
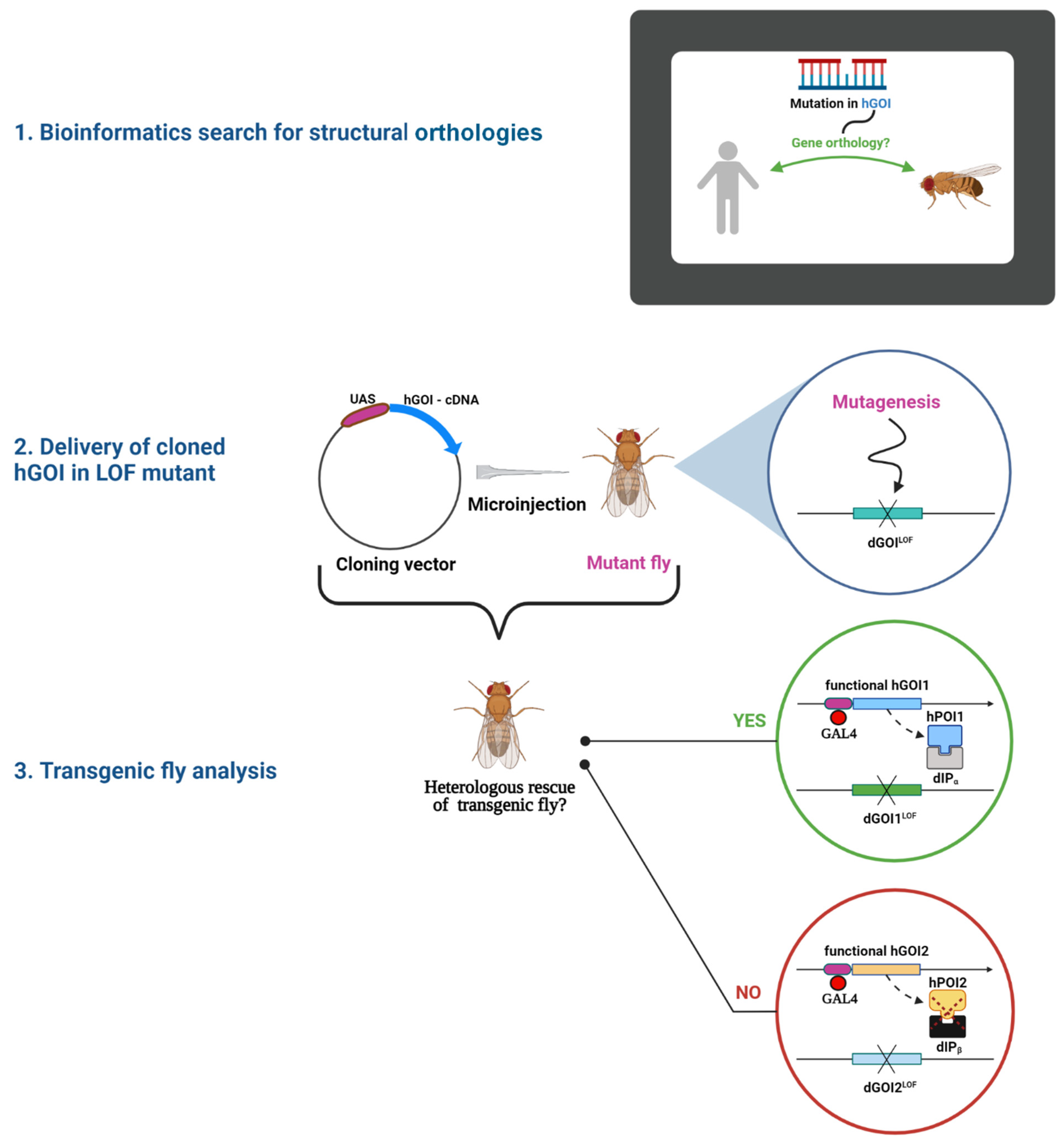
Inter-Species Rescue of Mutant Phenotype—The Standard for Genetic Analysis of Human Genetic Disorders in Drosophila melanogaster Model

 and
and 
Abstract
1. Introduction
2. Neurodegenerative and Neuromuscular Disorders
2.1. Parkinson’s Disease
2.2. Amyotrophic Lateral Sclerosis
2.3. Autism Spectrum Disorder
3. Cardiac Disorders
3.1. Congenital Heart Defects
3.2. Cardiomyopathy Phenotypes
3.3. Other Cardiac Disorders
4. Cancer
4.1. Validating Orthologs of Human Tumor Suppressors Using the Drosophila melanogaster Model
4.2. Elucidating the Role of Tumor Microenvironment and Host-Neoplastic Cells Competition in Gut Adenoma Development
4.3. Demonstrating the Species-Dependent Pathways of Notch Hyperactivation
5. Infectious Diseases
5.1. Molecular Mechanisms of Neuropathological Effects Caused by Zika Virus NS4A Protein
5.2. Elucidating the Molecular Players in the Cytotoxicity of Cholera Toxin
5.3. Molecular Mechanisms of Apoptosis Induced by Helicobacter pylori Cytotoxin-Associated Gene A
5.4. Discovering Novel Candidates for Assessing Genetic Susceptibility to Different Infections
5.5. Demonstrating the Functional Homology of Human Vasodilator-Stimulated Phosphoprotein (VASP) and Drosophila enabled
6. Discussion
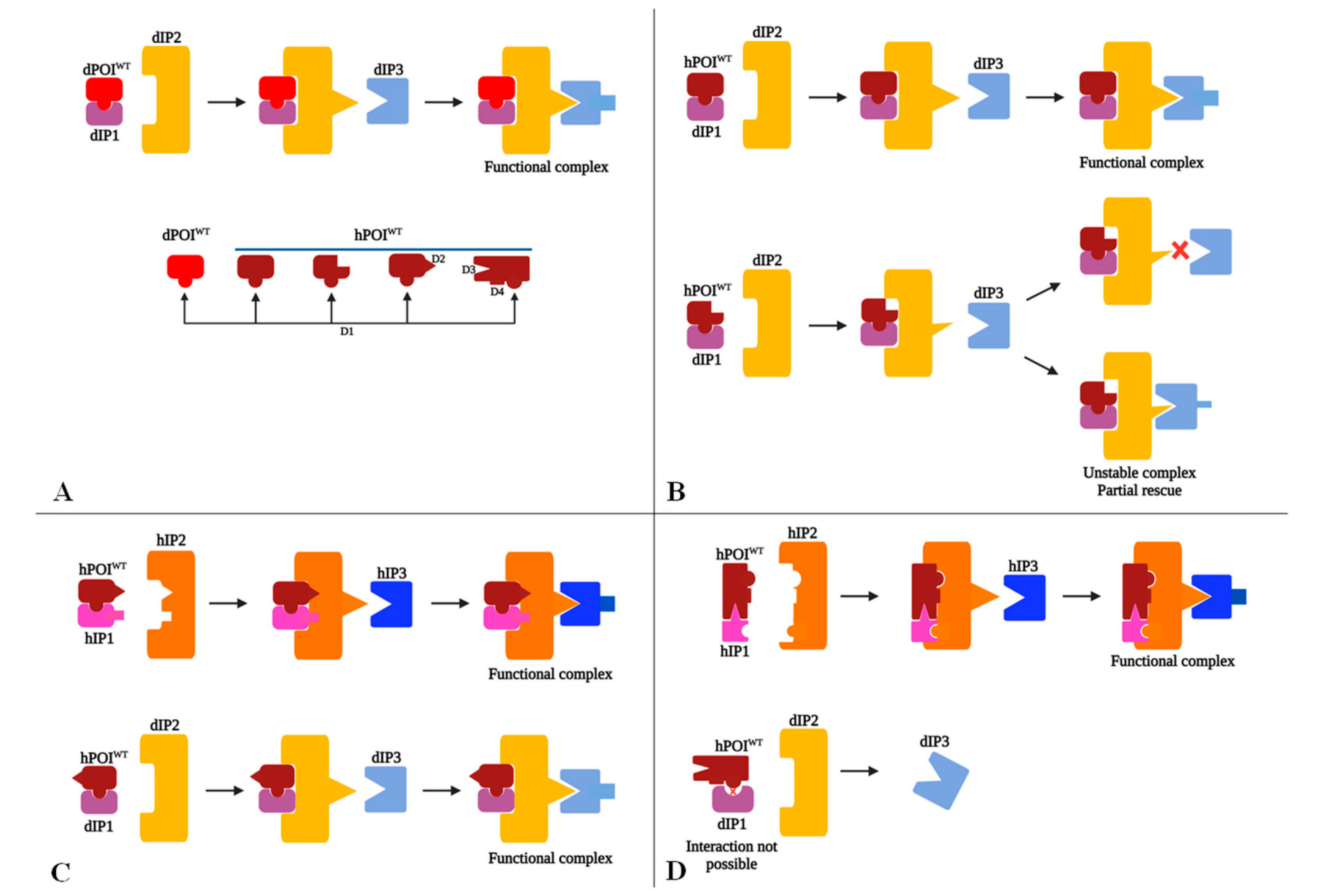
6.1. Issues When Modeling hGDs in D. melanogaster Regardless of Positive Heterologous Rescue Results
6.2. Possible Scenarios Accounting for Heterologous Rescue Failure or Partial Rescue in D. melanogaster
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- De Maria Marchiano, R.; Di Sante, G.; Piro, G.; Carbone, C.; Tortora, G.; Boldrini, L.; Pietragalla, A.; Daniele, G.; Tredicine, M.; Cesario, A.; et al. Translational research in the era of precision medicine: Where we are and where we will go. J. Pers. Med. 2021, 11, 216. [Google Scholar] [CrossRef] [PubMed]
- Beckingham, K.M.; Armstrong, J.D.; Texada, M.J.; Munjaal, R.; Baker, D.A. Drosophila melanogaster—The model organism of choice for the complex biology of multi-cellular organisms. Gravit. Space Biol. Bull. Publ. Am. Soc. Gravit. Space Biol. 2005, 18, 17–29. [Google Scholar]
- Adams, M.D.; Celniker, S.E.; Holt, R.A.; Evans, C.A.; Gocayne, J.D.; Amanatides, P.G.; Scherer, S.E.; Li, P.W.; Hoskins, R.A.; Galle, R.F.; et al. The genome sequence of Drosophila melanogaster. Science 2000, 287, 2185–2195. [Google Scholar] [CrossRef]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed]
