
Distinct Functional Alterations and Therapeutic Options of Two Pathological De Novo Variants of the T292 Residue of GABRA1 Identified in Children with Epileptic Encephalopathy and Neurodevelopmental Disorders

 ,
, 
 and
and
Abstract
:1. Introduction
2. Results
2.1. Phenotypic Comparisons of the Two Patients Carrying the GABRA1 Mutants
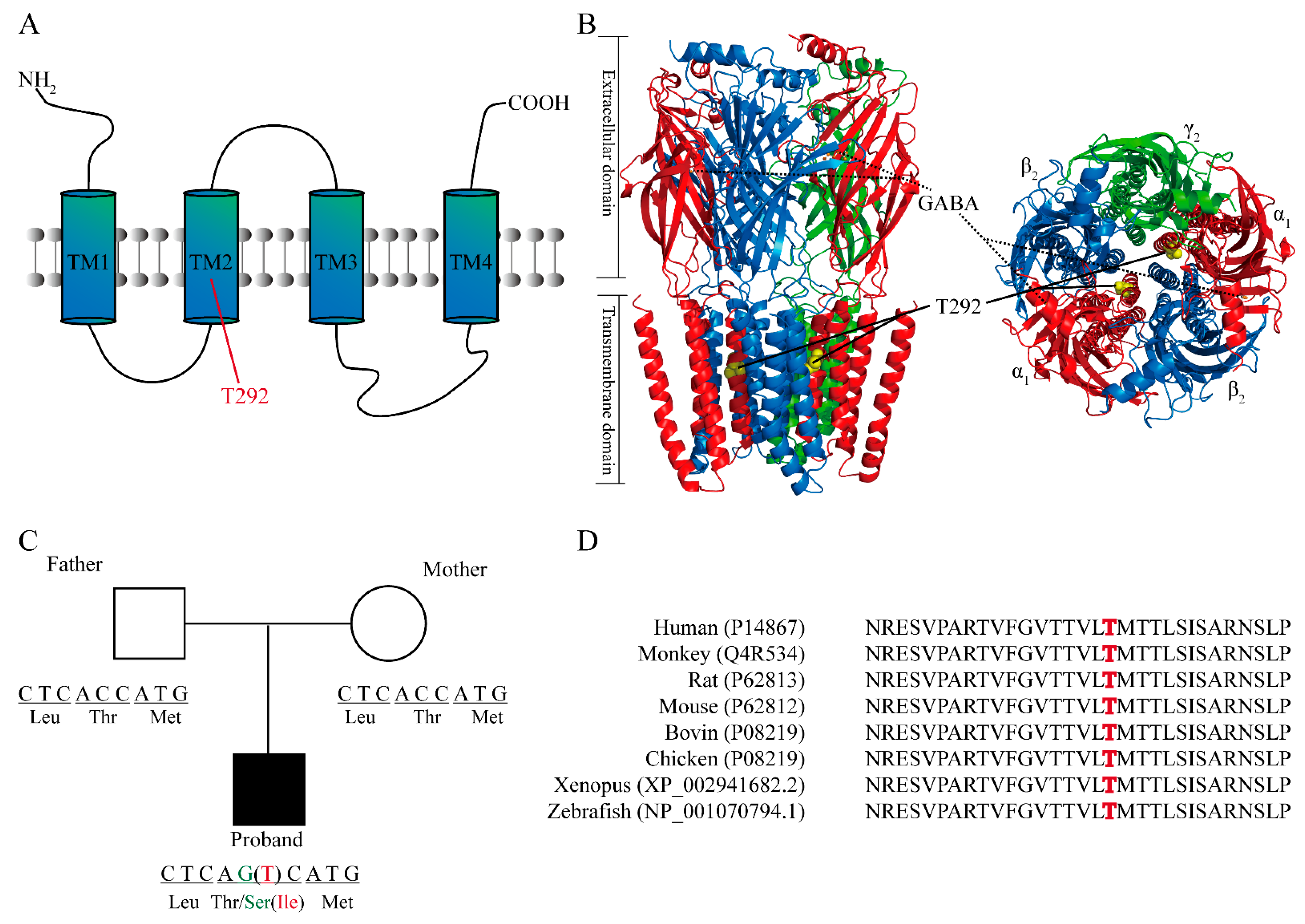
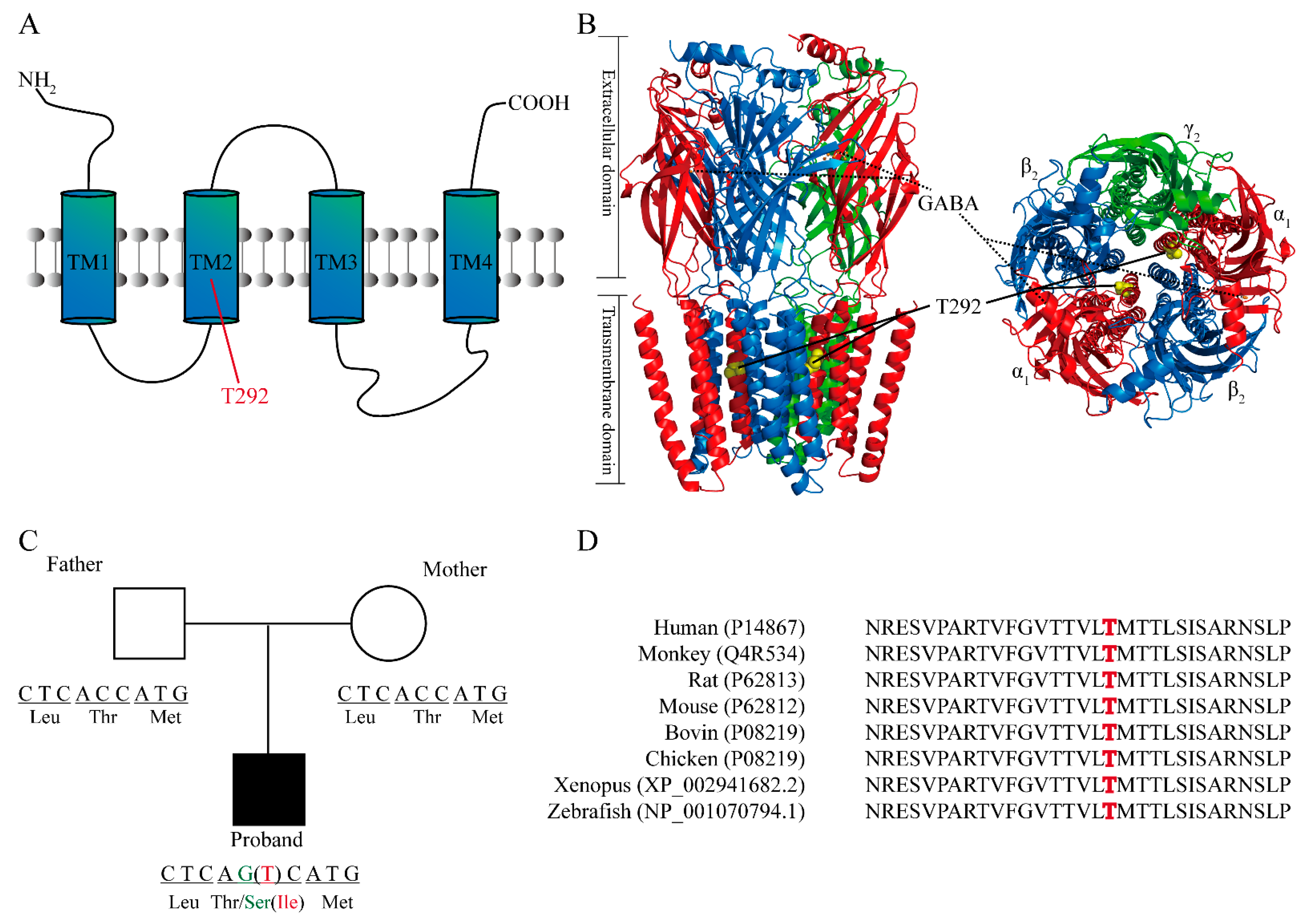
2.2. Structural Analysis and the Location of T292 Residue in GABRA1
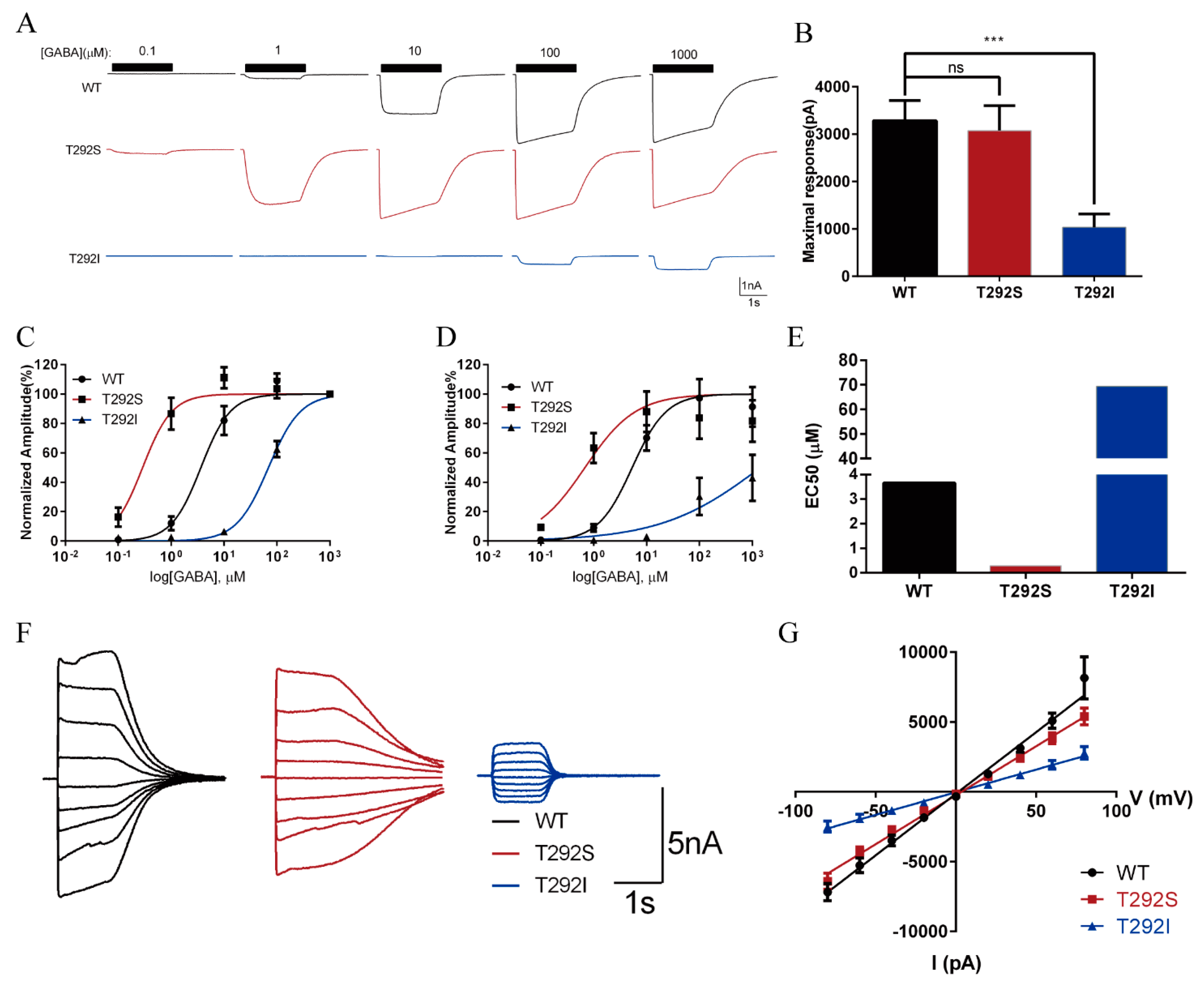
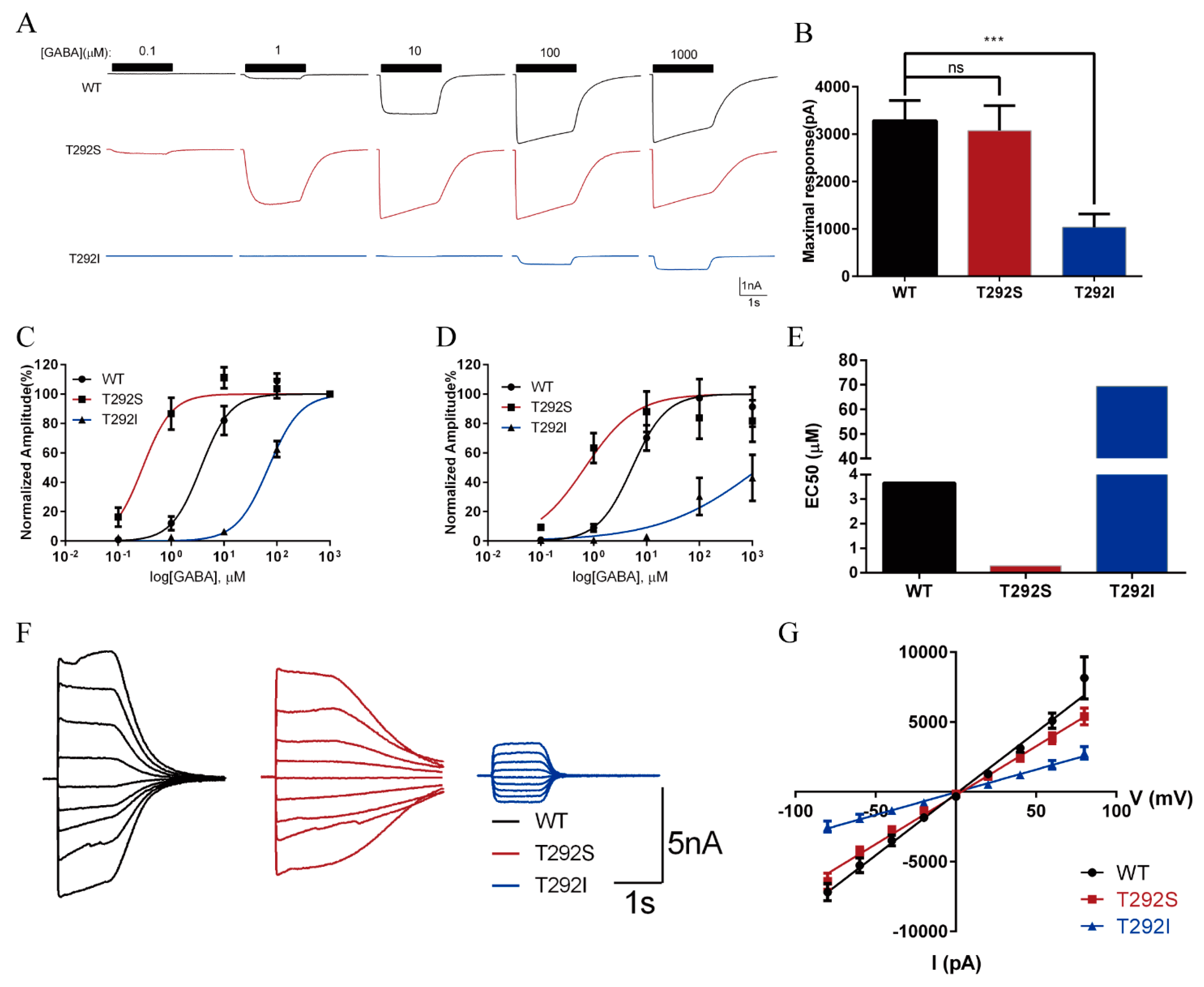
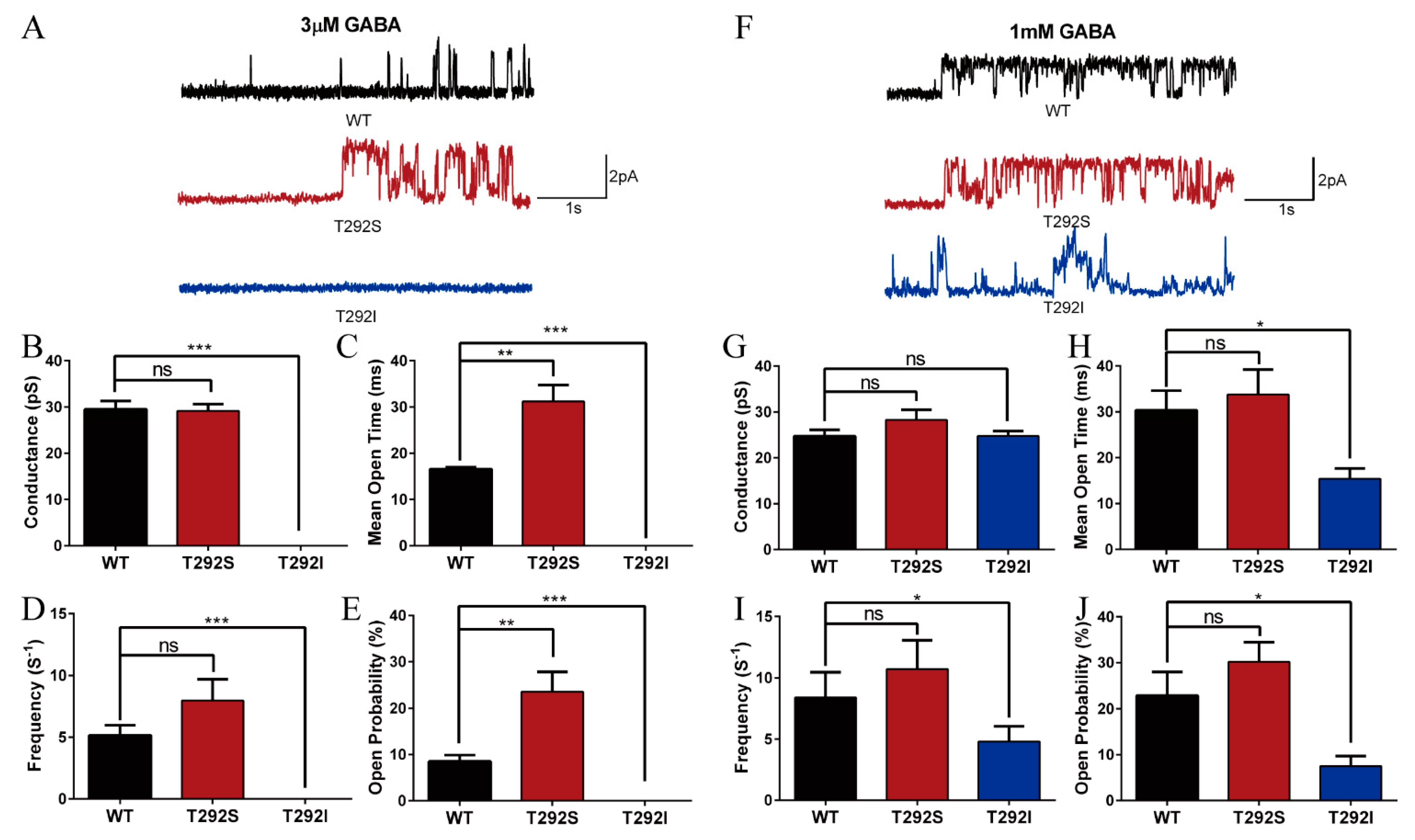
2.3. The T292S and T292I Variants Bidirectionally Affect GABA-Evoked Responses
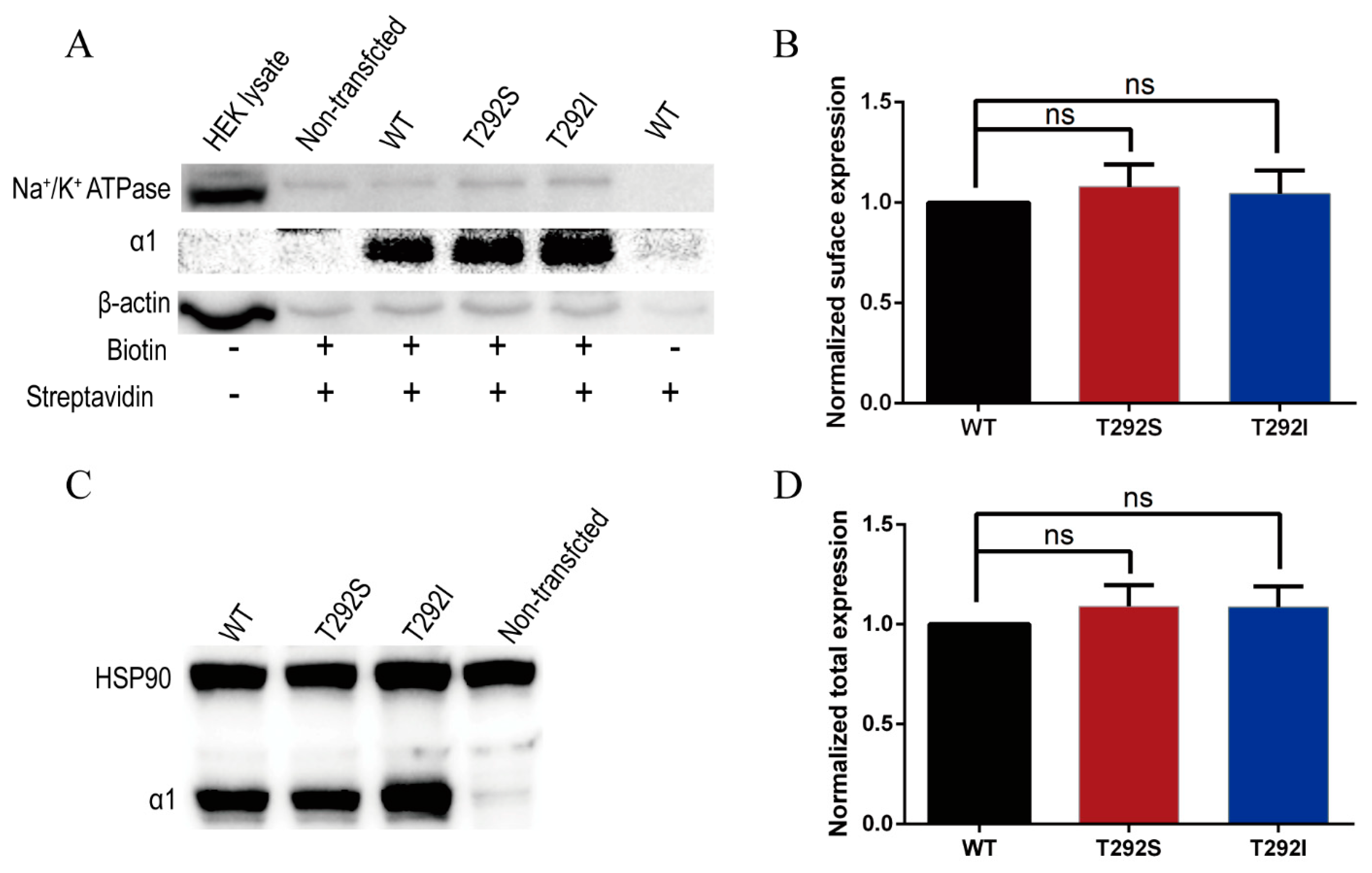
2.4. Neither T292S nor T292I Variants Affected GABAAR Total/Surface Expressions
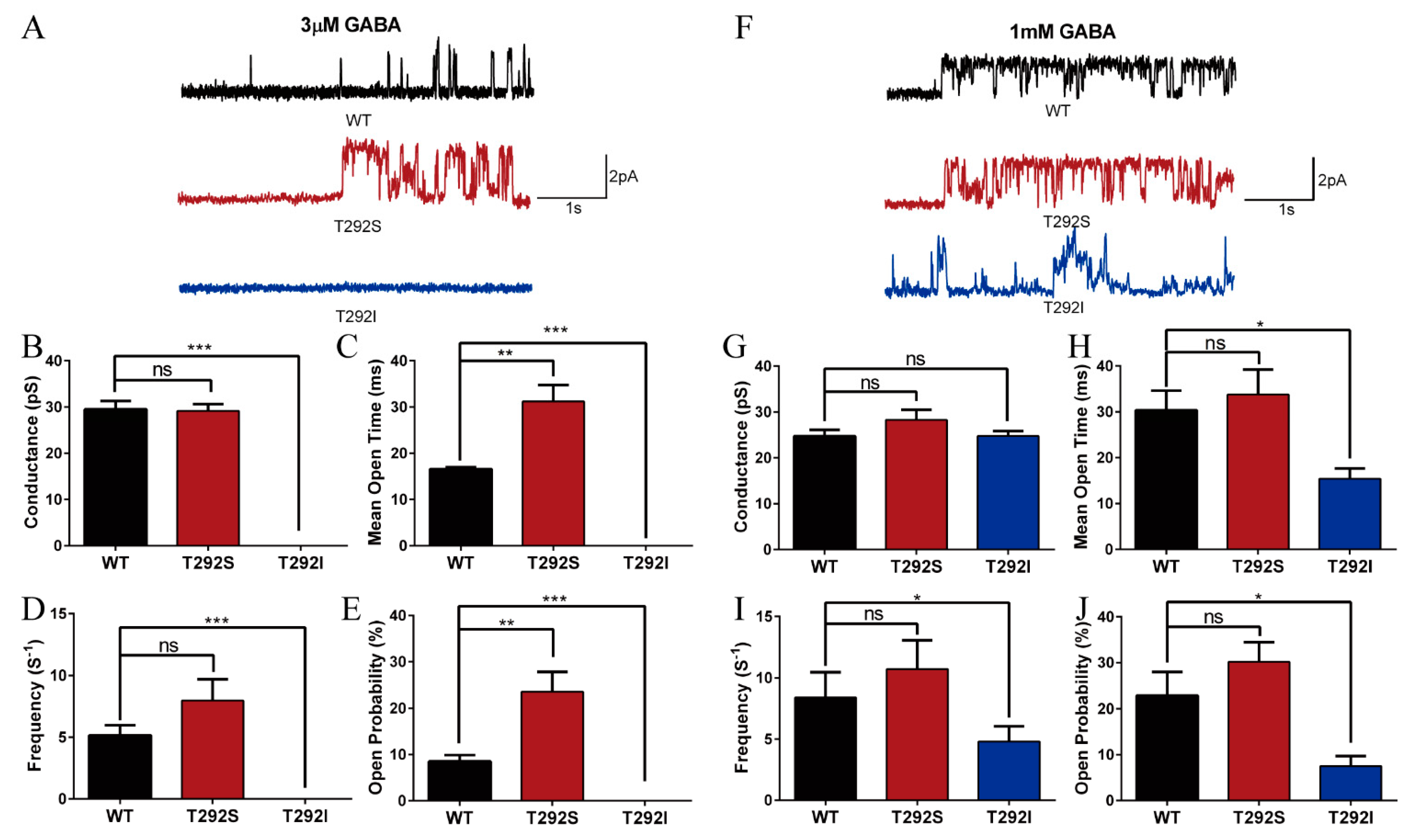
2.5. T292S and T292I Variants Bidirectionally Altered GABAAR Single Channel Properties
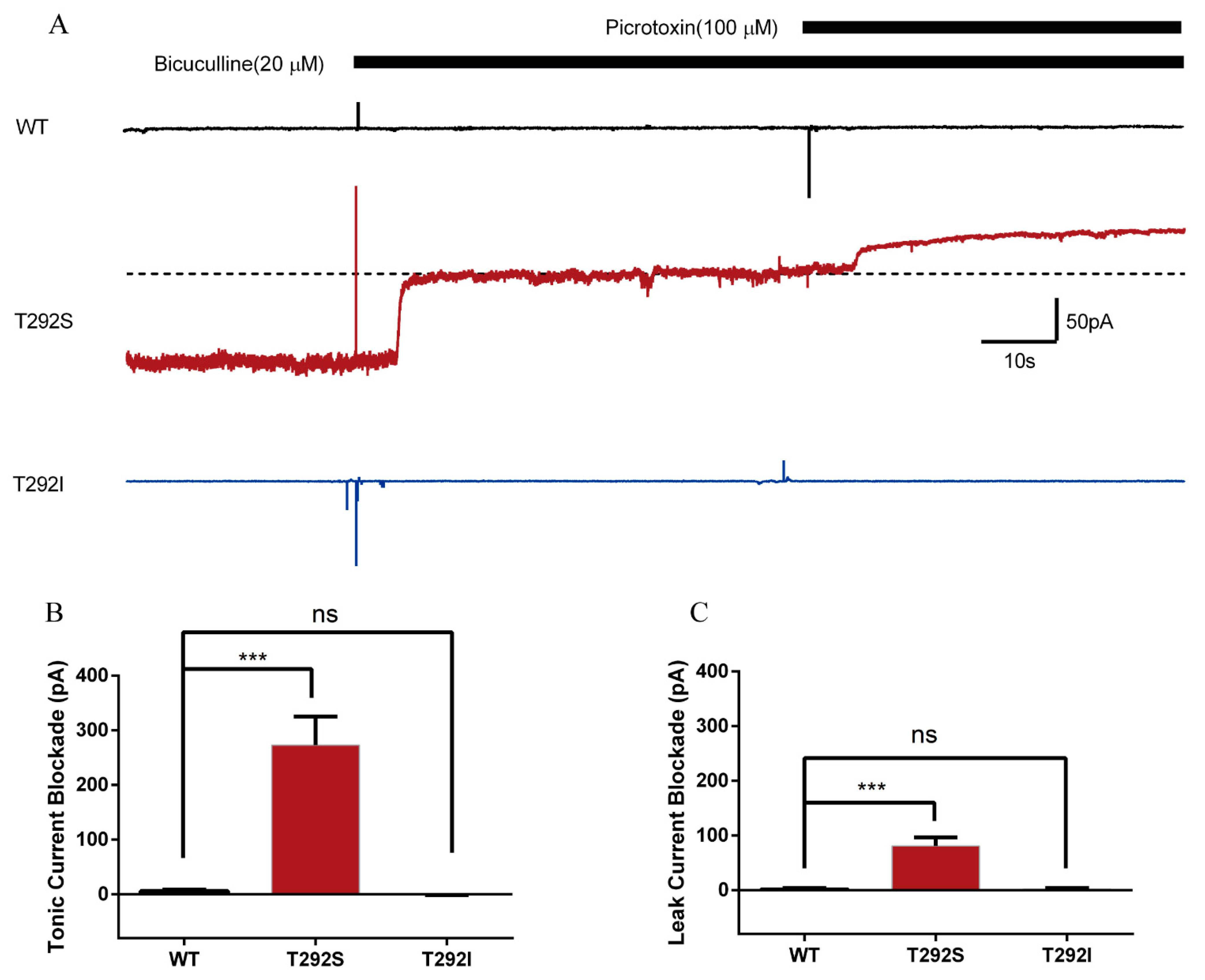
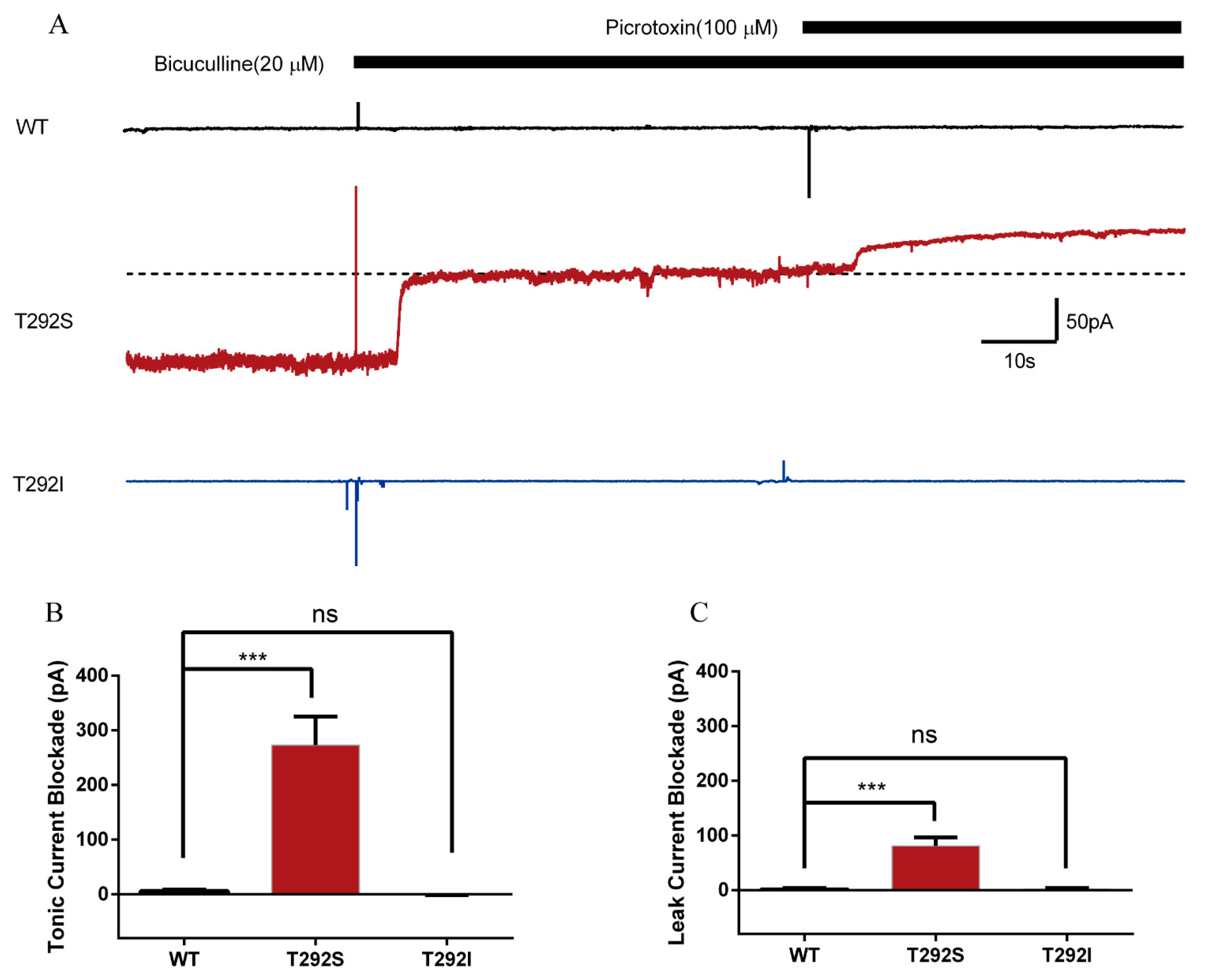
2.6. T292S and T292I Variants Resulted in Different Changes of GABAAR-Gated Tonic and Leak Currents
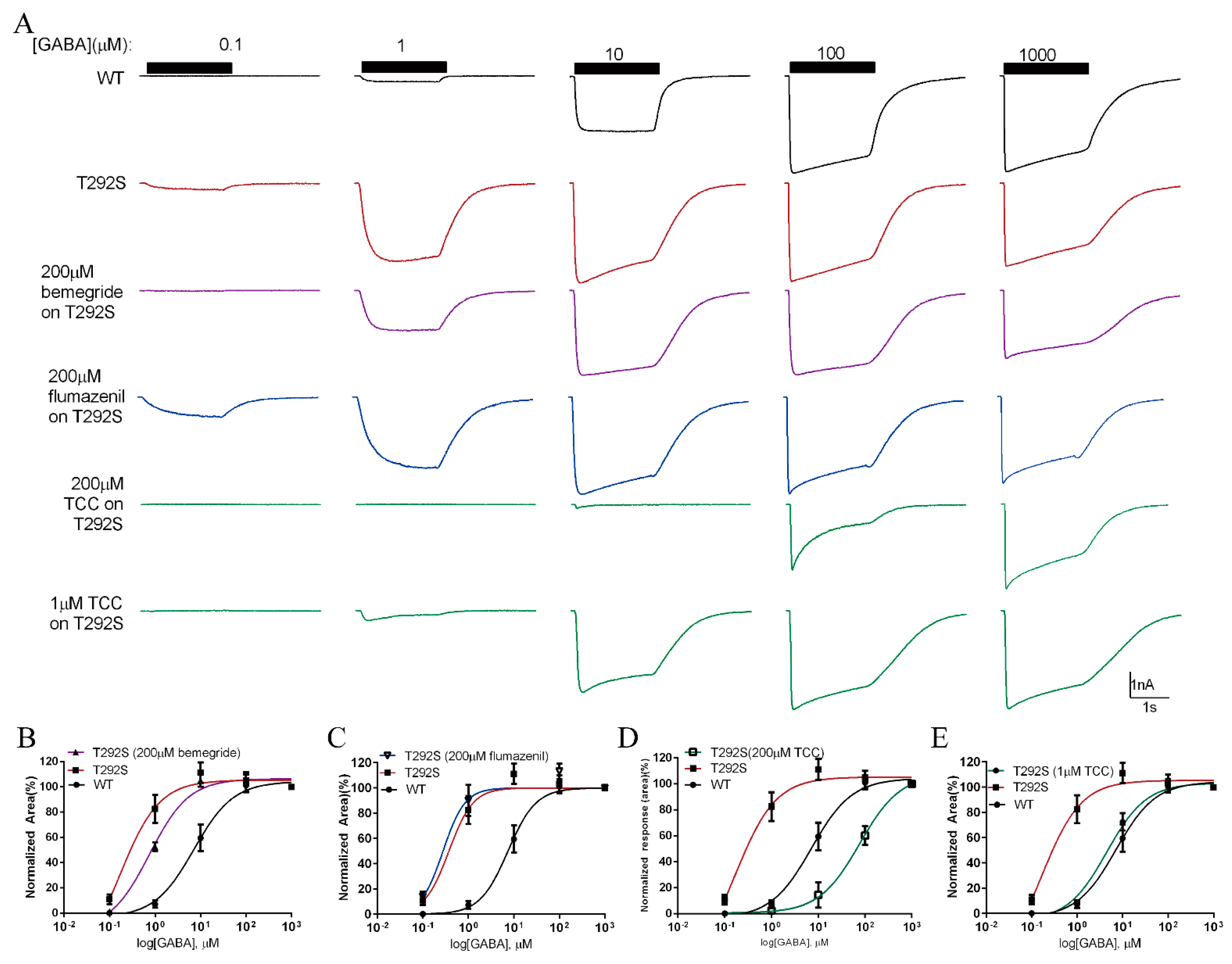
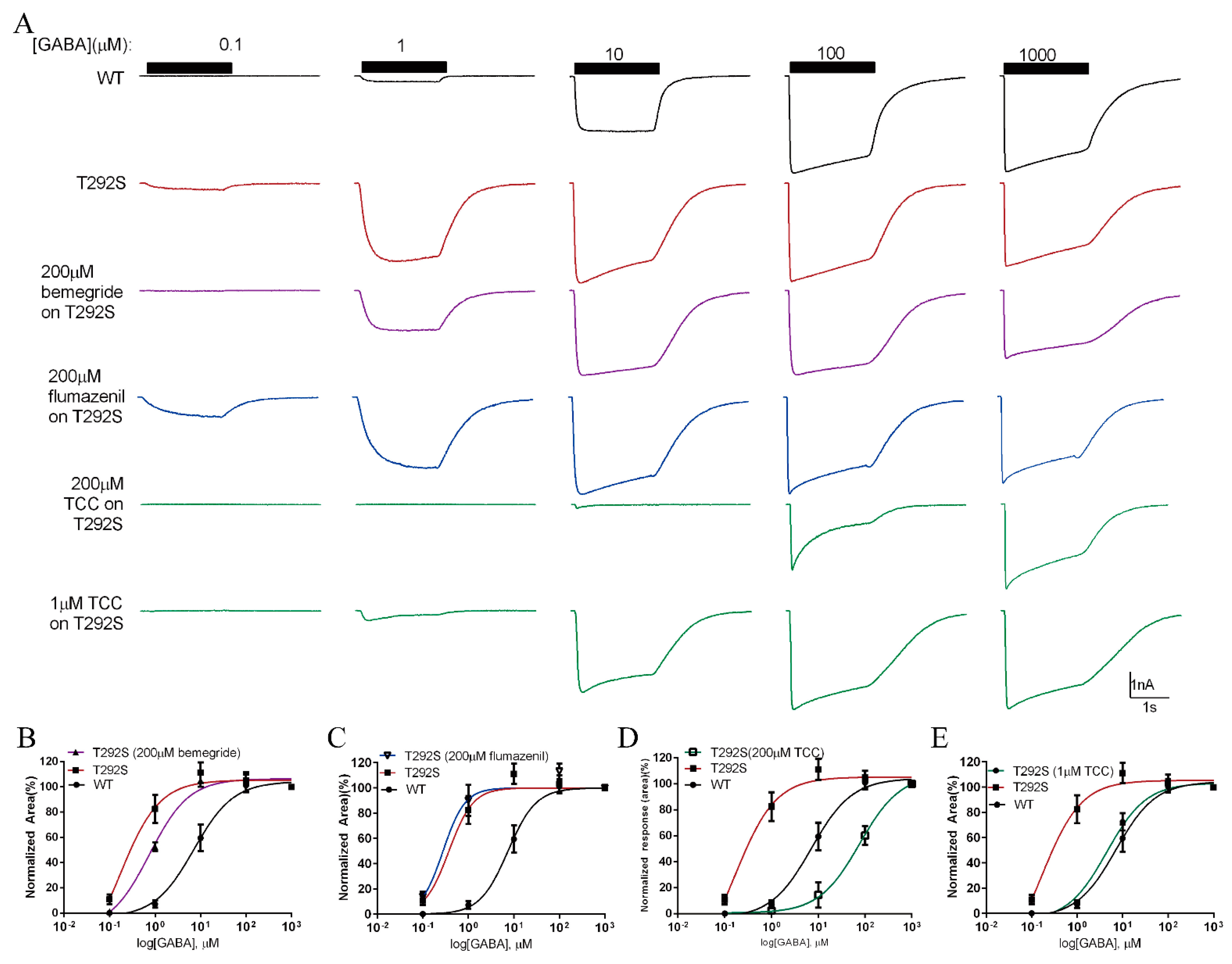
2.7. Thiocolchicoside Restores the Function of T292S GABAAR
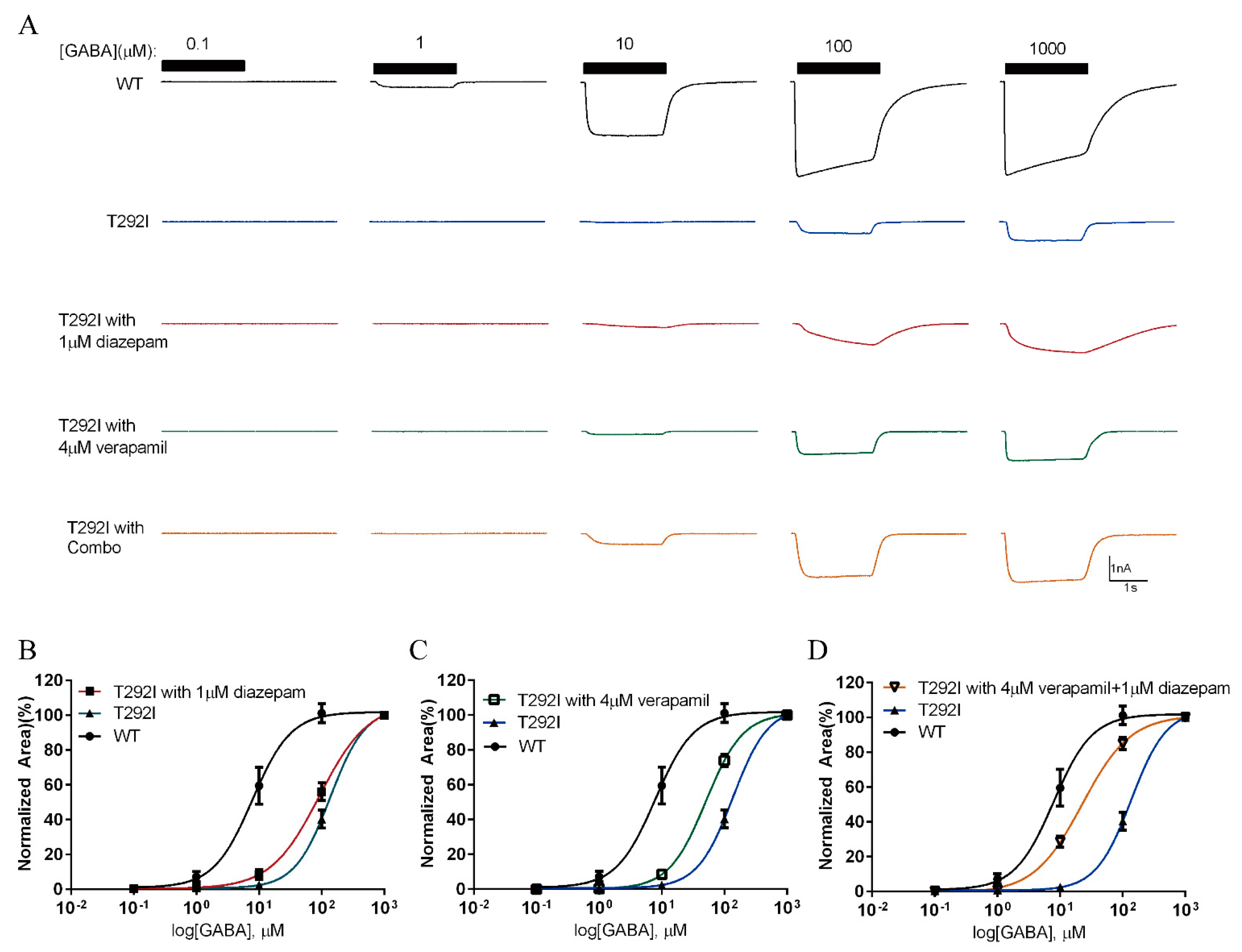
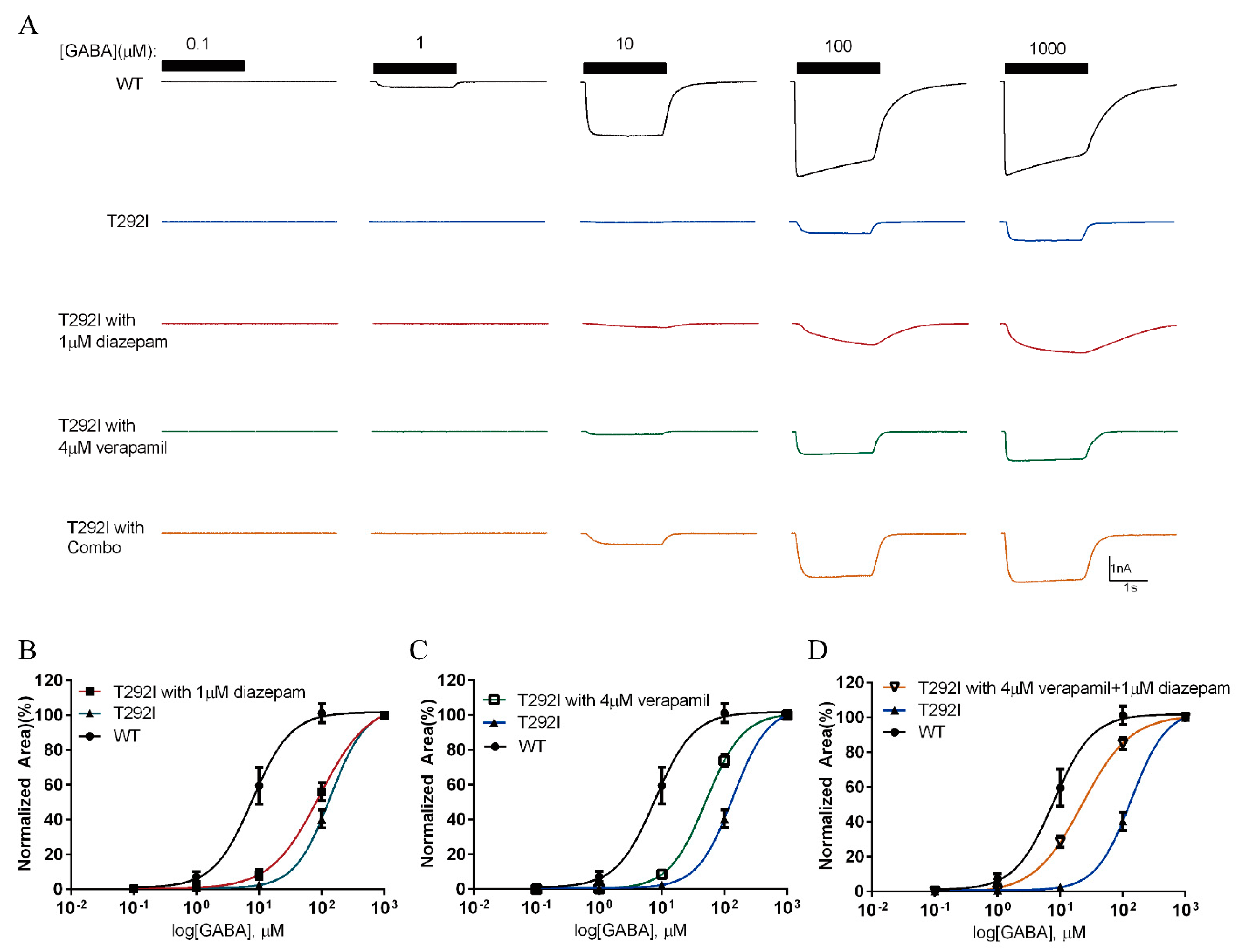
2.8. Combination of Verapamil and Diazepam Partially Rescues the Function of T292I GABAAR
3. Discussion
4. Materials and Methods
4.1. Structural Modeling of GABAA Receptor
4.2. Plasmid Construction
4.3. Cell Culture and Transfection
4.4. Western Blot and Surface Biotinylation
4.5. Electrophysiology
4.6. Chemicals
4.7. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thijs, R.D.; Surges, R.; O’Brien, T.J.; Sander, J.W. Epilepsy in adults. Lancet 2019, 393, 689–701. [Google Scholar] [CrossRef]
- Arnold, S.T.; Dodson, W.E. Epilepsy in children. Bailliere’s Clin. Neurol. 1996, 5, 783–802. [Google Scholar]
