationship |
| RF | random forest |
| ROS | reactive oxygen species |
| RyR | ryanodine-sensitive Ca2+ channel |
| SEN | sensitivity |
| SPE | specificity |
| THRα | thyroid hormone receptor α |
| THRβ | thyroid hormone receptor β |
| TN | true negative |
| TP | true positive |
| TPO | thyroperoxidase |
| TTR | transtyretrin serum binding protein |
| VGSC | voltage-gated sodium channel |
References
- Landrigan, P.J.; Lambertini, L.; Birnbaum, L.S. A Research Strategy to Discover the Environmental Causes of Autism and Neurodevelopmental Disabilities. Environ. Health Perspect. 2012, 120, a258–a260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landrigan, P.J.; Sonawane, B.; Butler, R.N.; Trasande, L.; Callan, R.; Droller, D. Early Environmental Origins of Neurodegenerative Disease in Later Life. Environ. Health Perspect. 2005, 113, 1230–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lein, P.; Silbergeld, E.; Locke, P.; Goldberg, A.M. In Vitro and Other Alternative Approaches to Developmental Neurotoxicity Testing (DNT). Environ. Toxicol. Pharmacol. 2005, 19, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Bal-Price, A.; Crofton, K.M.; Sachana, M.; Shafer, T.J.; Behl, M.; Forsby, A.; Hargreaves, A.; Landesmann, B.; Lein, P.J.; Louisse, J. Putative Adverse Outcome Pathways Relevant to Neurotoxicity. Crit. Rev. Toxicol. 2015, 45, 83–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stiles, J.; Jernigan, T.L. The Basics of Brain Development. Neuropsychol. Rev. 2010, 20, 327–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloom, B.; Cohen, R.A.; Freeman, G. Summary Health Statistics for US Children: National Health Interview Survey, 2009; National Center for Health Statistics: Hyattsville, MD, USA, 2010; Volume 247, pp. 1–82.
- Bandeen-Roche, K.; Glass, T.A.; Bolla, K.I. Cumulative Lead Dose and Cognitive Function in Older Adults. Altern. Med. Rev. 2010, 15, 112–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayan, P.; Ehsani, S.; Lindquist, S. Combating Neurodegenerative Disease with Chemical Probes and Model Systems. Nat. Chem. Biol. 2014, 10, 911–920. [Google Scholar] [CrossRef]
- Trippier, P.C.; Jansen Labby, K.; Hawker, D.D.; Mataka, J.J.; Silverman, R.B. Target-and Mechanism-Based Therapeutics for Neurodegenerative Diseases: Strength in Numbers. J. Med. Chem. 2013, 56, 3121–3147. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S. The Macroeconomics of Dementia—Will the World Economy Get Alzheimer’s Disease? Arch. Med. Res. 2012, 43, 705–709. [Google Scholar] [CrossRef]
- Grandjean, P.; Landrigan, P.J. Neurobehavioural Effects of Developmental Toxicity. Lancet Neurol. 2014, 13, 330–338. [Google Scholar] [CrossRef] [Green Version]
- Grandjean, P.; Landrigan, P.J. Developmental Neurotoxicity of Industrial Chemicals. Lancet 2006, 368, 2167–2178. [Google Scholar] [CrossRef]
- Bal-Price, A.; Pistollato, F.; Sachana, M.; Bopp, S.K.; Munn, S.; Worth, A. Strategies to Improve the Regulatory Assessment of Developmental Neurotoxicity (DNT) Using in Vitro Methods. Toxicol. Appl. Pharmacol. 2018, 354, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, R.; Crofton, K.M. Developmental Neurotoxicity Guideline Study: Issues with Methodology, Evaluation and Regulation. Congenit. Anom. 2012, 52, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Collins, F.S.; Gray, G.M.; Bucher, J.R. Transforming Environmental Health Protection. Science 2008, 319, 906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dearden, J.C. The History and Development of Quantitative Structure-Activity Relationships (QSARs). In Oncology: Breakthroughs in Research and Practice; IGI Global: Hershey, PA, USA, 2017; pp. 67–117. [Google Scholar]
