1. Introduction
Curcumin, a major active component of Turmeric, has long been used as a spice, and it possess a wide range of biological activities, including antibacterial and antifungal [
1], antioxidant, anticancer [
2,
3], antimicrobial [
4], and inflammatory properties [
5]. Curcumin has piqued the interest of many academics since then, and numerous papers have been published on the subject [
6,
7,
8]. It was first discovered by Vogel and Pelletier as a powder “yellow coloring matter” from rhizomes of
C. longa (Zingiberaceae family) [
9], and characterized and first synthesized by Milobedeska, Lampe et al. [
10,
11].
The chemical structure was investigated by Heger et al. [
12] as [(1
E, 6
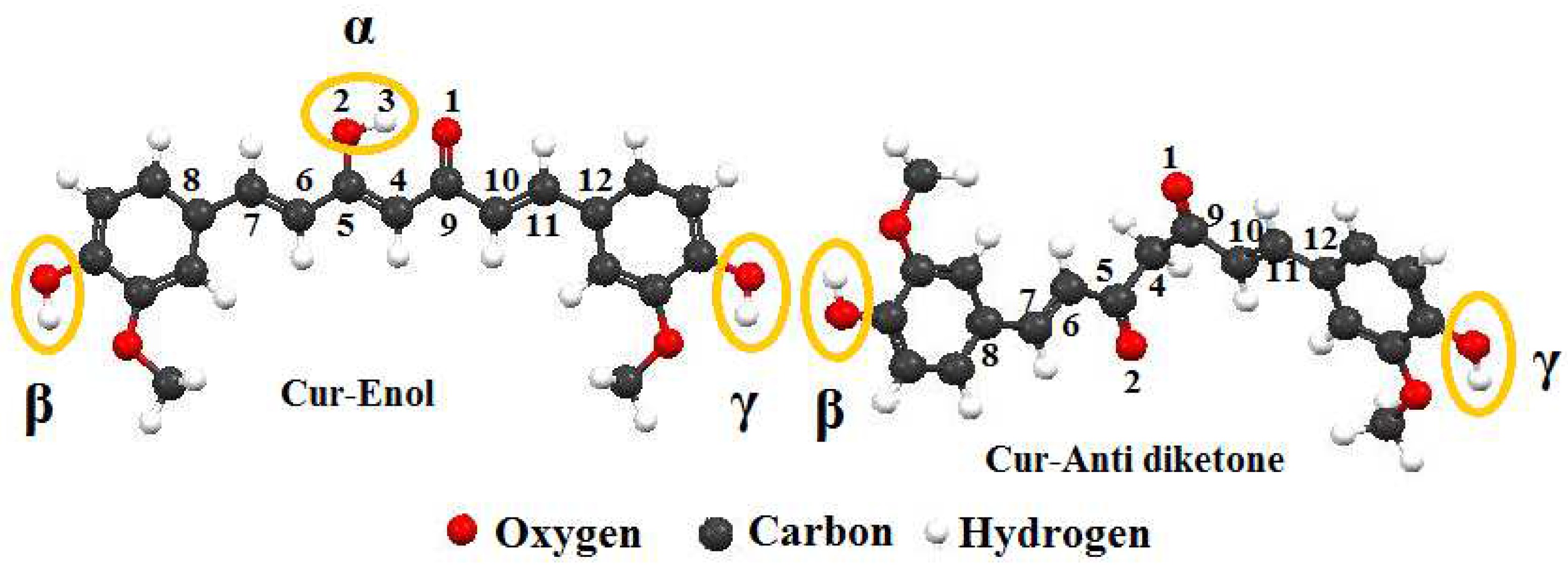
E)-1,7-bis(4-hydroxy-3-methoxy-phenyl)-1,6-heptadiene-3,5-dione]. Curcumin has a melting point of 183 °C, molecular formula of C
21H
20O
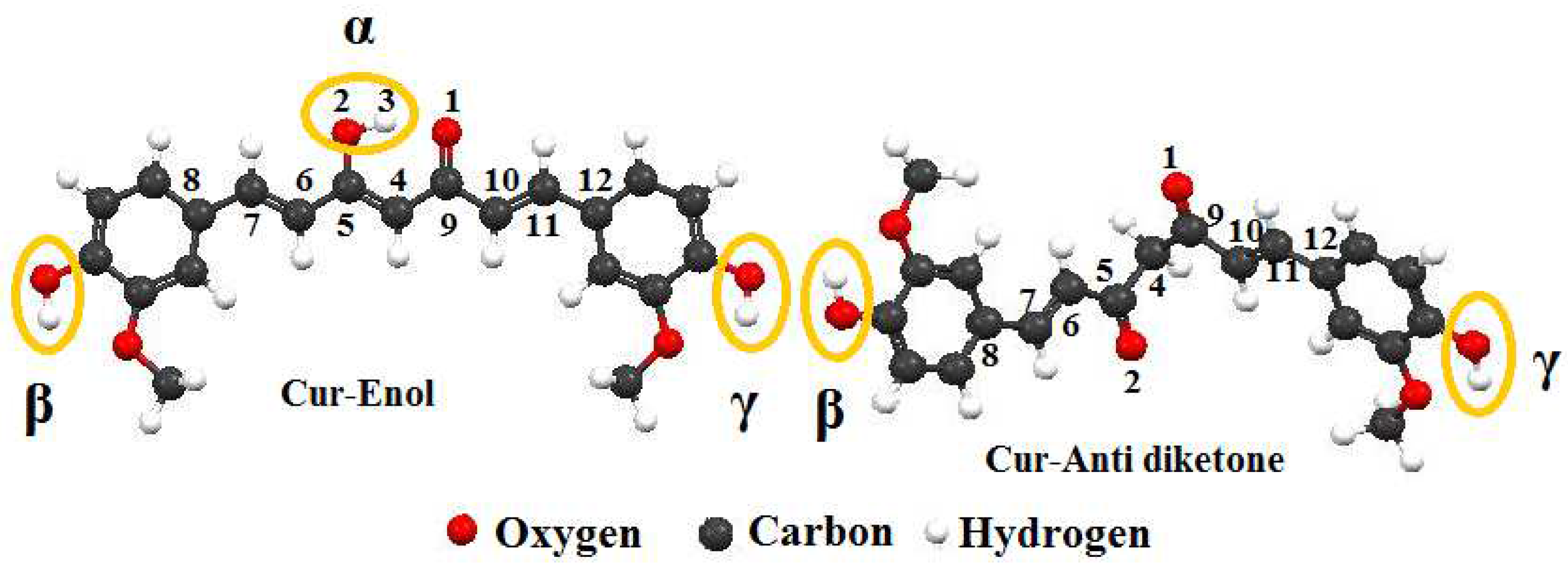
6, and molecular weight of 368.37 g/mol. Ketone and enol are the two tautomeric forms of curcumin that impact its complexation process, physicochemical, and biological characteristics [
13].
Polyphenols have gained attention essentially due to their antioxidant capabilities. They can trap free radicals and prevent lipid peroxidation by scavenging free radicals. They can also capture metal ions due to their chelating capabilities. Nevertheless, curcumin complexes remain little explored despite their promising properties particularly for Alzheimer’s disease [
14]. In general, the experimental identification of therapeutic characteristics of specific natural compounds requires a prior deep understanding, specifically for computational investigations. Examples include the solubility in water and other liquids of interest, especially when dealing with chemicals intended for human consumption. The insoluble nature of curcumin, for example, makes it challenging to utilize in medicinal applications.
The complexation of a chemical with other metals can drastically alter certain molecular features, resulting in new compounds with distinct properties that are potentially suitable for desired purposes. This is the case of curcumin and its complexes. Hence, the main purpose of this investigation is the study of the complexation behavior and the antioxidant properties of curcumin–metal