
Complex of HIV-1 Integrase with Cellular Ku Protein: Interaction Interface and Search for Inhibitors


 , , ,
, , ,
Abstract
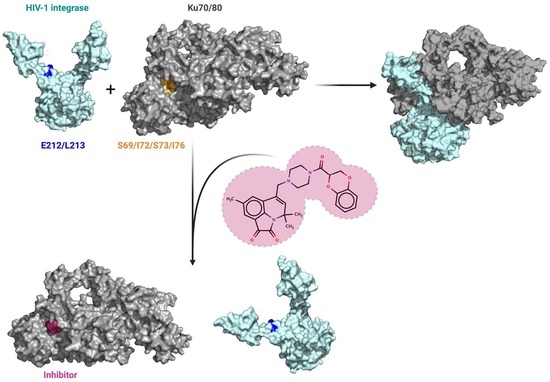
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
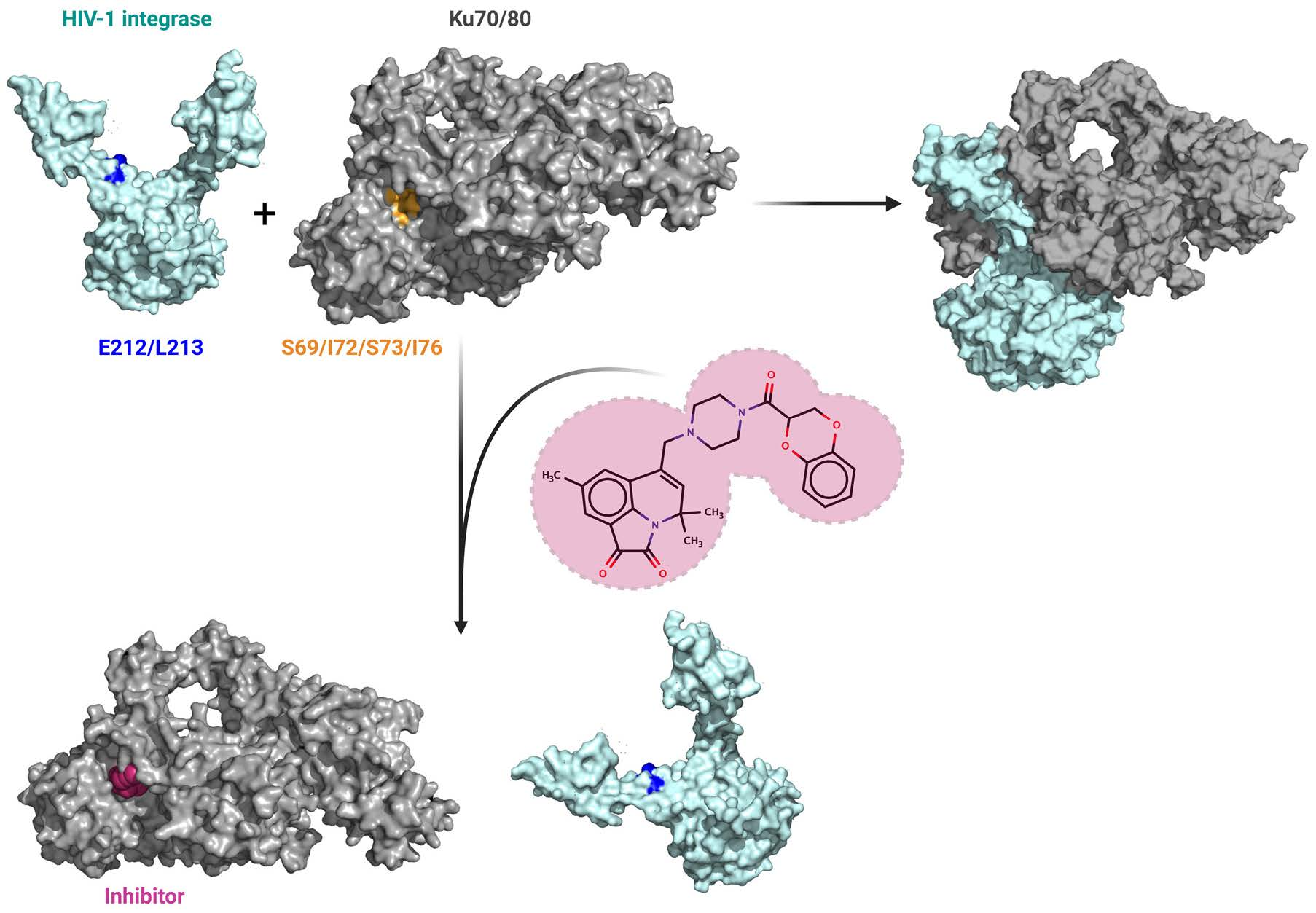
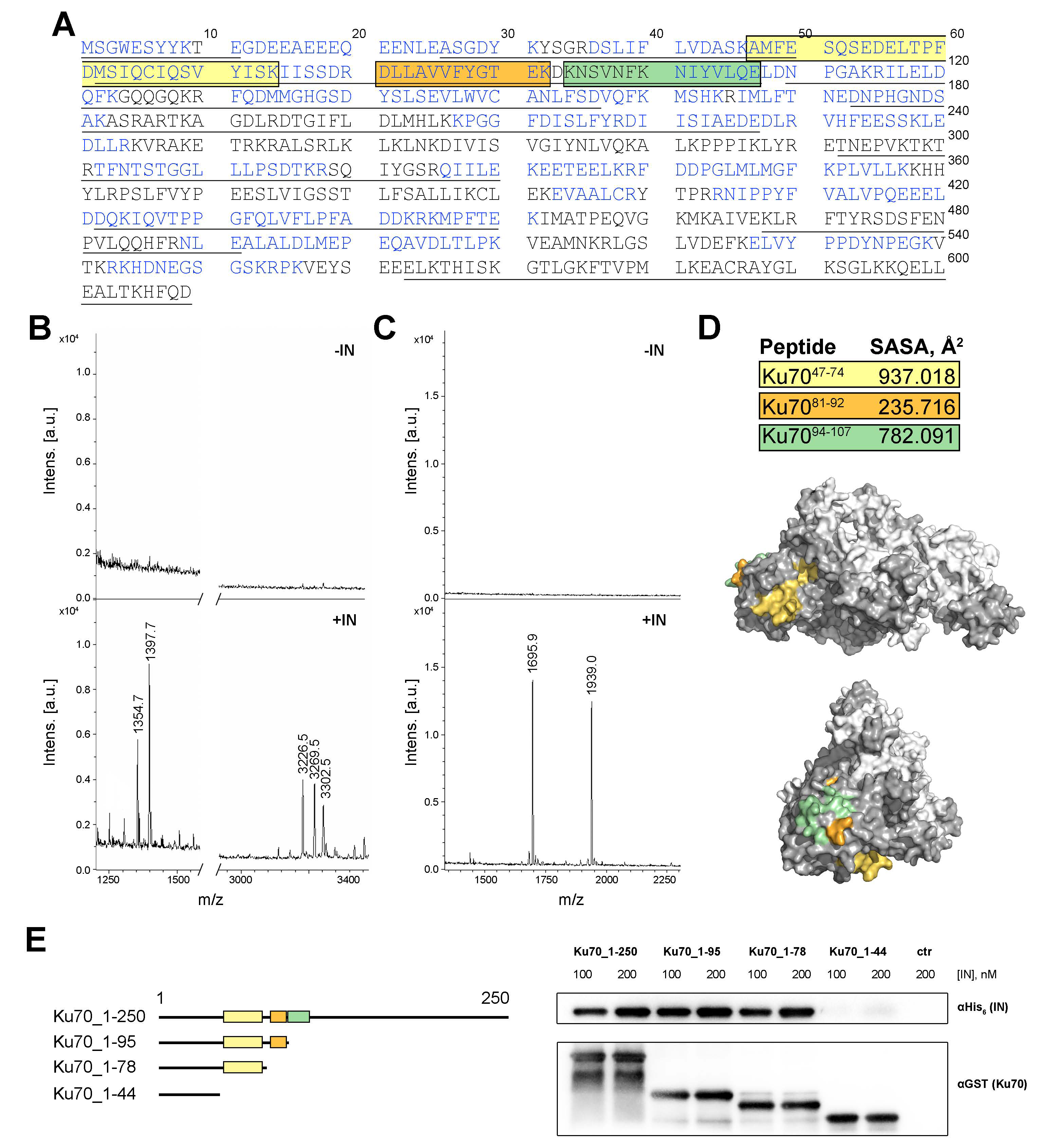
2.1. Peptide Fishing Assay Reveals Short Peptides of the Ku70 N-Terminus That Bind to HIV-1 Integrase
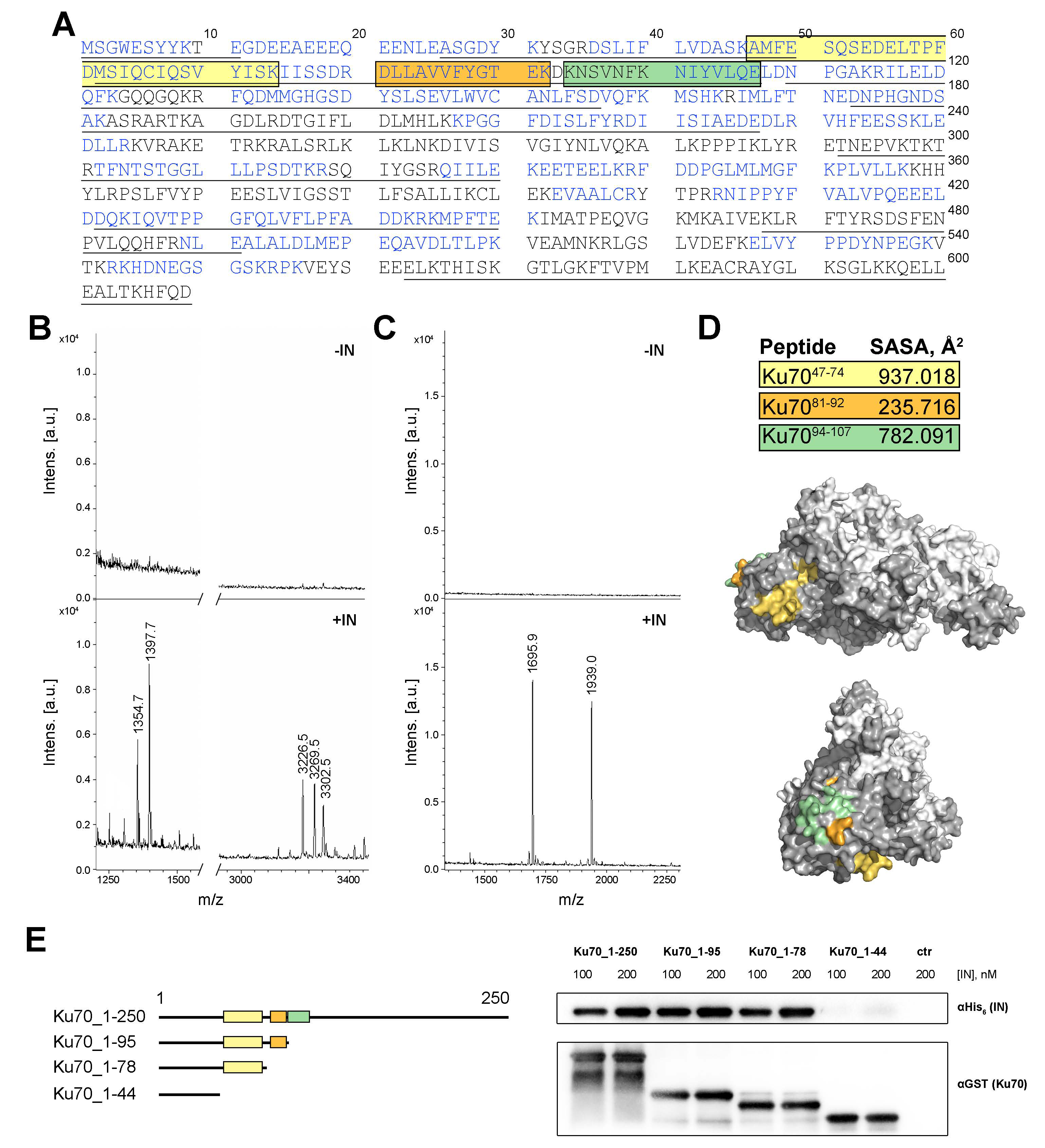
2.2. Ku7047–74 Peptide Is Essential for IN Binding
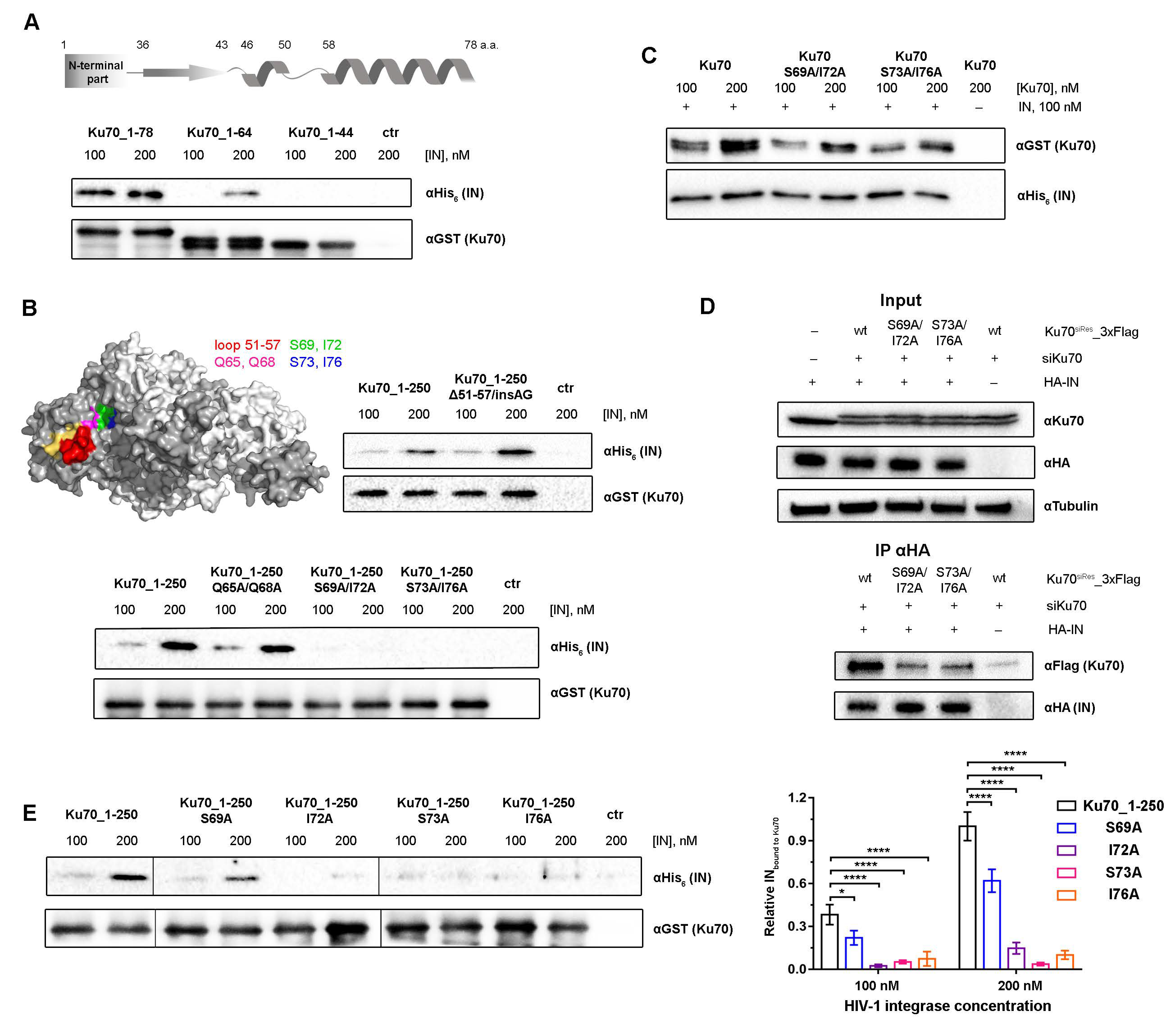
2.3. Amino Acids S69, I72, S73, and I76 of Ku70 Participate in the IN Binding
2.4. Double Substitutions S69A/I72A and S73A/L76A within Ku70 Impede Its Interaction with HIV-1 Integrase in 293T Cells
2.5. Elucidation of Ku70 Amino Acid Residues Involved in Integrase Binding
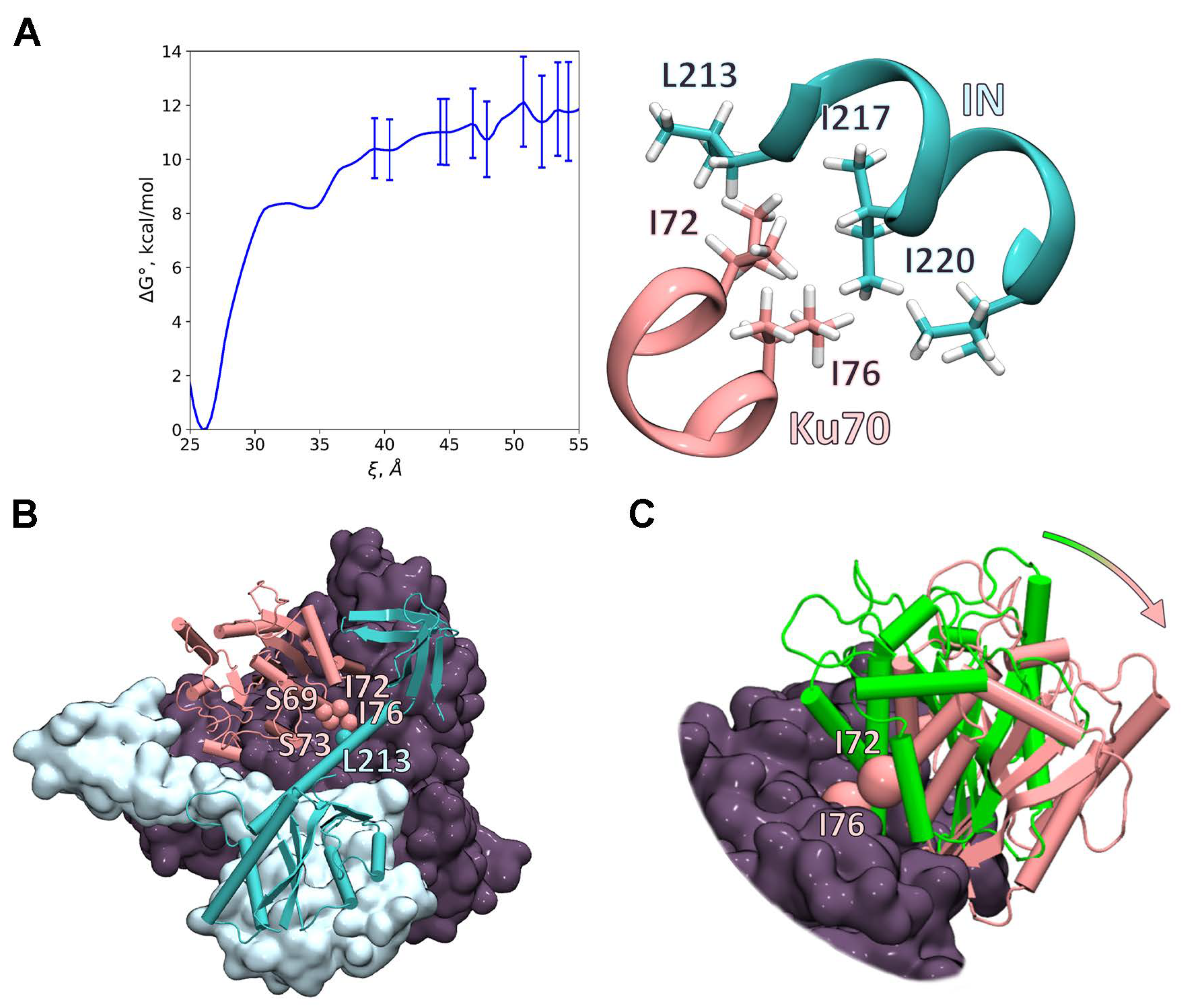
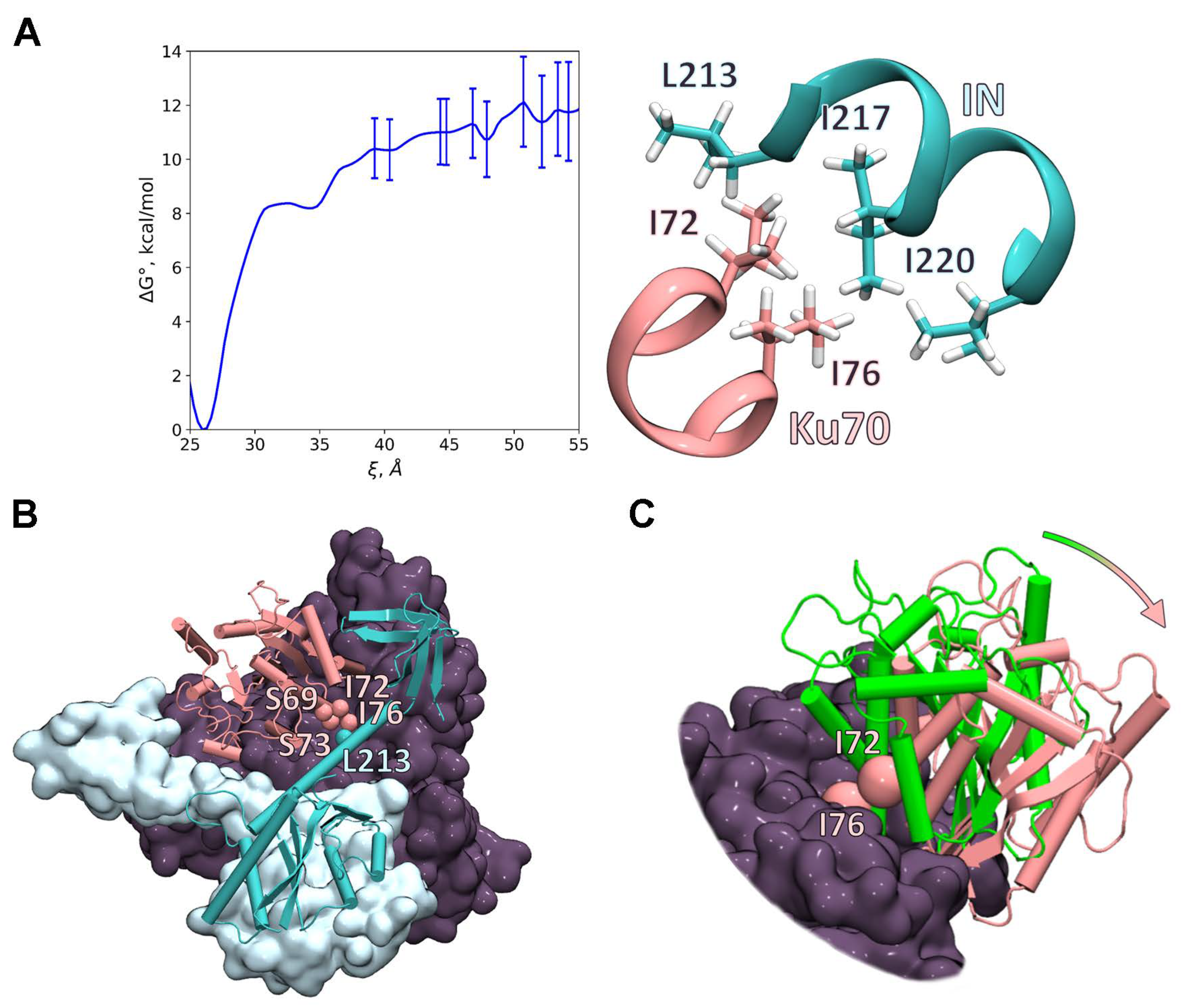
2.6. Investigation of the IN/Ku70 Complex Structure Using Molecular Dynamic Approach
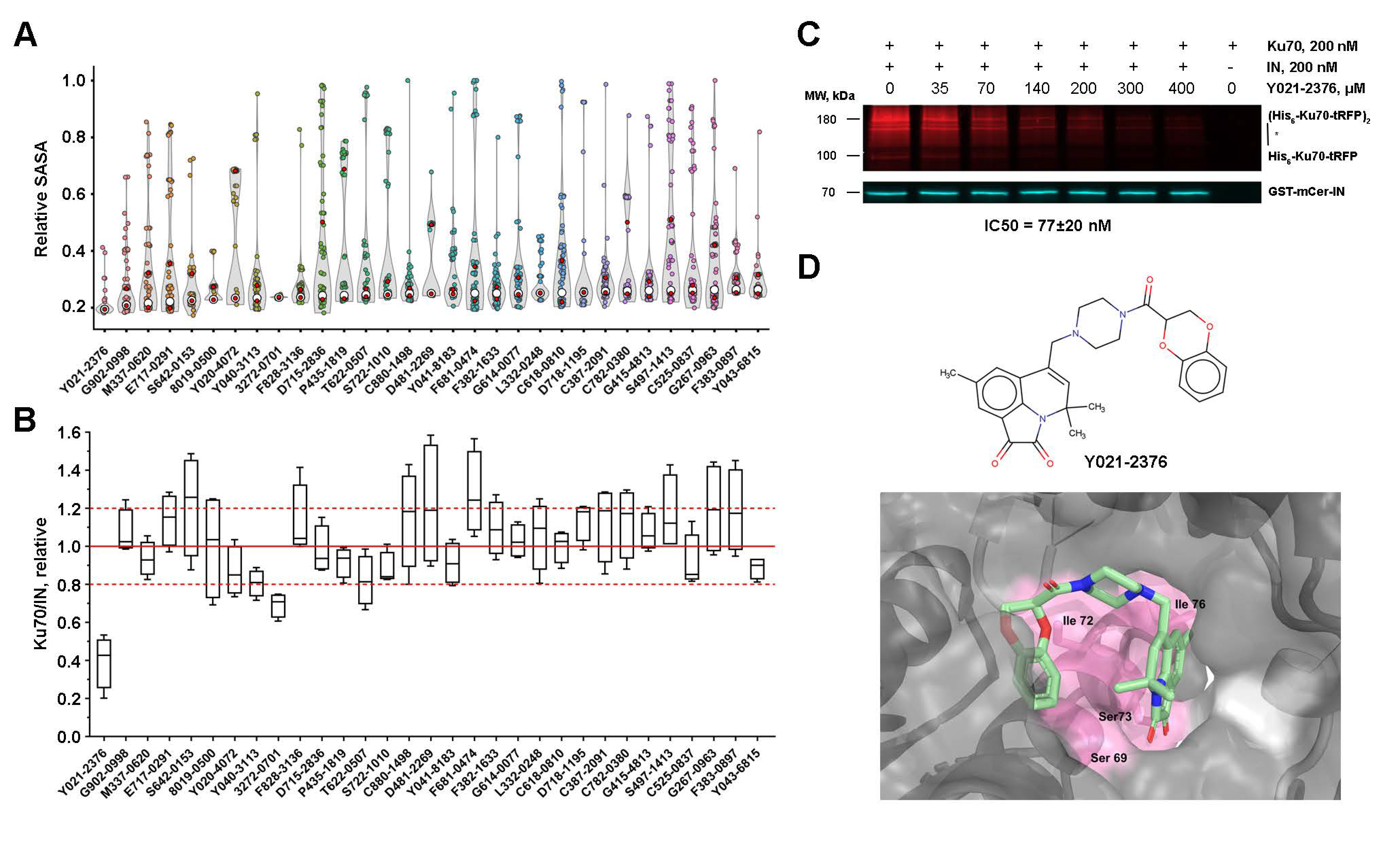
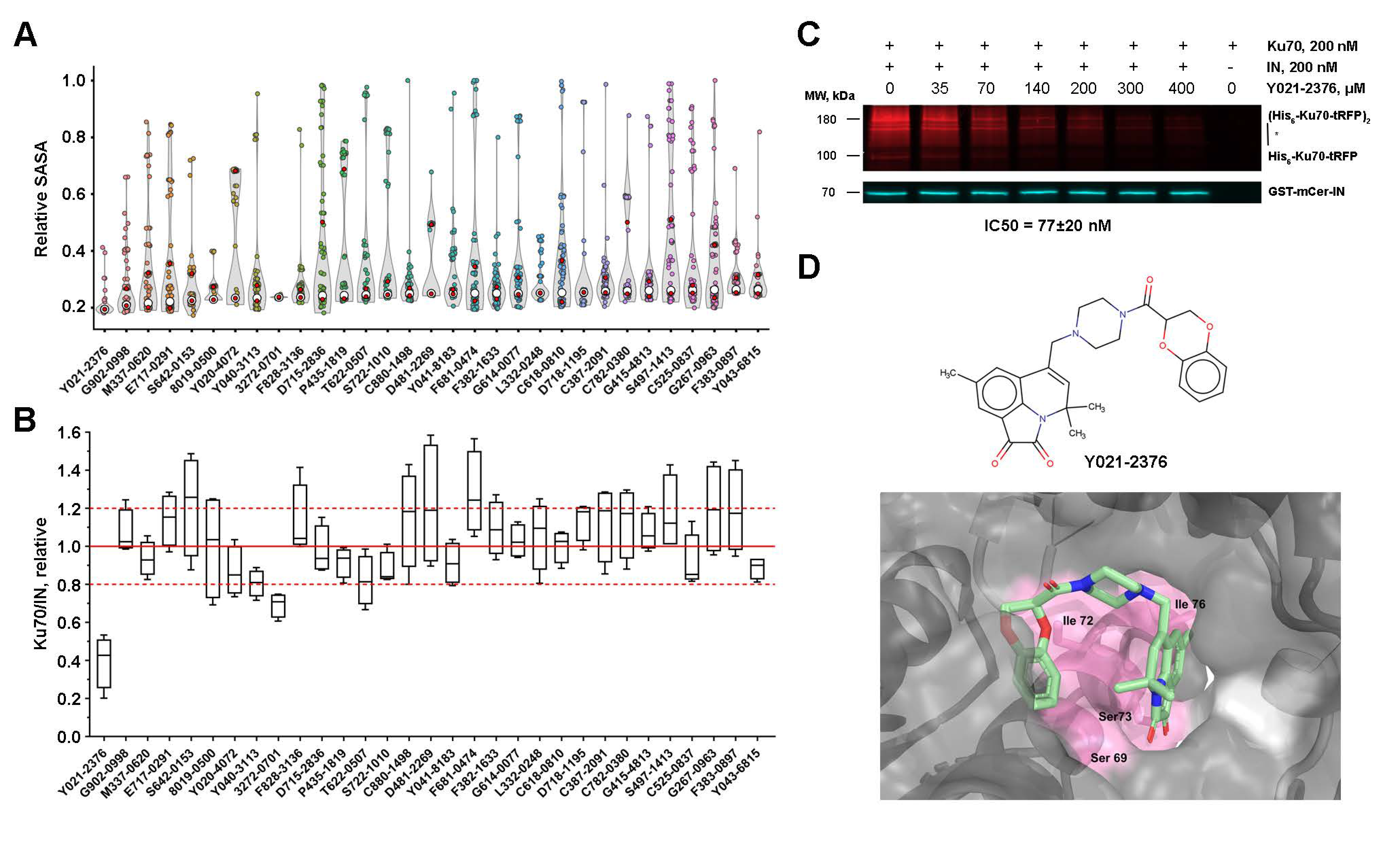
2.7. Low-Molecular-Weight Compounds Shielding Residues S69, I72, S73, and I76 in Ku70 Disrupt Its Interaction with IN
3. Discussion
4. Materials and Methods
4.1. Oligonucleotides and Plasmids
4.2. Recombinant Proteins Expression and Purification
4.3. Peptide Fishing Assay
4.4. Protein Binding Assays
4.5. Co-Immunoprecipitation
4.6. Western Blot Analysis
4.7. Fluorescence Gel Imaging
4.8. Fluorescence Imaging
4.9. Molecular Dynamic Simulations
4.10. Molecular Docking
4.10.1. System Configuration and Computational Details
4.10.2. Protein Model Preparation
4.10.3. Ligand Preparation
4.10.4. First Docking Iteration
4.10.5. Second Docking Iteration
4.10.6. SASA Calculation
4.10.7. In Silico PAINS Filtering
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Deeks, S.G.; Lewin, S.R.; Havlir, D.V. The end of AIDS: HIV infection as a chronic disease. Lancet 2013, 382, 1525–1533. [Google Scholar] [CrossRef] [Green Version]