- Ankley, G.T.; Bennett, R.S.; Erickson, R.J.; Hoff, D.J.; Hornung, M.W.; Johnson, R.D.; Mount, D.R.; Nichols, J.W.; Russom, C.L.; Schmieder, P.K. Adverse Outcome Pathways: A Conceptual Framework to Support Ecotoxicology Research and Risk Assessment. Environ. Toxicol. Chem. Int. J. 2010, 29, 730–741. [Google Scholar] [CrossRef]
- Vinken, M. The Adverse Outcome Pathway Concept: A Pragmatic Tool in Toxicology. Toxicology 2013, 312, 158–165. [Google Scholar] [CrossRef]
- Leist, M.; Ghallab, A.; Graepel, R.; Marchan, R.; Hassan, R.; Bennekou, S.H.; Limonciel, A.; Vinken, M.; Schildknecht, S.; Waldmann, T.; et al. Adverse Outcome Pathways: Opportunities, Limitations and Open Questions. Arch. Toxicol. 2017, 91, 3477–3505. [Google Scholar] [CrossRef] [Green Version]
- Allen, T.E.; Goodman, J.M.; Gutsell, S.; Russell, P.J. A History of the Molecular Initiating Event. Chem. Res. Toxicol. 2016, 29, 2060–2070. [Google Scholar] [CrossRef]
- Allen, T.E.; Goodman, J.M.; Gutsell, S.; Russell, P.J. Quantitative Predictions for Molecular Initiating Events Using Three-Dimensional Quantitative Structure–Activity Relationships. Chem. Res. Toxicol. 2019, 33, 324–332. [Google Scholar] [CrossRef]
- Benigni, R. Building Predictive Adverse Outcome Pathway Models: Role of Molecular Initiating Events and Structure–Activity Relationships. Appl. Vitr. Toxicol. 2017, 3, 265–270. [Google Scholar] [CrossRef]
- Cronin, M.T.; Richarz, A.-N. Relationship between Adverse Outcome Pathways and Chemistry-Based in Silico Models to Predict Toxicity. Appl. Vitr. Toxicol. 2017, 3, 286–297. [Google Scholar] [CrossRef]
- Gadaleta, D.; Manganelli, S.; Roncaglioni, A.; Toma, C.; Benfenati, E.; Mombelli, E. QSAR Modeling of ToxCast Assays Relevant to the Molecular Initiating Events of AOPs Leading to Hepatic Steatosis. J. Chem. Inf. Model. 2018, 58, 1501–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patlewicz, G.; Simon, T.W.; Rowlands, J.C.; Budinsky, R.A.; Becker, R.A. Proposing a Scientific Confidence Framework to Help Support the Application of Adverse Outcome Pathways for Regulatory Purposes. Regul. Toxicol. Pharmacol. 2015, 71, 463–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tollefsen, K.E.; Scholz, S.; Cronin, M.T.; Edwards, S.W.; de Knecht, J.; Crofton, K.; Garcia-Reyero, N.; Hartung, T.; Worth, A.; Patlewicz, G. Applying Adverse Outcome Pathways (AOPs) to Support Integrated Approaches to Testing and Assessment (IATA). Regul. Toxicol. Pharmacol. 2014, 70, 629–640. [Google Scholar] [CrossRef]
- Li, J.; Settivari, R.; LeBaron, M.J.; Marty, M.S. An Industry Perspective: A Streamlined Screening Strategy Using Alternative Models for Chemical Assessment of Developmental Neurotoxicity. Neurotoxicology 2019, 73, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Marx-Stoelting, P.; de LM Solano, M.; Aoyama, H.; Adams, R.H.; Bal-Price, A.; Buschmann, J.; Chahoud, I.; Clark, R.; Fang, T.; Fujiwara, M.; et al. 25th Anniversary of the Berlin Workshop on Developmental Toxicology: DevTox Database Update, Challenges in Risk Assessment of Developmental Neurotoxicity and Alternative Methodologies in Bone Development and Growth. Reprod. Toxicol. 