complexes based on Nickel, Copper, and Magnesium transition metals by density functional theory (DFT) techniques, i.e., time-dependent density functional theory (TDDFT) to simulate the absorption and emission spectra; quantum theory of atoms in molecules (QTAIM) to simultaneously explore the electron density and hydrogen bond interactions; extended transition state natural orbitals for chemical valence (ETS-NOCV) to evaluate the antioxidant activity; and COSMO-RS to calculate and analyze the solubility of curcumin and molecular interactions in various solvents. The results will be helpful to the experimentalists to synthesize curcumin complexes with more reliable properties.
2. Computational Methods
As part of the DFT (density functional theory) level computations, the geometric optimization calculations were conducted using Gaussian09 [
15] and the functional B3LYP (Becke three-parameter Lee–Yong–Parr) [
16]. All the atoms were based on the 6-311G (2d, 2p) basis. The polarizable continuum model (PCM) introduced the DMSO (dimethyl sulfoxide) solvent effect. Casida’s method obtained characteristics and absorption spectrum of the excited states [
17].
The theoretical study of curcumin-based transition metal compounds performed by Density Functional Theory (DFT) [
18,
19] using ADF software (Amsterdam Density Functional) [
20] was only used for the single-point calculations, the functional GGA: PW91 (Generalized Gradient Approximation) [
21], the Slater TZP (Triple zeta polarized) set of atomic bases [
22], valence orbitals of all atoms (4s and 3d for Ni, Cu, and Mg; 2s and 2p for O and C; and 1s for H. Relativistic effects were taken into account at the scalar level using the regular approximation of order zero (ZORA) [
23].
The integration parameter and the energy convergence criterion are: 6 and 10-6 a.u., respectively. Several DFT approaches can be used to describe the properties of chemical bonds; the quantum theory of atoms in molecules (QTAIM) is one of them. Hydrogen bonds were characterized using QTAIM [
24], which was connected with investigating electron density’s topology [
24,
25,
26]. Using the NOCV technique [
27], the dissociation energy of the phenolic hydrogen atoms could be calculated, allowing the antioxidant activity of the molecule and its complexes to be measured.