- Devinsky, O.; Vezzani, A.; O’Brien, T.J.; Jette, N.; Scheffer, I.E.; de Curtis, M.; Perucca, P. Epilepsy. Nat. Rev. Dis. Primers 2018, 4, 18024. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lin, Z.J.; Liu, L.; Xu, H.Q.; Shi, Y.W.; Yi, Y.H.; He, N.; Liao, W.P. Epilepsy-associated genes. Seizure 2017, 44, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, F.; Yan, L.M.; Su, T.; He, N.; Lin, Z.J.; Wang, J.; Shi, Y.W.; Yi, Y.H.; Liao, W.P. Ion Channel Genes and Epilepsy: Functional Alteration, Pathogenic Potential, and Mechanism of Epilepsy. Neurosci. Bull. 2017, 33, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Krampfl, K.; Maljevic, S.; Cossette, P.; Ziegler, E.; Rouleau, G.A.; Lerche, H.; Bufler, J. Molecular analysis of the A322D mutation in the GABA receptor alpha-subunit causing juvenile myoclonic epilepsy. Eur. J. Neurosci. 2005, 22, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, M.J.; Ding, L.; Maheshwari, A.; Macdonald, R.L. The GABAA receptor alpha1 subunit epilepsy mutation A322D inhibits transmembrane helix formation and causes proteasomal degradation. Proc. Natl. Acad. Sci. USA 2007, 104, 12999–13004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, L.; Feng, H.J.; Macdonald, R.L.; Botzolakis, E.J.; Hu, N.; Gallagher, M.J. GABA(A) receptor alpha1 subunit mutation A322D associated with autosomal dominant juvenile myoclonic epilepsy reduces the expression and alters the composition of wild type GABA(A) receptors. J. Biol. Chem. 2010, 285, 26390–26405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galanopoulou, A.S. GABA(A) receptors in normal development and seizures: Friends or foes? Curr. Neuropharmacol. 2008, 6, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braat, S.; Kooy, R.F. The GABAA Receptor as a Therapeutic Target for Neurodevelopmental Disorders. Neuron 2015, 86, 1119–1130. [Google Scholar] [CrossRef] [Green Version]