2021, 100, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Lenselink, E.B.; Ten Dijke, N.; Bongers, B.; Papadatos, G.; Van Vlijmen, H.W.; Kowalczyk, W.; Ijzerman, A.P.; Van Westen, G.J. Beyond the Hype: Deep Neural Networks Outperform Established Methods Using a ChEMBL Bioactivity Benchmark Set. J. Cheminform. 2017, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Bosc, N.; Atkinson, F.; Felix, E.; Gaulton, A.; Hersey, A.; Leach, A.R. Large Scale Comparison of QSAR and Conformal Prediction Methods and Their Applications in Drug Discovery. J. Cheminform. 2019, 11, 4. [Google Scholar] [CrossRef]
- Couratier, P.; Sindou, P.; Hugon, J.; Vallat, J.-M.; Dumas, M. Cell Culture Evidence for Neuronal Degeneration in Amyotrophic Lateral Sclerosis Being Linked to Glutamate AMPA/Kainate Receptors. Lancet 1993, 341, 265–268. [Google Scholar] [CrossRef]
- Weiss, J.; Yin, H.-Z.; Choi, D. Basal Forebrain Cholinergic Neurons Are Selectively Vulnerable to AMPA/Kainate Receptor-Mediated Neurotoxicity. Neuroscience 1994, 60, 659–664. [Google Scholar] [CrossRef]
- Muratov, E.N.; Bajorath, J.; Sheridan, R.P.; Tetko, I.V.; Filimonov, D.; Poroikov, V.; Oprea, T.I.; Baskin, I.I.; Varnek, A.; Roitberg, A.; et al. QSAR without Borders. Chem. Soc. Rev. 2020, 49, 3525–3564. [Google Scholar] [CrossRef] [PubMed]
- Polishchuk, P.G.; Muratov, E.N.; Artemenko, A.G.; Kolumbin, O.G.; Muratov, N.N.; Kuz’min, V.E. Application of Random Forest Approach to QSAR Prediction of Aquatic Toxicity. J. Chem. Inf. Model. 2009, 49, 2481–2488. [Google Scholar] [CrossRef] [PubMed]
- Svetnik, V.; Liaw, A.; Tong, C.; Culberson, J.C.; Sheridan, R.P.; Feuston, B.P. Random Forest: A Classification and Regression Tool for Compound Classification and QSAR Modeling. J. Chem. Inf. Comput. Sci. 2003, 43, 1947–1958. [Google Scholar] [CrossRef] [PubMed]
- Masjosthusmann, S.; Barenys, M.; El-Gamal, M.; Geerts, L.; Gerosa, L.; Gorreja, A.; Kühne, B.; Marchetti, N.; Tigges, J.; Viviani, B.; et al. Literature Review and Appraisal on Alternative Neurotoxicity Testing Methods. EFSA Support. Publ. 2018, 15, 1410E. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Polcher, A.; Joas, A. Systematic Literature Review on Parkinson’s Disease and Childhood Leukaemia and Mode of Actions for Pesticides. EFSA Support. Publ. 2016, 13, 955E. [Google Scholar] [CrossRef]
- Gadaleta, D.; Vuković, K.; Toma, C.; Lavado, G.J.; Karmaus, A.L.; Mansouri, K.; Kleinstreuer, N.C.; Benfenati, E.; Roncaglioni, A. SAR and QSAR Modeling of a Large Collection of LD 50 Rat Acute Oral Toxicity Data. J. Cheminform. 2019, 11, 58. [Google Scholar] [CrossRef] [Green Version]
- Škuta, C.; Cortés-Ciriano, I.; Dehaen, W.; Kříž, P.; van Westen, G.J.; Tetko, I.V.; Bender, A.; Svozil, D. QSAR-Derived Affinity Fingerprints (Part 1): Fingerprint Construction and Modeling Performance for Similarity Searching, Bioactivity Classification and Scaffold Hopping. J. Cheminform. 2020, 12, 39. [Google Scholar] [CrossRef]
- Rice, D.; Barone, S., Jr. Critical Periods of Vulnerability for the Developing Nervous System: Evidence from Humans and Animal Models. Environ. Health Perspect. 2000, 108 (Suppl. 3), 511–533. [Google Scholar]
- Cruz-Monteagudo, M.; Medina-Franco, J.L.; Perez-Castillo, Y.; Nicolotti, O.; Cordeiro, M.N.D.; Borges, F. Activity Cliffs in Drug Discovery: Dr Jekyll or Mr Hyde? Drug Discov. Today 2014, 19, 1069–1080. [Google Scholar] [CrossRef]