- Kuritzkes, D.R. Drug resistance in HIV-1. Curr. Opin. Virol. 2011, 1, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Pennings, P.S. HIV drug resistance: Problems and perspectives. Infect. Dis. Rep. 2013, 5, 21–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Günthard, H.F.; Calvez, V.; Paredes, R.; Pillay, D.; Shafer, R.W.; Wensing, A.M.; Jacobsen, D.M.; Richman, D.D.; Francisco, S. Human Immunodeficiency Virus Drug Resistance: 2018 Recommendations of the International Antiviral Society-USA Panel. Clin. Infect. Dis. 2019, 68, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Taltynov, O.; Desimmie, B.A.; Demeulemeester, J.; Christ, F.; Debyser, Z. Cellular Cofactors of Lentiviral Integrase: From Target Validation to Drug Discovery. Mol. Biol. Int. 2012, 2012, 863405. [Google Scholar] [CrossRef] [PubMed]
- Taltynov, O.; De Rijck, J.; Debyser, Z. Identification and Validation of HIV Cofactors. In Encyclopedia of AIDS; Springer: New York, NY, USA, 2018; pp. 1043–1047. [Google Scholar]
- Dürr, R.; Keppler, O.; Christ, F.; Crespan, E.; Garbelli, A.; Maga, G.; Dietrich, U. Targeting cellular cofactors in HIV therapy. Top. Med. Chem. 2015, 15, 183–222. [Google Scholar] [CrossRef]
- Lesbats, P.; Engelman, A.N.; Cherepanov, P. Retroviral DNA Integration. Chem. Rev. 2016, 116, 12730–12757. [Google Scholar] [CrossRef] [Green Version]
- Poeschla, E.M. Integrase, LEDGF/p75 and HIV replication. Cell. Mol. Life Sci. 2008, 65, 1403–1424. [Google Scholar] [CrossRef] [Green Version]
- Christ, F.; Debyser, Z. The LEDGF/p75 integrase interaction, a novel target for anti-HIV therapy. Virology 2013, 435, 102–109. [Google Scholar] [CrossRef] [Green Version]
- Le Rouzic, E.; Bonnard, D.; Chasset, S.; Bruneau, J.M.; Chevreuil, F.; Le Strat, F.; Nguyen, J.; Beauvoir, R.; Amadori, C.; Brias, J.; et al. Dual inhibition of HIV-1 replication by integrase-LEDGF allosteric inhibitors is predominant at the post-integration stage. Retrovirology 2013, 10, 144. [Google Scholar] [CrossRef] [Green Version]