- He, Q.; Nomura, T.; Xu, J.; Contractor, A. The developmental switch in GABA polarity is delayed in fragile X mice. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Duarte, S.T.; Armstrong, J.; Roche, A.; Ortez, C.; Pérez, A.; O’Callaghan Mdel, M.; Pereira, A.; Sanmartí, F.; Ormazábal, A.; Artuch, R.; et al. Abnormal expression of cerebrospinal fluid cation chloride cotransporters in patients with Rett syndrome. PLoS ONE 2013, 8, e68851. [Google Scholar] [CrossRef] [PubMed]
- Carvill, G.L.; Weckhuysen, S.; McMahon, J.M.; Hartmann, C.; Møller, R.S.; Hjalgrim, H.; Cook, J.; Geraghty, E.; O’Roak, B.J.; Petrou, S.; et al. GABRA1 and STXBP1: Novel genetic causes of Dravet syndrome. Neurology 2014, 82, 1245–1253. [Google Scholar] [CrossRef] [Green Version]
- Pirker, S.; Schwarzer, C.; Wieselthaler, A.; Sieghart, W.; Sperk, G. GABA(A) receptors: Immunocytochemical distribution of 13 subunits in the adult rat brain. Neuroscience 2000, 101, 815–850. [Google Scholar] [CrossRef]
- Collingridge, G.L.; Isaac, J.T.; Wang, Y.T. Receptor trafficking and synaptic plasticity. Nat. Rev. Neurosci. 2004, 5, 952–962. [Google Scholar] [CrossRef] [PubMed]
- Michels, G.; Moss, S.J. GABAA receptors: Properties and trafficking. Crit. Rev. Biochem. Mol. Biol. 2007, 42, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Sieghart, W.; Sperk, G. Subunit composition, distribution and function of GABA(A) receptor subtypes. Curr. Top. Med. Chem. 2002, 2, 795–816. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, R.L.; Olsen, R.W. GABAA receptor channels. Annu. Rev. Neurosci. 1994, 17, 569–602. [Google Scholar] [CrossRef]
- Reyes-Nava, N.G.; Yu, H.C.; Coughlin, C.R., 2nd; Shaikh, T.H.; Quintana, A.M. Abnormal expression of GABA(A) receptor subunits and hypomotility upon loss of gabra1 in zebrafish. Biol. Open 2020, 9, bio051367. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.F.; Chiu, M.; Chan, E.S.; Axerio-Cilies, P.; Lu, J.; Huh, L.; Connolly, M.B.; Guella, I.; Farrer, M.J.; Xu, Z.D.; et al. Pathophysiology of and therapeutic options for a GABRA1 variant linked to epileptic encephalopathy. Mol. Brain 2019, 12, 92. [Google Scholar] [CrossRef]