- Carlson, L.M.; Champagne, F.A.; Cory-Slechta, D.A.; Dishaw, L.; Faustman, E.; Mundy, W.; Segal, D.; Sobin, C.; Starkey, C.; Taylor, M. Potential Frameworks to Support Evaluation of Mechanistic Data for Developmental Neurotoxicity Outcomes: A Symposium Report. Neurotoxicol. Teratol. 2020, 78, 106865. [Google Scholar] [CrossRef]
- Fritsche, E.; Grandjean, P.; Crofton, K.M.; Aschner, M.; Goldberg, A.; Heinonen, T.; Hessel, E.V.; Hogberg, H.T.; Bennekou, S.H.; Lein, P.J. Consensus Statement on the Need for Innovation, Transition and Implementation of Developmental Neurotoxicity (DNT) Testing for Regulatory Purposes. Toxicol. Appl. Pharmacol. 2018, 354, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Villeneuve, D.L.; Crump, D.; Garcia-Reyero, N.; Hecker, M.; Hutchinson, T.H.; LaLone, C.A.; Landesmann, B.; Lettieri, T.; Munn, S.; Nepelska, M.; et al. Adverse Outcome Pathway (AOP) Development I: Strategies and Principles. Toxicol. Sci. 2014, 142, 312–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spînu, N.; Bal-Price, A.; Cronin, M.T.; Enoch, S.J.; Madden, J.C.; Worth, A.P. Development and Analysis of an Adverse Outcome Pathway Network for Human Neurotoxicity. Arch. Toxicol. 2019, 93, 2759–2772. [Google Scholar] [CrossRef] [Green Version]
- Schrattenholz, A.; Soskic, V. NMDA Receptors Are Not Alone: Dynamic Regulation of NMDA Receptor Structure and Function by Neuregulins and Transient Cholesterol-Rich Membrane Domains Leads to Disease-Specific Nuances of Glutamate-Signalling. Curr. Top. Med. Chem. 2006, 6, 663–686. [Google Scholar] [CrossRef] [PubMed]
- Toscano, C.D.; Guilarte, T.R. Lead Neurotoxicity: From Exposure to Molecular Effects. Brain Res. Rev. 2005, 49, 529–554. [Google Scholar] [CrossRef]
- Farina, M.; Rocha, J.B.; Aschner, M. Mechanisms of Methylmercury-Induced Neurotoxicity: Evidence from Experimental Studies. Life Sci. 2011, 89, 555–563. [Google Scholar] [CrossRef] [Green Version]
- Haorah, J.; Ramirez, S.H.; Floreani, N.; Gorantla, S.; Morsey, B.; Persidsky, Y. Mechanism of Alcohol-Induced Oxidative Stress and Neuronal Injury. Free. Radic. Biol. Med. 2008, 45, 1542–1550. [Google Scholar] [CrossRef] [Green Version]
- Valencia-Olvera, A.C.; Morán, J.; Camacho-Carranza, R.; Prospéro-García, O.; Espinosa-Aguirre, J.J. CYP2E1 Induction Leads to Oxidative Stress and Cytotoxicity in Glutathione-Depleted Cerebellar Granule Neurons. Toxicol. Vitr. 2014, 28, 1206–1214. [Google Scholar] [CrossRef]
- De la Vieja, A.; Dohan, O.; Levy, O.; Carrasco, N. Molecular Analysis of the Sodium/Iodide Symporter: Impact on Thyroid and Extrathyroid Pathophysiology. Physiol. Rev. 2000, 80, 1083–1105. [Google Scholar] [CrossRef]
- Dohan, O.; De la Vieja, A.; Paroder, V.; Riedel, C.; Artani, M.; Reed, M.; Ginter, C.S.; Carrasco, N. The Sodium/Iodide Symporter (NIS): Characterization, Regulation, and Medical Significance. Endocr. Rev. 2003, 24, 48–77. [Google Scholar] [CrossRef] [Green Version]
- Darvesh, S.; Hopkins, D.A.; Geula, C. Neurobiology of Butyrylcholinesterase. Nat. Rev. Neurosci. 2003, 4, 131–138. [Google Scholar] [CrossRef]
- US Environmental Protection Agency. The Use of Data on Cholinesterase Inhibition for Risk Assessments of Organophosphorous and Carbamate Pesticides; US Environmental Protection Agency: Washington, DC, USA, 2000.