- Dunham, M.J.; Fowler, D.M. Contemporary, yeast-based approaches to understanding human genetic variation. Curr. Opin. Genet. Dev. 2013, 23, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.M.; Hong, E.L.; Amundsen, C.; Balakrishnan, R.; Binkley, G.; Chan, E.T.; Christie, K.R.; Costanzo, M.C.; Dwight, S.S.; Engel, S.R.; et al. Saccharomyces Genome Database: The genomics resource of budding yeast. Nucleic Acids Res. 2012, 40, D700–D705. [Google Scholar] [CrossRef] [PubMed]
- Larkin, A.; Marygold, S.J.; Antonazzo, G.; Attrill, H.; Dos Santos, G.; Garapati, P.V.; Goodman, J.L.; Gramates, L.S.; Millburn, G.; Strelets, V.B.; et al. FlyBase: Updates to the Drosophila melanogaster knowledge base. Nucleic Acids Res. 2021, 49, D899–D907. [Google Scholar] [CrossRef]
- Bellen, H.J.; Wangler, M.F.; Yamamoto, S. The fruit fly at the interface of diagnosis and pathogenic mechanisms of rare and common human diseases. Hum. Mol. Genet. 2019, 28, R207–R214. [Google Scholar] [CrossRef]
- Specchia, V.; Puricella, A.; D’Attis, S.; Massari, S.; Giangrande, A.; Bozzetti, M.P. Drosophila melanogaster as a Model to Study the Multiple Phenotypes, Related to Genome Stability of the Fragile-X Syndrome. Front. Genet. 2019, 10, 10. [Google Scholar] [CrossRef]
- Bellen, H.J.; Yamamoto, S. Morgan’s legacy: Fruit flies and the functional annotation of conserved genes. Cell 2015, 163, 12–14. [Google Scholar] [CrossRef] [PubMed]
- Rorth, P. Gal4 in the Drosophila female germline. Mech. Dev. 1998, 78, 113–118. [Google Scholar] [CrossRef]
- Wangler, M.F.; Yamamoto, S.; Chao, H.-T.; Posey, J.E.; Westerfield, M.; Postlethwait, J.; Members of the Undiagnosed Diseases Network (UDN); Hieter, P.; Boycott, K.M.; Campeau, P.M.; et al. Model Organisms Facilitate Rare Disease Diagnosis and Therapeutic Research. Genetics 2017, 207, 9–27. [Google Scholar] [CrossRef]
- Harnish, J.M.; Deal, S.L.; Chao, H.T.; Wangler, M.F.; Yamamoto, S. In Vivo Functional Study of Disease-associated Rare Human Variants Using Drosophila. J. Vis. Exp. JoVE 2019, 150, e59658. [Google Scholar] [CrossRef]
- Lu, C.; Vihtelic, T.S.; Hyde, D.R.; Li, T. A neuronal-specific mammalian homolog of the Drosophila retinal degeneration B gene with expression restricted to the retina and dentate gyrus. J. Neurosci. Off. J. Soc. Neurosci. 1999, 19, 7317–7325. [Google Scholar] [CrossRef]
- Leiserson, W.M.; Harkins, E.W.; Keshishian, H. Fray, a Drosophila serine/threonine kinase homologous to mammalian PASK, is required for axonal ensheathment. Neuron 2000, 28, 793–806. [Google Scholar] [CrossRef]