- Christ, F.; Voet, A.; Marchand, A.; Nicolet, S.; Desimmie, B.A.; Marchand, D.; Bardiot, D.; Van Der Veken, N.J.; Van Remoortel, B.; Strelkov, S.V.; et al. Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nat. Chem. Biol. 2010, 6, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Kessl, J.J.; Kutluay, S.B.; Townsend, D.; Rebensburg, S.; Slaughter, A.; Larue, R.C.; Shkriabai, N.; Bakouche, N.; Fuchs, J.R.; Bieniasz, P.D.; et al. HIV-1 Integrase Binds the Viral RNA Genome and Is Essential during Virion Morphogenesis. Cell 2016, 166, 1257–1268.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessl, J.J.; Jena, N.; Koh, Y.; Taskent-Sezgin, H.; Slaughter, A.; Feng, L.; de Silva, S.; Wu, L.; Le Grice, S.F.J.; Engelman, A.; et al. Multimode, Cooperative Mechanism of Action of Allosteric HIV-1 Integrase Inhibitors. J. Biol. Chem. 2012, 287, 16801–16811. [Google Scholar] [CrossRef] [Green Version]
- Tsiang, M.; Jones, G.S.; Niedziela-Majka, A.; Kan, E.; Lansdon, E.B.; Huang, W.; Hung, M.; Samuel, D.; Novikov, N.; Xu, Y.; et al. New Class of HIV-1 Integrase (IN) Inhibitors with a Dual Mode of Action. J. Biol. Chem. 2012, 287, 21189–21203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenwick, C.; Amad, M.; Bailey, M.D.; Bethell, R.; Bös, M.; Bonneau, P.; Cordingley, M.; Coulombe, R.; Duan, J.; Edwards, P.; et al. Preclinical Profile of BI 224436, a Novel HIV-1 Non-Catalytic-Site Integrase Inhibitor. Antimicrob. Agents Chemother. 2014, 58, 3233–3244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Slaughter, A.; Jena, N.; Feng, L.; Kessl, J.J.; Fadel, H.J.; Malani, N.; Male, F.; Wu, L.; Poeschla, E.; et al. A New Class of Multimerization Selective Inhibitors of HIV-1 Integrase. PLoS Pathog. 2014, 10, e1004171. [Google Scholar] [CrossRef] [Green Version]
- Choi, E.; Mallareddy, J.R.; Lu, D.; Kolluru, S. Recent advances in the discovery of small-molecule inhibitors of HIV-1 integrase. Future Sci. 2018, 4, FSO338. [Google Scholar] [CrossRef] [Green Version]