- Holland, E.B.; Feng, W.; Zheng, J.; Dong, Y.; Li, X.; Lehmler, H.-J.; Pessah, I.N. An Extended Structure–Activity Relationship of Nondioxin-like PCBs Evaluates and Supports Modeling Predictions and Identifies Picomolar Potency of PCB 202 towards Ryanodine Receptors. Toxicol. Sci. 2017, 155, 170–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul Friedman, K.; Watt, E.D.; Hornung, M.W.; Hedge, J.M.; Judson, R.S.; Crofton, K.M.; Houck, K.A.; Simmons, S.O. Tiered High-Throughput Screening Approach to Identify Thyroperoxidase Inhibitors within the ToxCast Phase I and II Chemical Libraries. Toxicol. Sci. 2016, 151, 160–180. [Google Scholar] [CrossRef] [Green Version]
- Zoeller, R.; Rovet, J. Timing of Thyroid Hormone Action in the Developing Brain: Clinical Observations and Experimental Findings. J. Neuroendocrinol. 2004, 16, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Bernal, J. Thyroid Hormone Receptors in Brain Development and Function. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Westerholz, S.; De Lima, A.; Voigt, T. Regulation of Early Spontaneous Network Activity and GABAergic Neurons Development by Thyroid Hormone. Neuroscience 2010, 168, 573–589. [Google Scholar] [CrossRef]
- Crofton, K.M.; Zoeller, R.T. Mode of Action: Neurotoxicity Induced by Thyroid Hormone Disruption during Development—Hearing Loss Resulting from Exposure to PHAHs. Crit. Rev. Toxicol. 2005, 35, 757–769. [Google Scholar] [CrossRef] [PubMed]
- Van Maele-Fabry, G.; Hoet, P.; Vilain, F.; Lison, D. Occupational Exposure to Pesticides and Parkinson’s Disease: A Systematic Review and Meta-Analysis of Cohort Studies. Environ. Int. 2012, 46, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Goldin, A.L. Resurgence of Sodium Channel Research. Annu. Rev. Physiol. 2001, 63, 871–894. [Google Scholar] [CrossRef] [Green Version]
- Soderlund, D.M. Molecular Mechanisms of Pyrethroid Insecticide Neurotoxicity: Recent Advances. Arch. Toxicol. 2012, 86, 165–181. [Google Scholar] [CrossRef] [Green Version]
- McGonigle, I.; Lummis, S.C. Molecular Characterization of Agonists That Bind to an Insect GABA Receptor. Biochemistry 2010, 49, 2897–2902. [Google Scholar] [CrossRef] [PubMed]
- Gong, P.; Hong, H.; Perkins, E.J. Ionotropic GABA Receptor Antagonism-Induced Adverse Outcome Pathways for Potential Neurotoxicity Biomarkers. Biomark. Med. 2015, 9, 1225–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL Database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef] [PubMed]
- Bento, A.P.; Gaulton, A.; Hersey, A.; Bellis, L.J.; Chambers, J.; Davies, M.; Krüger, F.A.; Light, Y.; Mak, L.; McGlinchey, S. The ChEMBL Bioactivity Database: An Update. Nucleic Acids Res. 2014, 42, D1083–D1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadaleta, D.; d’Alessandro, L.; Marzo, M.; Benfenati, E.; Roncaglioni, A. Quantitative Structure-Activity Relationship Modeling of the Amplex Ultrared Assay to Predict Thyroperoxidase Inhibitory Activity. Front. Pharmacol. 2021, 12, 713037. [Google Scholar] [CrossRef]
- Enoch, S.J.; Madden, J.C.; Cronin, M.T.D. Identification of Mechanisms of Toxic Action for Skin Sensitisation Using a SMARTS Pattern Based Approach. SAR QSAR Environ. Res. 2008, 19, 555–578. [Google Scholar] [CrossRef]
- Gadaleta, D.; Lombardo, A.; Toma, C.; Benfenati, E. A New Semi-Automated Workflow for Chemical Data Retrieval and Quality Checking for Modeling Applications. J. Cheminform. 2018, 10, 60. [Google Scholar] [CrossRef] [Green Version]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Thiel, K.; Wiswedel, B. KNIME-the Konstanz Information Miner: Version 2.0 and Beyond. AcM SIGKDD Explor. Newsl. 2009, 11, 26–31. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Liaw, A.; Breiman, L. Using Random Forest to Learn Imbalanced Data. Univ. Calif. Berkeley 2004, 110, 24. [Google Scholar]
- Dal Pozzolo, A.; Boracchi, G.; Caelen, O.; Alippi, C.; Bontempi, G. Credit Card Fraud Detection and Concept-Drift Adaptation with Delayed Supervised Information. In Proceedings of the 2015 International Joint Conference on Neural Networks (IJCNN), Killarney, Ireland, 12–16 July 2015; pp. 1–8. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Kode: DRAGON 7.0.8. 2017. Available online: https://chm.kode-solutions.net/products_dragon.php (accessed on 7 March 2022).