- Hernandez, C.C.; XiangWei, W.; Hu, N.; Shen, D.; Shen, W.; Lagrange, A.H.; Zhang, Y.; Dai, L.; Ding, C.; Sun, Z.; et al. Altered inhibitory synapses in de novo GABRA5 and GABRA1 mutations associated with early onset epileptic encephalopathies. Brain A J. Neurol. 2019, 142, 1938–1954. [Google Scholar] [CrossRef]
- Butler, K.M.; Moody, O.A.; Schuler, E.; Coryell, J.; Alexander, J.J.; Jenkins, A.; Escayg, A. De novo variants in GABRA2 and GABRA5 alter receptor function and contribute to early-onset epilepsy. Brain A J. Neurol. 2018, 141, 2392–2405. [Google Scholar] [CrossRef] [PubMed]
- Jacob, T.C.; Moss, S.J.; Jurd, R. GABA(A) receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nat. Rev. Neurosci. 2008, 9, 331–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janve, V.S.; Hernandez, C.C.; Verdier, K.M.; Hu, N.; Macdonald, R.L. Epileptic encephalopathy de novo GABRB mutations impair γ-aminobutyric acid type A receptor function. Ann. Neurol. 2016, 79, 806–825. [Google Scholar] [CrossRef] [PubMed]
- Johannesen, K.; Marini, C.; Pfeffer, S.; Møller, R.S.; Dorn, T.; Niturad, C.E.; Gardella, E.; Weber, Y.; Søndergård, M.; Hjalgrim, H.; et al. Phenotypic spectrum of GABRA1: From generalized epilepsies to severe epileptic encephalopathies. Neurology 2016, 87, 1140–1151. [Google Scholar] [CrossRef]
- Kodera, H.; Ohba, C.; Kato, M.; Maeda, T.; Araki, K.; Tajima, D.; Matsuo, M.; Hino-Fukuyo, N.; Kohashi, K.; Ishiyama, A.; et al. De novo GABRA1 mutations in Ohtahara and West syndromes. Epilepsia 2016, 57, 566–573. [Google Scholar] [CrossRef] [Green Version]
- Maljevic, S.; Krampfl, K.; Cobilanschi, J.; Tilgen, N.; Beyer, S.; Weber, Y.G.; Schlesinger, F.; Ursu, D.; Melzer, W.; Cossette, P.; et al. A mutation in the GABA(A) receptor alpha(1)-subunit is associated with absence epilepsy. Ann. Neurol. 2006, 59, 983–987. [Google Scholar] [CrossRef]
- Fisher, J.L. A mutation in the GABAA receptor alpha 1 subunit linked to human epilepsy affects channel gating properties. Neuropharmacology 2004, 46, 629–637. [Google Scholar] [CrossRef]