According to the relevant research [
28,
29], the ETS technique was included in the NOCV method [
30,
31]. Covalent bonds [
32], intramolecular agnostic interactions [
29,
31], and intermolecular hydrogen bonds [
33] may all be determined using the combined ETS-NOCV technique. The binding interactions were investigated using a Morokuma-type energy decomposition described by Ziegler and Rauk [
34]. According to the stated approach, the total binding energy (Δ
E) is defined as:
ΔEpauli indicates the repulsive four-electron interactions between the occupied orbitals, and ΔEels represents the classical electrostatic interaction between the molecule fragments in the complex in their final locations. Finally, the term ΔEorb stabilizes interactions between occupied and empty molecular orbitals of the two fragments. It should be emphasized that the last term ΔEorb contains the combination of occupied and vacant orbitals in the same fragment (polarization of internal fragments).
The strain density can be expressed as a sum of eigenvectors,
corresponding to the eigenvalues
and
with the same absolute value:
where
M denotes the total number of molecular orbitals on the fragments.
Specifically, all occupied and empty molecular orbitals of the two fragments are included in the summation. Its eigenvalue
indicates the number of electrons that are moved from the anti-bonding orbitals,
. As a function of the eigenvalues of NOCV (
), the orbital interaction term (Δ
Eorb) is represented in the combined ETS-NOCV scheme as follows:
The diagonal elements of the Kohn–Sham matrix on NOCVs in the transition state (TS) are and .
Equations (2) and (3) show that
can be visualized, and its energetic contributions to the overall bond energy are provided [
26]. COSMO-RS is a completely predictive model for thermodynamic characteristics of fluids and solutions using statistical thermodynamics and quantum chemistry [
35]. Polarization occurs when the charge density is high due to the solvent at the cavity surface. The total energy may be calculated by calculating the forces that result.
COSMO-RS combines statistical thermodynamics with quantum chemistry to provide an utterly predictive model for the thermodynamic characteristics of fluids and solutions. The charge density of the solvent at the cavity’s surface expresses its polarization effects. The