- Kosnik, M.B.; Strickland, J.D.; Marvel, S.W.; Wallis, D.J.; Wallace, K.; Richard, A.M.; Reif, D.M.; Shafer, T.J. Concentration–Response Evaluation of ToxCast Compounds for Multivariate Activity Patterns of Neural Network Function. Arch. Toxicol. 2020, 94, 469–484. [Google Scholar] [CrossRef] [PubMed]
- Strickland, J.D.; Martin, M.T.; Richard, A.M.; Houck, K.A.; Shafer, T.J. Screening the ToxCast Phase II Libraries for Alterations in Network Function Using Cortical Neurons Grown on Multi-Well Microelectrode Array (MwMEA) Plates. Arch. Toxicol. 2018, 92, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Altman, N.S. An Introduction to Kernel and Nearest-Neighbor Nonparametric Regression. Am. Stat. 1992, 46, 175–185. [Google Scholar]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Dudek, A.Z.; Arodz, T.; Gálvez, J. Computational Methods in Developing Quantitative Structure-Activity Relationships (QSAR): A Review. Comb. Chem. High Throughput Screen. 2006, 9, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Mao, J.; Mohiuddin, K.M. Artificial Neural Networks: A Tutorial. Computer 1996, 29, 31–44. [Google Scholar] [CrossRef] [Green Version]
- Daylight Chemical Information Systems, Inc. 6. Fingerprints—Screening and Similarity. 2019. Available online: https://www.daylight.com/dayhtml/doc/theory/theory.finger.html (accessed on 26 January 2022).
- Todeschini, R.; Consonni, V. Handbook of Molecular Descriptors; John Wiley & Sons: Hoboken, NJ, USA, 2008; Volume 11. [Google Scholar]
- Blaauboer, B.J. The Integration of Data on Physico-Chemical Properties, in Vitro-Derived Toxicity Data and Physiologically Based Kinetic and Dynamic as Modelling a Tool in Hazard and Risk Assessment. A Commentary. Toxicol. Lett. 2003, 138, 161–171. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| MIE | TP | FP | TN | FN | SEN | SPE | BA | MCC | AUC |
|---|---|---|---|---|---|---|---|---|---|
| AChE | 555.6 | 63.0 | 887.2 | 68.0 | 0.89 | 0.93 | 0.91 | 0.83 | 0.96 |
| AMPAR | 13.4 | 20.0 | 651.0 | 1.2 | 0.92 | 0.97 | 0.94 | 0.60 | 0.99 |
| CAR | 9.0 | 6.8 | 668.6 | 1.2 | 0.88 | 0.99 | 0.94 | 0.72 | 0.95 |
| CYP2E1 | 4.0 | 35.2 | 645.2 | 1.0 | 0.80 | 0.95 | 0.87 | 0.29 | 0.90 |
| GABAR | 20.0 | 11.0 | 649.0 | 6.0 | 0.77 | 0.98 | 0.88 | 0.69 | 0.96 |
| KAR | 4.4 | 17.4 | 663.0 | 0.6 | 0.88 | 0.97 | 0.93 | 0.42 | 0.97 |
| NADHOX | 15.2 | 4.4 | 665.4 | 0.4 | 0.97 | 0.99 | 0.98 | 0.87 | 1.00 |
| NIS | 11.0 | 0.6 | 673.4 | 0.4 | 0.97 | 1.00 | 0.98 | 0.96 | 1.00 |
| NMDAR | 50.0 | 27.8 | 604.4 | 3.4 | 0.94 | 0.96 | 0.95 | 0.75 | 0.98 |
| PXR | 35.8 | 35.8 | 601.8 | 13.0 | 0.73 | 0.94 | 0.84 | 0.57 | 0.92 |
| RYR | 11.0 | 0.6 | 673.6 | 0.2 | 0.98 | 1.00 | 0.99 | 0.96 | 0.99 |
| THRα | 60.0 | 23.4 | 599.2 | 2.8 | 0.96 | 0.96 | 0.96 | 0.81 | 0.99 |
| THRβ | 110.2 | 37.8 | 500.0 | 38.4 | 0.74 | 0.93 | 0.84 | 0.67 | 0.93 |
| TTR | 14.8 | 44.0 | 624.0 | 3.8 | 0.80 | 0.93 | 0.87 | 0.42 | 0.94 |
| VGSC | 28.4 | 12.8 | 639.2 | 5.0 | 0.85 | 0.98 | 0.92 | 0.76 | 0.97 |
| Classifier | Variable | TP | FP | TN | FN | NC | SEN | SPE | BA | MCC | AUC |
|---|---|---|---|---|---|---|---|---|---|---|---|