- Balakrishnan, M.; Yant, S.R.; Tsai, L.; O’Sullivan, C.; Bam, R.A.; Tsai, A.; Niedziela-Majka, A.; Stray, K.M.; Sakowicz, R.; Cihlar, T. Non-Catalytic Site HIV-1 Integrase Inhibitors Disrupt Core Maturation and Induce a Reverse Transcription Block in Target Cells. PLoS ONE 2013, 8, e74163. [Google Scholar] [CrossRef]
- Vranckx, L.; Demeulemeester, J.; Saleh, S.; Boll, A.; Vansant, G.; Schrijvers, R.; Weydert, C.; Battivelli, E.; Verdin, E.; Cereseto, A.; et al. LEDGIN-mediated Inhibition of Integrase–LEDGF/p75 Interaction Reduces Reactivation of Residual Latent HIV. EBioMedicine 2016, 8, 248–264. [Google Scholar] [CrossRef] [Green Version]
- Vansant, G.; Vranckx, L.S.; Zurnic, I.; Van Looveren, D.; Van De Velde, P.; Nobles, C.; Gijsbers, R.; Christ, F.; Debyser, Z. Impact of LEDGIN treatment during virus production on residual HIV-1 transcription. Retrovirology 2019, 16, 8. [Google Scholar] [CrossRef]
- Bruggemans, A.; Vansant, G.; Balakrishnan, M.; Mitchell, M.L.; Cai, R.; Christ, F.; Debyser, Z. GS-9822, a preclinical LEDGIN candidate, displays a block-and-lock phenotype in cell culture. Antimicrob. Agents Chemother. 2021, 65, e02328-20. [Google Scholar] [CrossRef] [PubMed]
- Lingappa, J.R.; Lingappa, V.R.; Reed, J.C. Addressing Antiretroviral Drug Resistance with Host-Targeting Drugs—First Steps towards Developing a Host-Targeting HIV-1 Assembly Inhibitor. Viruses 2021, 13, 451. [Google Scholar] [CrossRef] [PubMed]
- Anisenko, A.N.; Knyazhanskaya, E.S.; Zalevsky, A.O.; Agapkina, J.Y.; Sizov, A.I.; Zatsepin, T.S.; Gottikh, M.B. Characterization of HIV-1 integrase interaction with human Ku70 protein and initial implications for drug targeting. Sci. Rep. 2017, 7, 5649. [Google Scholar] [CrossRef] [PubMed]
- Knyazhanskaya, E.; Anisenko, A.; Shadrina, O.; Kalinina, A.; Zatsepin, T.; Zalevsky, A.; Mazurov, D.; Gottikh, M. NHEJ pathway is involved in post-integrational DNA repair due to Ku70 binding to HIV-1 integrase. Retrovirology 2019, 16, 30. [Google Scholar] [CrossRef]