resulting force was quantified to calculate the total energy. The COSMO-RS model is one of the most outstanding forecasters of solubility, particularly for tiny compounds, despite its lack of popularity [
36].
4. Conclusions
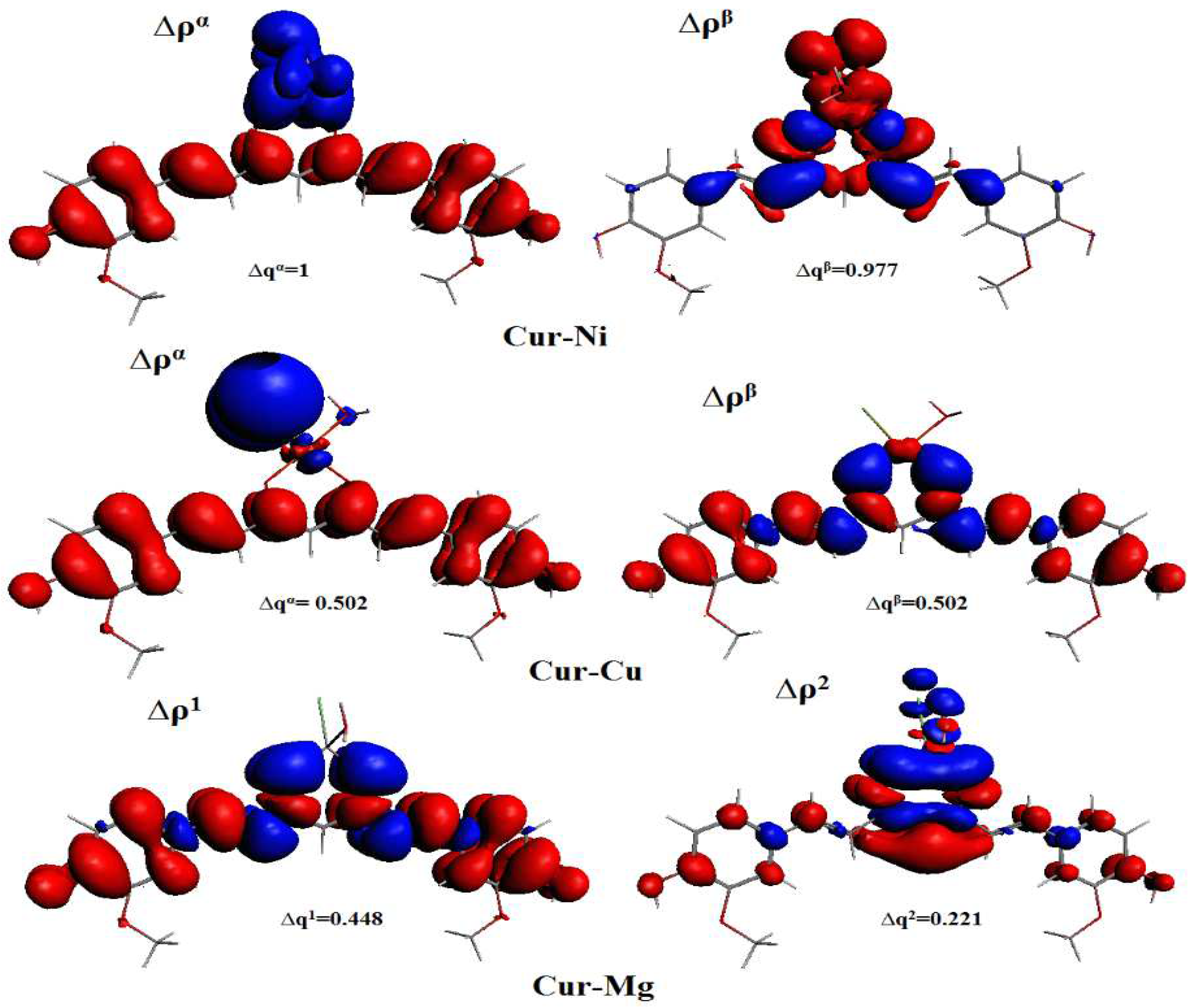
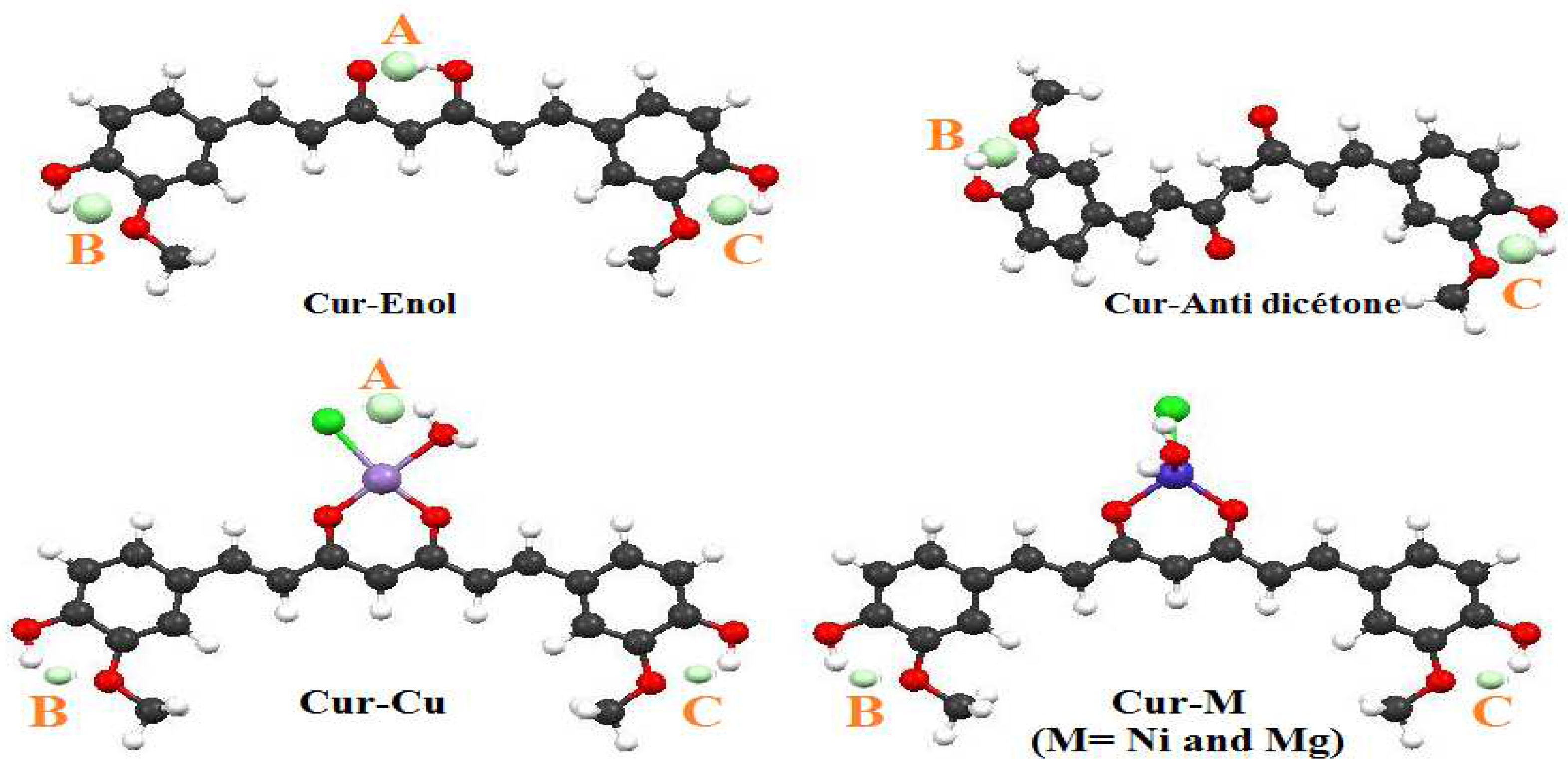
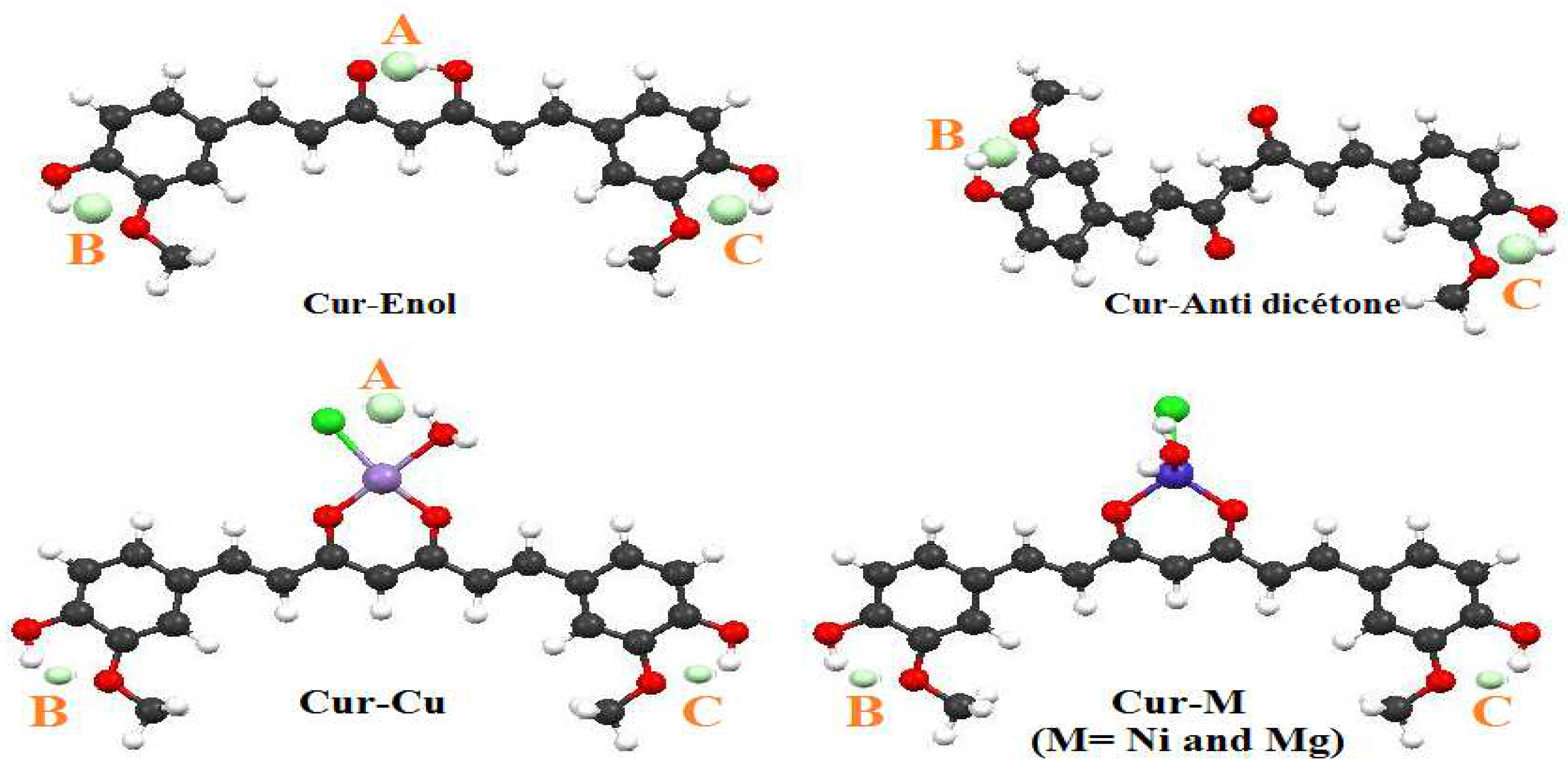
DFT quantum calculations highlighted interesting properties of curcumin and its complexes, i.e., Cur-M (M = Ni, Cu, and Mg). The enolic form of curcumin (Cur-Enol) was found to be energetically more stable and biologically more active compared with its Cur-Anti-diketone tautomer. Furthermore, the antioxidant properties of Cur-Enol calculated by both ETS-NOCV and BDE methods were found to be better compared with Cur-Anti-diketone and its complexes. A combined ETS with the NOCV approach was also conducted to study the nature of curcumin-metal (Cur-M) bonding, and the results showed that all Cur-M bonding had a primarily ionic character (Cur-Ni: 71%, Cur-Cu: 57%, and Cur-Mg: 91%). QTAIM results showed the existence of two hydrogen bonds for Cur-Enol and Cur-Cu, which explained their stability compared to Cur-Mg and Cur-Ni.