- Maurya, B.; Surabhi, S.; Pandey, P.; Mukherjee, A.; Mutsuddi, M. Insights into Human Neurodegeneration: Lessons Learnt from Drosophila; Springer: New York, NY, USA, 2019; pp. 373–403. [Google Scholar] [CrossRef]
- Qiao, H.H.; Wang, F.; Xu, R.G.; Sun, J.; Zhu, R.; Mao, D.; Ren, X.; Wang, X.; Jia, Y.; Peng, P.; et al. An efficient and multiple target transgenic RNAi technique with low toxicity in Drosophila. Nat. Commun. 2018, 9, 4160. [Google Scholar] [CrossRef]
- McGurk, L.; Berson, A.; Bonini, N.M. Drosophila as an In Vivo Model for Human Neurodegenerative Disease. Genetics 2015, 201, 377–402. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, T.E.; Taylor, J.P. Flightless flies: Drosophila models of neuromuscular disease. Ann. N. Y. Acad. Sci. 2010, 1184, e1–e20. [Google Scholar] [CrossRef]
- Rubin, G.M.; Yandell, M.D.; Wortman, J.R.; Gabor Miklos, G.L.; Nelson, C.R.; Hariharan, I.K.; Fortini, M.E.; Li, P.W.; Apweiler, R.; Fleischmann, W.; et al. Comparative genomics of the eukaryotes. Science 2000, 287, 2204–2215. [Google Scholar] [CrossRef]
- Brody, T. The Interactive Fly: Gene networks, development and the Internet. Trends Genet. 1999, 15, 333–334. [Google Scholar] [CrossRef]
- Yang, Y.; Nishimura, I.; Imai, Y.; Takahashi, R.; Lu, B. Parkin suppresses dopaminergic neuronselective neurotoxicity induced by Pael-R in Drosophila. Neuron 2003, 37, 911–924. [Google Scholar] [CrossRef]
- Burchell, V.S.; Nelson, D.E.; Sanchez-Martinez, A.; Delgado-Camprubi, M.; Ivatt, R.M.; Pogson, J.H.; Randle, S.J.; Wray, S.; Lewis, P.A.; Houlden, H.; et al. The Parkinson’s disease–linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat. Neurosci. 2013, 16, 1257–1265. [Google Scholar] [CrossRef]
- Paisán-Ruiz, C.; Guevara, R.; Federoff, M.; Hanagasi, H.; Sina, F.; Elahi, E.; Schneider, S.A.; Schwingenschuh, P.; Bajaj, N.; Emre, M.; et al. Early-onset L-dopa-responsive parkinsonism with pyramidal signs due to ATP13A2, PLA2G6, FBXO7 and spatacsin mutations. Mov. Disord. 2010, 25, 1791–1800. [Google Scholar] [CrossRef] [PubMed]
- Baughman, J.M.; Nilsson, R.; Gohil, V.M.; Arlow, D.H.; Gauhar, Z.; Mootha, V.K. A computational screen for regulators of oxidative phosphorylation implicates SLIRP in mitochondrial RNA homeostasis. PLoS Genet. 2009, 5, e1000590. [Google Scholar] [CrossRef]
- Meng, H.; Yamashita, C.; Shiba-Fukushima, K.; Inoshita, T.; Funayama, M.; Sato, S.; Hatta, T.; Natsume, T.; Umitsu, M.; Takagi, J.; et al. Loss of Parkinson’s disease-associated protein CHCHD2 affects mitochondrial crista structure and destabilizes cytochrome c. Nat. Commun. 2017, 8, 15500. [Google Scholar] [CrossRef]