- Passos, D.O.; Li, M.; Yang, R.; Rebensburg, S.V.; Ghirlando, R.; Jeon, Y.; Shkriabai, N.; Kvaratskhelia, M.; Craigie, R.; Lyumkis, D. Cryo-EM structures and atomic model of the HIV-1 strand transfer complex intasome. Science 2017, 355, 89–92. [Google Scholar] [CrossRef] [Green Version]
- Deprez, E.; Tauc, P.; Leh, H.; Mouscadet, J.F.; Auclair, C.; Brochon, J.C. Oligomeric states of the HIV-1 integrase as measured by time-resolved fluorescence anisotropy. Biochemistry 2000, 39, 9275–9284. [Google Scholar] [CrossRef]
- Galkin, S.; Rozina, A.; Zalevsky, A.; Gottikh, M.; Anisenko, A. A Fluorescent Assay to Search for Inhibitors of HIV-1 Integrase Interactions with Human Ku70 Protein, and Its Application for Characterization of Oligonucleotide Inhibitors. Biomolecules 2020, 10, 1236. [Google Scholar] [CrossRef]
- Chen, J.C.-H.; Krucinski, J.; Miercke, L.J.W.; Finer-Moore, J.S.; Tang, A.H.; Leavitt, A.D.; Stroud, R.M. Crystal structure of the HIV-1 integrase catalytic core and C-terminal domains: A model for viral DNA binding. Proc. Natl. Acad. Sci. USA 2000, 97, 8233–8238. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Chen, X.; Wang, H.; Jurado, K.A.; Engelman, A.N.; Craigie, R. A Peptide Derived from Lens Epithelium-Derived Growth Factor Stimulates HIV-1 DNA Integration and Facilitates Intasome Structural Studies. J. Mol. Biol. 2020, 432, 2055–2066. [Google Scholar] [CrossRef]
- Passos, D.O.; Li, M.; Jóźwik, I.K.; Zhao, X.Z.; Santos-Martins, D.; Yang, R.; Smith, S.J.; Jeon, Y.; Forli, S.; Hughes, S.H.; et al. Structural basis for strand-transfer inhibitor binding to HIV intasomes. Science 2020, 367, 810–814. [Google Scholar] [CrossRef]