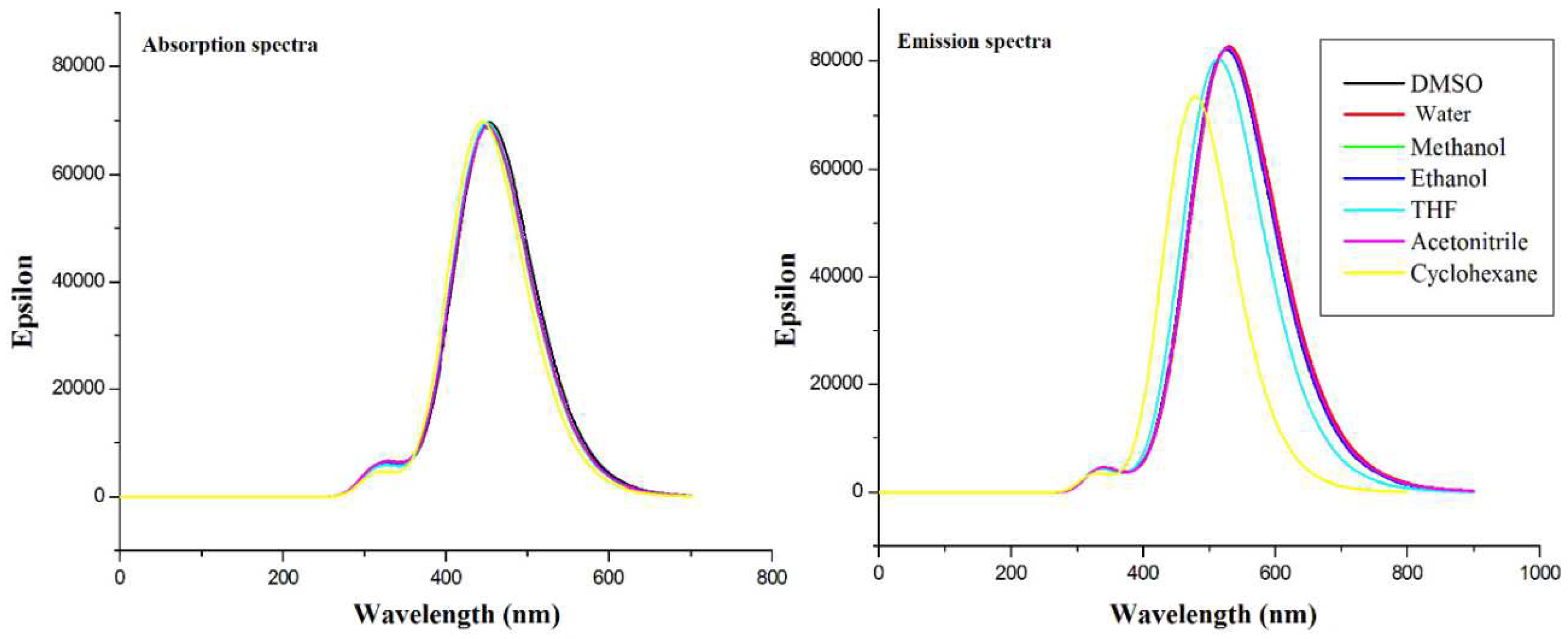
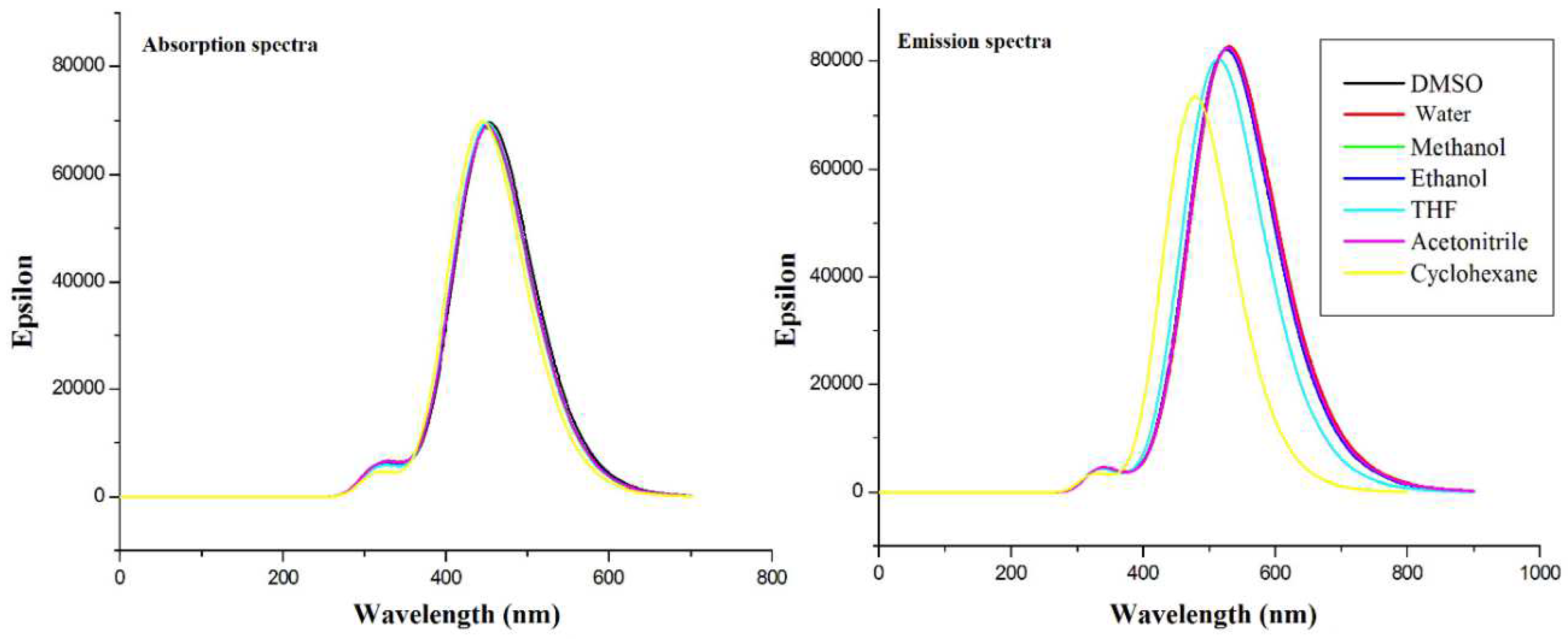
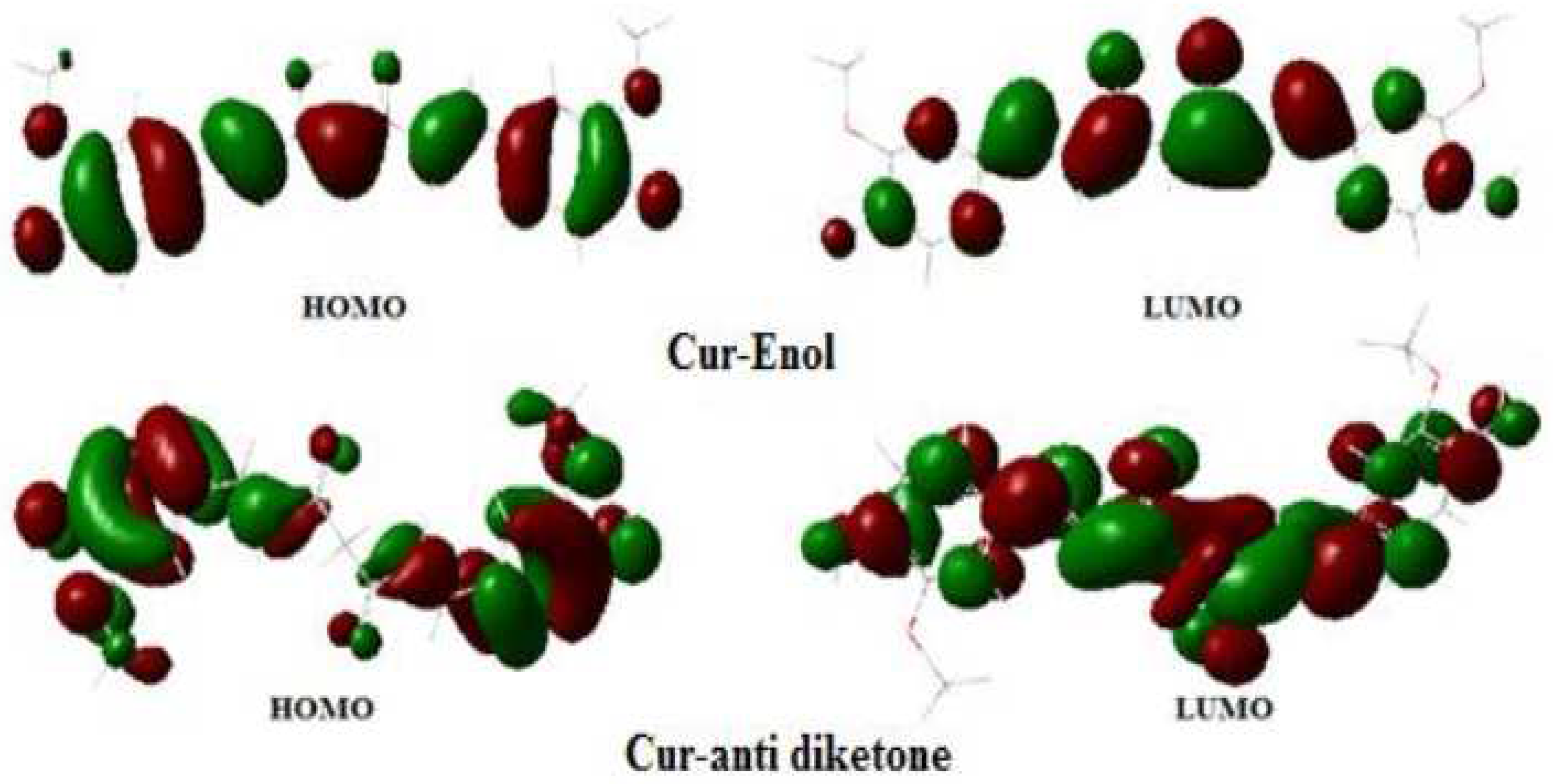
The TDDFT calculated absorption and emission transitions of curcumin gave peaks that were similar to the published experimental findings with some shifts. The first excitation had the highest probability and was of the HOMO-LUMO type ( electronic transition). Solvatochromism existed in both absorption and emission and was found to be weak. We concluded that the HOMO–LUMO transition in Cur-Enol in all solvents was the most present in the UV-vis region in absorption and emission.