| K-NN | MIE predictions | 30.5 | 11.4 | 19.6 | 7.5 | 0.0 | 0.80 | 0.63 | 0.72 | 0.44 | 0.76 |
| Descriptors | 29.5 | 11.5 | 19.5 | 8.5 | 0.0 | 0.78 | 0.63 | 0.70 | 0.41 | 0.76 | |
| Fingerprints | 14.6 | 2.3 | 28.5 | 22.2 | 1.4 | 0.40 | 0.92 | 0.66 | 0.37 | 0.75 | |
| MLP-NNET | MIE predictions | 29.4 | 9.4 | 21.6 | 8.6 | 0.0 | 0.77 | 0.70 | 0.74 | 0.47 | 0.78 |
| Descriptors | 30.2 | 9.8 | 21.2 | 7.8 | 0.0 | 0.79 | 0.68 | 0.74 | 0.48 | 0.79 | |
| Fingerprints | 28.1 | 12.4 | 18.6 | 9.9 | 0.0 | 0.74 | 0.60 | 0.67 | 0.34 | 0.69 | |
| RF | MIE predictions | 31.1 | 11.6 | 19.2 | 6.4 | 0.7 | 0.83 | 0.62 | 0.73 | 0.47 | 0.77 |
| Descriptors | 32.9 | 6.4 | 24.4 | 4.9 | 0.4 | 0.87 | 0.79 | 0.83 | 0.66 | 0.91 | |
| Fingerprints | 32.9 | 11.9 | 18.8 | 4.8 | 0.5 | 0.87 | 0.61 | 0.74 | 0.51 | 0.80 |
| ID | MIE | Target | Reference |
|---|---|---|---|
| A | Binding of agonist, Ionotropic glutamate receptors | Glutamate [NMDA] receptor | [45] |
| A | Binding of agonist, Ionotropic glutamate receptors | Glutamate receptor ionotropic kainate | [45] |
| A | Binding of agonist, Ionotropic glutamate receptors | Glutamate receptor ionotropic AMPA | [45] |
| B | Binding of antagonist, NMDA receptors | Glutamate [NMDA] receptor | [45] |
| C | Binding of inhibitor, NADH-ubiquinone oxidoreductase (complex I) | Mitochondrial complex I (NADH dehydrogenase) | [45] |
| D | Binding, SH/SeH proteins involved in protection against oxidative stress | Aspecific1 | [45] |
| E | CYP2E1 Activation | Cytochrome P450 2E1 | [45] |
| F | Inhibition, Na+/I− symporter (NIS) | Sodium/iodide cotransporter | [45] |
| G | Thyroperoxidase, Inhibition | Thyroid peroxidase 1 | [45] |
| H | Protein Adduct Formation | Aspecific 2 | [45] |
| I | Binding of inhibitors to acetylcholinesterase (AChE) | Acetylcholinesterase | [27] |
| L | Binding of non-dioxin-like polychlorinated biphenyls with ryanodine receptor (RyR) | Ryanodine receptors 1, 2 and 3 | [27] |
| M | Interaction uncouplers with oxidative phosphorylation | Aspecific 3 | [27] |
| N | Binding of redox cycling chemicals with NADH-quinone oxidoreductase | Mitochondrial complex I (NADH dehydrogenase) | [27] |
| O | Binding of redox cycling chemicals with NADH cytochrome b5 reductase | NADH-cytochrome b5 reductase | [27] |
| P | Xenobiotic nuclear receptor activation | Pregnane X receptor | [27] |
| P | Xenobiotic nuclear receptor activation | Nuclear receptor subfamily 1 group I member 3 (Constitutive Androstane Receptor) | [27] |
| Q | Interference with thyroid serum binding protein | Transthyretin | [27] |
| R | Deiodinase inhibition | Deiodinase 4 | [27] |
| S | Thyroid receptor binding | Thyroid hormone receptor beta | [27] |
| S | Thyroid receptor binding | Thyroid hormone receptor alpha | [27] |
| T | Thyroid hormone transporter interference | Monocarboxylate transporter 8 4 | [27] |
| T | Thyroid hormone transporter interference | Monocarboxylate transporter 10 4 | [27] |
| T | Thyroid hormone transporter interference | Solute carrier organic anion transporter family member 1C1 4 | [27] |