- Landschulz, W.H.; Johnson, P.F.; McKnight, S.L. The Leucine Zipper: A Hypothetical Structure Common to a New Class of DNA Binding Proteins. Science 1988, 240, 1759–1764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, J.R.; Corpina, R.A.; Goldberg, J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature 2001, 412, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Nemoz, C.; Ropars, V.; Frit, P.; Gontier, A.; Drevet, P.; Yu, J.; Guerois, R.; Pitois, A.; Comte, A.; Delteil, C.; et al. XLF and APLF bind Ku80 at two remote sites to ensure DNA repair by non-homologous end joining. Nat. Struct. Mol. Biol. 2018, 25, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, M.; Nguyen, B.-Y.; Gotuzzo, E.; Mendo, F.; Ratanasuwan, W.; Kovacs, C.; Prada, G.; Morales-Ramirez, J.O.; Crumpacker, C.S.; Isaacs, R.D.; et al. Rapid and Durable Antiretroviral Effect of the HIV-1 Integrase Inhibitor Raltegravir as Part of Combination Therapy in Treatment-Naive Patients With HIV-1 Infection. JAIDS J. Acquir. Immune Defic. Syndr. 2007, 46, 125–133. [Google Scholar] [CrossRef] [Green Version]
- Anstett, K.; Brenner, B.; Mesplede, T.; Wainberg, M.A. HIV drug resistance against strand transfer integrase inhibitors. Retrovirology 2017, 14, 36. [Google Scholar] [CrossRef]
- Jiang, J.; Xu, X.; Guo, W.; Su, J.; Huang, J.; Liang, B.; Chen, H.; Zang, N.; Liao, Y.; Ye, L.; et al. Dolutegravir(DTG, S/GSK1349572) combined with other ARTs is superior to RAL- or EFV-based regimens for treatment of HIV-1 infection: A meta-analysis of randomized controlled trials. AIDS Res. Ther. 2016, 13, 30. [Google Scholar] [CrossRef] [Green Version]
- Swindells, S.; Andrade-Villanueva, J.-F.; Richmond, G.J.; Rizzardini, G.; Baumgarten, A.; Masiá, M.; Latiff, G.; Pokrovsky, V.; Bredeek, F.; Smith, G.; et al. Long-Acting Cabotegravir and Rilpivirine for Maintenance of HIV-1 Suppression. N. Engl. J. Med. 2020, 382, 1112–1123. [Google Scholar] [CrossRef]
- Oliveira, M.; Ibanescu, R.-I.; Anstett, K.; Mésplède, T.; Routy, J.-P.; Robbins, M.A.; Brenner, B.G. Selective resistance profiles emerging in patient-derived clinical isolates with cabotegravir, bictegravir, dolutegravir, and elvitegravir. Retrovirology 2018, 15, 56. [Google Scholar] [CrossRef] [Green Version]