- Mori, A.; Hatano, T.; Inoshita, T.; Shiba-Fukushima, K.; Koinuma, T.; Meng, H.; Kubo, S.-I.; Spratt, S.; Cui, C.; Yamashita, C.; et al. Parkinson’s disease-associated iPLA2-VIA/PLA2G6 regulates neuronal functions and α-synuclein stability through membrane remodeling. Proc. Natl. Acad. Sci. USA 2019, 116, 20689–20699. [Google Scholar] [CrossRef]
- Zhang, B.; Egli, D.; Georgiev, O.; Schaffner, W. The Drosophila homolog of mammalian zinc finger factor MTF-1 activates transcription in response to heavy metals. Mol. Cell. Biol. 2001, 21, 4505–4514. [Google Scholar] [CrossRef]
- Ghoshal, K.; Majumder, S.; Zhu, Q.; Hunzeker, J.; Datta, J.; Shah, M.; Sheridan, J.F.; Jacob, S.T. Influenza virus infection induces metallothionein gene expression in the mouse liver and lung by overlapping but distinct molecular mechanisms. Mol. Cell. Biol. 2001, 21, 8301–8317. [Google Scholar] [CrossRef]
- Saini, N.; Georgiev, O.; Schaffner, W. The parkin mutant phenotype in the fly is largely rescued by metal-responsive transcription factor (MTF-1). Mol. Cell. Biol. 2011, 31, 2151–2161. [Google Scholar] [CrossRef]
- Balamurugan, K.; Egli, D.; Selvaraj, A.; Zhang, B.; Georgiev, O.; Schaffner, W. Metal-responsive transcription factor (MTF-1) and heavy metal stress response in Drosophila and mammalian cells: A functional comparison. Biol. Chem. 2001, 385, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Morais, V.A.; Verstreken, P.; Roethig, A.; Smet, J.; Snellinx, A.; Vanbrabant, M.; Haddad, D.; Frezza, C.; Mandemakers, W.; Vogt-Weisenhorn, D.; et al. Parkinson’s disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol. Med. 2009, 1, 99–111. [Google Scholar] [CrossRef]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Longinetti, E.; Fang, F. Epidemiology of amyotrophic lateral sclerosis: An update of recent literature. Curr. Opin. Neurol. 2019, 32, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Abramzon, Y.A.; Fratta, P.; Traynor, B.J.; Chia, R. The Overlapping Genetics of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Front. Neurosci. 2020, 14, 42. [Google Scholar] [CrossRef]
- Elamin, M.; Bede, P.; Byrne, S.; Jordan, N.; Gallagher, L.; Wynne, B.; O’Brien, C.; Phukan, J.; Lynch, C.; Pender, N.; et al. Cognitive changes predict functional decline in ALS: A population-based longitudinal study. Neurology 2013, 80, 1590–1597. [Google Scholar] [CrossRef]