| U | Binding of pyrethroids to voltage-gated sodium channels (VGSC) | Sodium channel protein type N alpha subunit | [27] |
| V | Binding of antagonist to γ-aminobutyric acid receptor GABAAR | GABA-A receptor; alpha-1/beta-2/gamma-2 | [27] |
| Target | Code | CheMBL ID | Species | MIE | ACT | INA |
|---|---|---|---|---|---|---|
| Acetylcholinesterase | AChE | CHEMBL220 | Human | I | 3076 | 4793 |
| Glutamate receptor ionotropic AMPA | AMPAR | CHEMBL2096670 | Human | A | 73 | 3355 |
| Nuclear receptor subfamily 1 group I member 3 (Constitutive Androstane Receptor) | CAR | CHEMBL5503 | Human | P | 51 | 3377 |
| Cytochrome P450 2E1 | CYP2E1 | CHEMBL5281 | Human | E | 25 | 3402 |
| GABA-A receptor; alpha-1/beta-2/gamma-2 | GABAR | CHEMBL2095172 | Human | V | 129 | 3298 |
| Glutamate receptor ionotropic kainate | KAR | CHEMBL2109241 | Human | A | 25 | 3402 |
| Mitochondrial complex I (NADH dehydrogenase) | NADHOX | CHEMBL614865 | Bos taurus | C, N | 78 | 3349 |
| Sodium/iodide cotransporter | NIS | CHEMBL2331047 | Human | F | 56 | 3371 |
| Glutamate [NMDA] receptor | NMDAR | CHEMBL2094124 | Human | A, B | 267 | 3161 |
| Pregnane X receptor | PXR | CHEMBL3401 | Human | P | 244 | 3188 |
| Ryanodine receptors 1 | RYR | CHEMBL2062 CHEMBL4403 CHEMBL1846 | Human | L | 56 | 3371 |
| Thyroid hormone receptor alpha | THRα | CHEMBL1860 | Human | S | 311 | 3116 |
| Thyroid hormone receptor beta | THRβ | CHEMBL1947 | Human | S | 728 | 2704 |
| Transthyretin | TTR | CHEMBL3194 | Human | Q | 93 | 3340 |
| Sodium channel protein type N alpha subunit 2 | VGSC | CHEMBL1845 CHEMBL4187 CHEMBL5163 CHEMBL5202 | Human | U | 167 | 3260 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gadaleta, D.; Spînu, N.; Roncaglioni, A.; Cronin, M.T.D.; Benfenati, E. Prediction of the Neurotoxic Potential of Chemicals Based on Modelling of Molecular Initiating Events Upstream of the Adverse Outcome Pathways of (Developmental) Neurotoxicity. Int. J. Mol. Sci. 2022, 23, 3053. https://doi.org/10.3390/ijms23063053
Gadaleta D, Spînu N, Roncaglioni A, Cronin MTD, Benfenati E. Prediction of the Neurotoxic Potential of Chemicals Based on Modelling of Molecular Initiating Events Upstream of the Adverse Outcome Pathways of (Developmental) Neurotoxicity. International Journal of Molecular Sciences. 2022; 23(6):3053. https://doi.org/10.3390/ijms23063053
Chicago/Turabian StyleGadaleta, Domenico, Nicoleta Spînu, Alessandra Roncaglioni, Mark T. D. Cronin, and Emilio Benfenati. 2022. "Prediction of the Neurotoxic Potential of Chemicals Based on Modelling of Molecular Initiating Events Upstream of the Adverse Outcome Pathways of (Developmental) Neurotoxicity" International Journal of Molecular Sciences 23, no. 6: 3053. https://doi.org/10.3390/ijms23063053
APA StyleGadaleta, D., Spînu, N., Roncaglioni, A., Cronin, M. T. D., & Benfenati, E. (2022). Prediction of the Neurotoxic Potential of Chemicals Based on Modelling of Molecular Initiating Events Upstream of the Adverse Outcome Pathways of (Developmental) Neurotoxicity. International Journal of Molecular Sciences, 23(6), 3053. https://doi.org/10.3390/ijms23063053