- Ji, X.; Li, Z. Medicinal chemistry strategies toward host targeting antiviral agents. Med. Res. Rev. 2020, 40, 1519–1557. [Google Scholar] [CrossRef]
- Engelman, A. The Roles of Cellular Factors in Retroviral Integration. In Cellular Factors Involved in Early Steps of Retroviral Replication; Young, J.A.T., Ed.; Springer: Berlin/Heidelberg, Germany, 2003; pp. 209–238. [Google Scholar]
- Debyser, Z.; Christ, F.; De Rijck, J.; Gijsbers, R. Host factors for retroviral integration site selection. Trends Biochem. Sci. 2015, 40, 108–116. [Google Scholar] [CrossRef]
- Park, J.; Fu, Z.; Frangaj, A.; Liu, J.; Mosyak, L.; Shen, T.; Slavkovich, V.N.; Ray, K.M.; Taura, J.; Cao, B.; et al. Structure of human GABAB receptor in an inactive state. Nature 2020, 584, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Moll, J.R. Designed heterodimerizing leucine zippers with a ranger of pIs and stabilities up to 10−15 M. Protein Sci. 2001, 10, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Knyazhanskaya, E.S.; Smolov, M.A.; Kondrashina, O.V.; Gottikh, M.B. Relative Comparison of Catalytic Characteristics of human Foamy Virus and hIV-1 Integrases. Acta Nat. 2009, 1, 78–80. [Google Scholar] [CrossRef] [Green Version]
- Anisenko, A.N.; Knyazhanskaya, E.S.; Zatsepin, T.S.; Gottikh, M.B. Human Ku70 protein binds hairpin RNA and double stranded DNA through two different sites. Biochimie 2017, 132, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Leshchinskaya, I.B.; Shakirov, E.V.; Itskovitch, E.L.; Balaban, N.P.; Mardanova, A.M.; Sharipova, M.R.; Viryasov, M.B.; Rudenskaya, G.N.; Stepanov, V.M. Glutamyl endopeptidase of Bacillus intermedius, strain 3-19. FEBS Lett. 1997, 404, 241–244. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ1 and χ2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Santos-Martins, D.; Solis-Vasquez, L.; Tillack, A.F.; Sanner, M.F.; Koch, A.; Forli, S. Accelerating AutoDock 4 with GPUs and Gradient-Based Local Search. J. Chem. Theory Comput. 