- Layalle, S.; They, L.; Ourghani, S.; Raoul, C.; Soustelle, L. Amyotrophic Lateral Sclerosis Genes in Drosophila melanogaster. Int. J. Mol. Sci. 2021, 22, 904. [Google Scholar] [CrossRef]
- Picher-Martel, V.; Valdmanis, P.N.; Gould, P.V.; Julien, J.P.; Dupre, N. From animal models to human disease: A genetic approach for personalized medicine in ALS. Acta Neuropathol. Commun. 2016, 4, 70. [Google Scholar] [CrossRef]
- Liguori, F.; Amadio, S.; Volonté, C. Fly for ALS: Drosophila modeling on the route to amyotrophic lateral sclerosis modifiers. Cell. Mol. Life Sci. 2021, 78, 6143–6160. [Google Scholar] [CrossRef]
- Mockett, R.J.; Radyuk, S.N.; Benes, J.J.; Orr, W.C.; Sohal, R.S. Phenotypic effects of familial amyotrophic lateral sclerosis mutant Sod alleles in transgenic Drosophila. Proc. Natl. Acad. Sci. USA 2003, 100, 301–306. [Google Scholar] [CrossRef]
- Parkes, T.L.; Elia, A.J.; Dickinson, D.; Hilliker, A.J.; Phillips, J.P.; Boulianne, G.L. Extension of Drosophila lifespan by overexpression of human SOD1 in motorneurons. Nat. Genet. 1998, 19, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Tollervey, J.R.; Curk, T.; Rogelj, B.; Briese, M.; Cereda, M.; Kayikci, M.; Konig, J.; Hortobagyi, T.; Nishimura, A.L.; Zupunski, V.; et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 2011, 14, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Alami, N.H.; Smith, R.B.; Carrasco, M.A.; Williams, L.A.; Winborn, C.S.; Han, S.S.; Kiskinis, E.; Winborn, B.; Freibaum, B.D.; Kanagaraj, A.; et al. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 2014, 81, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Honda, D.; Ishigaki, S.; Iguchi, Y.; Fujioka, Y.; Udagawa, T.; Masuda, A.; Ohno, K.; Katsuno, M.; Sobue, G. The ALS/FTLD-related RNA-binding proteins TDP-43 and FUS have common downstream RNA targets in cortical neurons. FEBS Open Bio 2013, 4, 1–10. [Google Scholar] [CrossRef]
- Baldwin, K.R.; Godena, V.K.; Hewitt, V.L.; Whitworth, A.J. Axonal transport defects are a common phenotype in Drosophila models of ALS. Hum. Mol. Genet. 2016, 25, 2378–2392. [Google Scholar] [CrossRef]
- Wang, J.W.; Brent, J.R.; Tomlinson, A.; Shneider, N.A.; McCabe, B.D. The ALS-associated proteins FUS and TDP-43 function together to affect Drosophila locomotion and life span. J. Clin. Investig. 2011, 121, 4118–4126. [Google Scholar] [CrossRef]
- Arlington, V.A. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; The American Psychiatric Association: Washington, DC, USA, 2013. [Google Scholar]