The theoretical descriptors BDE and PI, for which the antioxidant activity showed that the compound Cur-Mg was the best antioxidant, presented the values of BDE and PI as the lowest on two sites with (143.36–143.35) and 115.45 kcal/mol, respectively. This result was confirmed by the highest value of the HOMO structure.
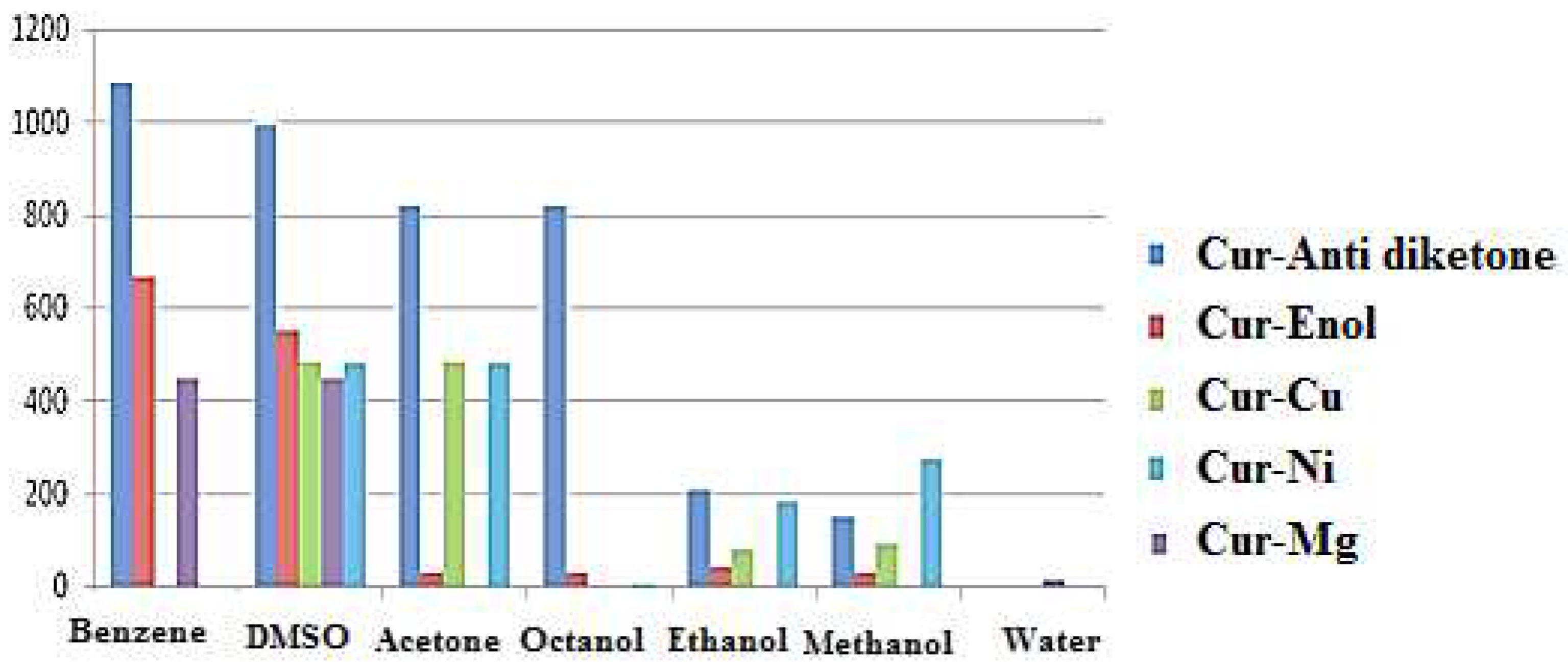
Solubility calculations demonstrated that both Cur-Enol and Cur-Anti-diketone tautomers were soluble in DMSO but insoluble in water. Only Cur-Mg had a low water solubility (14 g/L), indicating that complexation promoted curcumin water solubilization and improved the therapeutic effects.


 ,
, 



 : critical point).
: critical point).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}