2021, 17, 1060–1073. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ropp, P.J.; Spiegel, J.O.; Walker, J.L.; Green, H.; Morales, G.A.; Milliken, K.A.; Ringe, J.J.; Durrant, J.D. Gypsum-DL: An open-source program for preparing small-molecule libraries for structure-based virtual screening. J. Cheminform. 2019, 11, 34. [Google Scholar] [CrossRef] [PubMed]
- Mitternacht, S. FreeSASA: An open source C library for solvent accessible surface area calculations. F1000Research 2016, 5, 189. [Google Scholar] [CrossRef]
- Saubern, S.; Guha, R.; Baell, J.B. KNIME Workflow to Assess PAINS Filters in SMARTS Format. Comparison of RDKit and Indigo Cheminformatics Libraries. Mol. Inform. 2011, 30, 847–850. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ilgova, E.; Galkin, S.; Khrenova, M.; Serebryakova, M.; Gottikh, M.; Anisenko, A. Complex of HIV-1 Integrase with Cellular Ku Protein: Interaction Interface and Search for Inhibitors. Int. J. Mol. Sci. 2022, 23, 2908. https://doi.org/10.3390/ijms23062908
Ilgova E, Galkin S, Khrenova M, Serebryakova M, Gottikh M, Anisenko A. Complex of HIV-1 Integrase with Cellular Ku Protein: Interaction Interface and Search for Inhibitors. International Journal of Molecular Sciences. 2022; 23(6):2908. https://doi.org/10.3390/ijms23062908
Chicago/Turabian StyleIlgova, Ekaterina, Simon Galkin, Maria Khrenova, Marina Serebryakova, Marina Gottikh, and Andrey Anisenko. 2022. "Complex of HIV-1 Integrase with Cellular Ku Protein: Interaction Interface and Search for Inhibitors" International Journal of Molecular Sciences 23, no. 6: 2908. https://doi.org/10.3390/ijms23062908
APA StyleIlgova, E., Galkin, S., Khrenova, M., Serebryakova, M., Gottikh, M., & Anisenko, A. (2022). Complex of HIV-1 Integrase with Cellular Ku Protein: Interaction Interface and Search for Inhibitors. International Journal of Molecular Sciences, 23(6), 2908. https://doi.org/10.3390/ijms23062908