- Lord, C.; Brugha, T.S.; Charman, T.; Cusack, J.; Dumas, G.; Frazier, T.; Jones, E.J.H.; Jones, R.M.; Pickles, A.; State, M.W.; et al. Autism spectrum disorder. Nat. Rev. Dis. Prim. 2020, 6, 5. [Google Scholar] [CrossRef]
- Iovene, M.R.; Bombace, F.; Maresca, R.; Sapone, A.; Iardino, P.; Picardi, A.; Marotta, R.; Schiraldi, C.; Siniscalco, D.; Serra, N.; et al. Intestinal Dysbiosis and Yeast Isolation in Stool of Subjects with Autism Spectrum Disorders. Mycopathologia 2016, 182, 349–363. [Google Scholar] [CrossRef]
- Fulceri, F.; Morelli, M.; Santocchi, E.; Cena, H.; Del Bianco, T.; Narzisi, A.; Calderoni, S.; Muratori, F. Gastrointestinal symptoms and behavioral problems in preschoolers with autism spectrum disorder. Dig. Liver Dis. 2016, 48, 248–254. [Google Scholar] [CrossRef]
- Marler, S.; Ferguson, B.J.; Lee, E.B.; Peters, B.; Williams, K.C.; McDonnell, E.; Macklin, E.A.; Levitt, P.; Margolis, K.G.; Beversdorf, D.Q.; et al. Association of rigid-compulsive behavior with functional constipation in autism spectrum disorder. J. Autism Dev. Disord. 2017, 47, 1673–1681. [Google Scholar] [CrossRef]
- Abrahams, B.S.; Arking, D.E.; Campbell, D.B.; Mefford, H.C.; Morrow, E.M.; Weiss, L.A.; Menashe, I.; Wadkins, T.; Banerjee-Basu, S.; Packer, A. SFARI Gene 2.0: A community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol. Autism 2013, 4, 36. [Google Scholar] [CrossRef] [PubMed]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584.e23. [Google Scholar] [CrossRef] [PubMed]
- Salim, S.; Banu, A.; Alwa, A.; Gowda, S.B.M.; Mohammad, F. The gut-microbiota-brain axis in autism: What Drosophila models can offer? J. Neurodev. Disord. 2021, 13, 37. [Google Scholar] [CrossRef]
- Hu, Y.; Flockhart, I.; Vinayagam, A.; Bergwitz, C.; Berger, B.; Perrimon, N.; Mohr, S.E. An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinform. 2011, 12, 357. [Google Scholar] [CrossRef] [PubMed]
- Clifford, S.; Dissanayake, C.; Bui, Q.M.; Huggins, R.; Taylor, A.K.; Loesch, D.Z. Autism spectrum phenotype in males and females with fragile X full mutation and premutation. J. Autism Dev. Disord. 2006, 37, 738–747. [Google Scholar] [CrossRef]
- Coffee, R.L., Jr.; Tessier, C.R.; Woodruff, E.A., 3rd; Broadie, K. Fragile X mental retardation protein has a unique, evolutionarily conserved neuronal function not shared with FXR1P or FXR2P. Dis. Model. Mech. 2010, 3, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1948. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G.; Dasouki, M.J.; Zhou, X.P.; Talebizadeh, Z.; Brown, M.; Takahashi, T.N.; Miles, J.H.; Wang, C.H.; Stratton, R.; Pilarski, R.; et al. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J. Med. Genet. 2005, 42, 318–321. [Google Scholar] [CrossRef]