- Lachance-Touchette, P.; Choudhury, M.; Stoica, A.; Di Cristo, G.; Cossette, P. Single-cell genetic expression of mutant GABAA receptors causing Human genetic epilepsy alters dendritic spine and GABAergic bouton formation in a mutation-specific manner. Front. Cell. Neurosci. 2014, 8, 317. [Google Scholar] [CrossRef] [Green Version]
- Carta, M.; Murru, L.; Botta, P.; Talani, G.; Sechi, G.; De Riu, P.; Sanna, E.; Biggio, G. The muscle relaxant thiocolchicoside is an antagonist of GABAA receptor function in the central nervous system. Neuropharmacology 2006, 51, 805–815. [Google Scholar] [CrossRef]
- Allen, A.S.; Berkovic, S.F.; Cossette, P.; Delanty, N.; Dlugos, D.; Eichler, E.E.; Epstein, M.P.; Glauser, T.; Goldstein, D.B.; Han, Y.; et al. De novo mutations in epileptic encephalopathies. Nature 2013, 501, 217–221. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, C.C.; Kong, W.; Hu, N. Altered Channel Conductance States and Gating of GABA(A) Receptors by a Pore Mutation Linked to Dravet Syndrome. eNeuro 2017, 4, e025-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Akabas, M.H. Identification of channel-lining residues in the M2 membrane-spanning segment of the GABA(A) receptor alpha1 subunit. J. Gen. Physiol. 1996, 107, 195–205. [Google Scholar] [CrossRef] [Green Version]
- Vithlani, M.; Terunuma, M.; Moss, S.J. The dynamic modulation of GABA(A) receptor trafficking and its role in regulating the plasticity of inhibitory synapses. Physiol. Rev. 2011, 91, 1009–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradley, C.A.; Taghibiglou, C.; Collingridge, G.L.; Wang, Y.T. Mechanisms involved in the reduction of GABAA receptor alpha1-subunit expression caused by the epilepsy mutation A322D in the trafficking-competent receptor. J. Biol. Chem. 2008, 283, 22043–22050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, G.A. Advantages of an antagonist: Bicuculline and other GABA antagonists. Br. J. Pharmacol. 2013, 169, 328–336. [Google Scholar] [CrossRef] [Green Version]