- Mester, J.L.; Ghosh, R.; Pesaran, T.; Huether, R.; Karam, R.; Hruska, K.S.; Costa, H.A.; Lachlan, K.; Ngeow, J.; Barnholtz-Sloan, J.; et al. Gene-specific criteria for PTEN variant curation: Recommendations from the ClinGen PTEN Expert Panel. Hum. Mutat. 2018, 39, 1581–1592. [Google Scholar] [CrossRef]
- Sun, H.; Lesche, R.; Li, D.M.; Liliental, J.; Zhang, H.; Gao, J.; Gavrilova, N.; Mueller, B.; Liu, X.; Wu, H. PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5,-trisphosphate and Akt/protein kinase B signaling pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 6199–6204. [Google Scholar] [CrossRef]
- Ganguly, P.; Madonsela, L.; Chao, J.T.; Loewen, C.J.R.; O’Connor, T.P.; Verheyen, E.M.; Allan, D.W. A scalable Drosophila assay for clinical interpretation of human PTEN variants in suppression of PI3K/AKT induced cellular proliferation. PLoS Genet. 2021, 17, e1009774. [Google Scholar] [CrossRef]
- Pelc, K.; Cheron, G.; Dan, B. Behavior and neuropsychiatric manifestations in Angelman syndrome. Neuropsychiatr. Dis. Treat. 2008, 4, 577–584. [Google Scholar] [PubMed]
- Ratiu, A.C.; Ecovoiu, A.A.; Graur, M.; Gavrila, L. A second site lethal mutation masked the real phenotype of EP(3)3214 transgenic line. Bull. USAMV Anim. Sci. Biotechnol. 2008, 65, 475. [Google Scholar]
- Ratiu, A.C.; Neagu, A.; Mihalache, M.R.; Lazar, V. Long-term administration of omega-3 fatty acids alleviates Angelman syndrome-like phenotype in an Ube3a mutant strain of Drosophila melanogaster. Biointerface Res. Appl. Chem. 2015, 5, 996–1002. [Google Scholar]
- Chakraborty, M.; Paul, B.K.; Nayak, T.; Das, A.; Jana, N.R.; Bhutani, S. The E3 ligase ube3a is required for learning in Drosophila melanogaster. Biochem. Biophys. Res. Commun. 2015, 462, 71–77. [Google Scholar] [CrossRef]
- Chai, A.; Withers, J.; Koh, Y.H.; Parry, K.; Bao, H.; Zhang, B.; Budnik, V.; Pennetta, G. hVAPB, the causative gene of a heterogeneous group of motor neuron diseases in humans, is functionally interchangeable with its Drosophila homologue DVAP-33A at the neuromuscular junction. Hum. Mol. Genet. 2007, 17, 266–280. [Google Scholar] [CrossRef]
- Besson, M.T.; Dupont, P.; Fridell, Y.W.; Liévens, J.C. Increased energy metabolism rescues glia-induced pathology in a Drosophila model of Huntington’s disease. Hum. Mol. Genet. 2010, 19, 3372–3382. [Google Scholar] [CrossRef]