- Olsen, R.W. Allosteric ligands and their binding sites define γ-aminobutyric acid (GABA) type A receptor subtypes. Adv. Pharmacol. 2015, 73, 167–202. [Google Scholar] [CrossRef]
- Mistry, D.K.; Cottrell, G.A. Actions of steroids and bemegride on the GABAA receptor of mouse spinal neurones in culture. Exp. Physiol. 1990, 75, 199–209. [Google Scholar] [CrossRef] [Green Version]
- Safavynia, S.A.; Keating, G.; Speigel, I.; Fidler, J.A.; Kreuzer, M.; Rye, D.B.; Jenkins, A.; García, P.S. Effects of γ-Aminobutyric Acid Type A Receptor Modulation by Flumazenil on Emergence from General Anesthesia. Anesthesiology 2016, 125, 147–158. [Google Scholar] [CrossRef] [Green Version]
- Olsen, R.W. GABA(A) receptor: Positive and negative allosteric modulators. Neuropharmacology 2018, 136, 10–22. [Google Scholar] [CrossRef]
- Steudle, F.; Rehman, S.; Bampali, K. A novel de novo variant of GABRA1 causes increased sensitivity for GABA in vitro. Sci. Rep. 2020, 10, 2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cossette, P.; Liu, L.; Brisebois, K.; Dong, H.; Lortie, A.; Vanasse, M.; Saint-Hilaire, J.M.; Carmant, L.; Verner, A.; Lu, W.Y.; et al. Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nat. Genet. 2002, 31, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tong, L.; Song, S.; Niu, Y.; Li, J.; Wu, X.; Zhang, J.; Zai, C.C.; Luo, F.; Wu, J.; et al. Novel and de novo mutations in pediatric refractory epilepsy. Mol. Brain 2018, 11, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, M.; Ohmori, I.; Nakahori, T.; Ouchida, M.; Ohtsuka, Y. Mutation screen of GABRA1, GABRB2 and GABRG2 genes in Japanese patients with absence seizures. Neurosci. Lett. 2005, 383, 220–224. [Google Scholar] [CrossRef]
- Hernandez, C.C.; Tian, X.; Hu, N.; Shen, W.; Catron, M.A.; Yang, Y.; Chen, J. Dravet syndrome-associated mutations in GABRA1, GABRB2 and GABRG2 define the genetic landscape of defects of GABA(A) receptors. Brain Commun. 2021, 3, fcab033. [Google Scholar] [CrossRef]
- Geffrey, A.L.; Pollack, S.F.; Bruno, P.L.; Thiele, E.A. Drug-drug interaction between clobazam and cannabidiol in children with refractory epilepsy. Epilepsia 2015, 56, 1246–1251. [Google Scholar] [CrossRef]
- Sills, G.J.; Rogawski, M.A. Mechanisms of action of currently used antiseizure drugs. Neuropharmacology 2020, 168, 107966. [Google Scholar] [CrossRef]
- Greenfield, L.J., Jr. Molecular mechanisms of antiseizure drug activity at GABAA receptors. Seizure 2013, 22, 589–600. [Google Scholar] [CrossRef] [Green Version]
- Löscher, W.; Potschka, H.; Sisodiya, S.M.; Vezzani, A. Drug Resistance in Epilepsy: Clinical Impact, Potential Mechanisms, and New Innovative Treatment Options. Pharmacol. Rev. 2020, 72, 606–638. [Google Scholar] [CrossRef]
- Absalom, N.L.; Liao, V.W.Y.; Kothur, K.; Indurthi, D.C.; Bennetts, B.; Troedson, C.; Mohammad, S.S. Gain-of-function GABRB3 variants identified in vigabatrin-hypersensitive epileptic encephalopathies. Brain Commun. 2020, 2, fcaa162. [Google Scholar] [CrossRef]
- Billakota, S.; Devinsky, O.; Kim, K.W. Why we urgently need improved epilepsy therapies for adult patients. Neuropharmacology 2020, 170, 107855. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Gharpure, A.; Teng, J.; Zhuang, Y. Shared structural mechanisms of general anaesthetics and benzodiazepines. Nature 2020, 585, 303–308. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient 1 (Present Study) | Patient 2 [19,31] | |
|---|---|---|
| Onset age | 6 months (developmental delay) | 3 months (epilepsy) |
| Detection method | Whole exome sequencing | Whole exome sequencing |
| Nucleotide change | NM000806.5: c.875 C>G | NM000806.5: c.875 C>T |
| Protein change | p.Thr292Ser | p.Thr292Ile |
| Inheritance | De novo | De novo |
| Clinical features | No diagnosed seizure events Developmental delay | Lennox–Gastaut syndrome with light-sensitive myoclonic epilepsy and generalized tonic–clonic seizures |
| EEG | a few abnormal discharge | N/A |
| MRI | Normal | N/A |
| Functional alterations | Agonist sensitivity  Maximum response: normal EC50  Surface/total expression: normal Channel open probability Tonic and leak current | Agonist sensitivity Maximum response EC50 Surface/total expression: normal Channel open probability Tonic and leak current: normal |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, W.; Ge, Y.; Lu, J.; Melo, J.; So, Y.W.; Juneja, R.; Liu, L.; Wang, Y.T. Distinct Functional Alterations and Therapeutic Options of Two Pathological De Novo Variants of the T292 Residue of GABRA1 Identified in Children with Epileptic Encephalopathy and Neurodevelopmental Disorders. Int. J. Mol. Sci. 2022, 23, 2723. https://doi.org/10.3390/ijms23052723
Chen W, Ge Y, Lu J, Melo J, So YW, Juneja R, Liu L, Wang YT. Distinct Functional Alterations and Therapeutic Options of Two Pathological De Novo Variants of the T292 Residue of GABRA1 Identified in Children with Epileptic Encephalopathy and Neurodevelopmental Disorders. International Journal of Molecular Sciences. 2022; 23(5):2723. https://doi.org/10.3390/ijms23052723
Chicago/Turabian StyleChen, Wenlin, Yang Ge, Jie Lu, Joshua Melo, Yee Wah So, Romi Juneja, Lidong Liu, and Yu Tian Wang. 2022. "Distinct Functional Alterations and Therapeutic Options of Two Pathological De Novo Variants of the T292 Residue of GABRA1 Identified in Children with Epileptic Encephalopathy and Neurodevelopmental Disorders" International Journal of Molecular Sciences 23, no. 5: 2723. https://doi.org/10.3390/ijms23052723
APA StyleChen, W., Ge, Y., Lu, J., Melo, J., So, Y. W., Juneja, R., Liu, L., & Wang, Y. T. (2022). Distinct Functional Alterations and Therapeutic Options of Two Pathological De Novo Variants of the T292 Residue of GABRA1 Identified in Children with Epileptic Encephalopathy and Neurodevelopmental Disorders. International Journal of Molecular Sciences, 23(5), 2723. https://doi.org/10.3390/ijms23052723