- Rui, Y.N.; Xu, Z.; Patel, B.; Chen, Z.; Chen, D.; Tito, A.; David, G.; Sun, Y.; Stimming, E.F.; Bellen, H.J.; et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 2015, 17, 262–275. [Google Scholar] [CrossRef]
- Tsai, P.I.; Lin, C.H.; Hsieh, C.H.; Papakyrikos, A.M.; Kim, M.J.; Napolioni, V.; Schoor, C.; Couthouis, J.; Wu, R.M.; Wszolek, Z.K.; et al. PINK1 Phosphorylates MIC60/Mitofilin to Control Structural Plasticity of Mitochondrial Crista Junctions. Mol. Cell 2018, 69, 744–756.e6. [Google Scholar] [CrossRef]
- Poole, A.C.; Thomas, R.E.; Andrews, L.A.; McBride, H.M.; Whitworth, A.J.; Pallanck, L.J. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2008, 105, 1638–1643. [Google Scholar] [CrossRef]
- Wang, C.; Lu, R.; Ouyang, X.; Ho, M.W.; Chia, W.; Yu, F.; Lim, K.L. Drosophila overexpressing parkin R275W mutant exhibits dopaminergic neuron degeneration and mitochondrial abnormalities. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 8563–8570. [Google Scholar] [CrossRef] [PubMed]
- Soukup, S.F.; Kuenen, S.; Vanhauwaert, R.; Manetsberger, J.; Hernández-Díaz, S.; Swerts, J.; Schoovaerts, N.; Vilain, S.; Gounko, N.V.; Vints, K.; et al. LRRK2-Dependent EndophilinA Phosphoswitch Is Critical for Macroautophagy at Presynaptic Terminals. Neuron 2016, 92, 829–844. [Google Scholar] [CrossRef]
- Chuang, C.-L.; Lu, Y.-N.; Wang, H.-C.; Chang, H.-Y. Genetic dissection reveals that Akt is the critical kinase downstream of LRRK2 to phosphorylate and inhibit FOXO1, and promotes neuron survival. Hum. Mol. Genet. 2014, 23, 5649–5658. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Wong, C.; Gao, S.M.; Zhang, R.; Sun, R.; Li, Y.; Song, Y. The retromer complex safeguards against neural progenitor-derived tumorigenesis by regulating Notch receptor trafficking. eLife 2018, 7, e38181. [Google Scholar] [CrossRef] [PubMed]
- Bolkan, B.J.; Kretzschmar, D. Loss of Tau results in defects in photoreceptor development and progressive neuronal degeneration in Drosophila. Dev. Neurobiol. 2014, 74, 1210–1225. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-H.; Giampetruzzi, A.; Tran, H.; Fallini, C.; Gao, F.-B.; Landers, J.E. A Drosophila model of ALS reveals a partial loss of function of causative human PFN1 mutants. Hum. Mol. Genet. 2017, 26, 2146–2155. [Google Scholar] [CrossRef][Green Version]
- Johnson, A.E.; Shu, H.; Hauswirth, A.G.; Tong, A.; Davis, G.W. VCP-dependent muscle degeneration is linked to defects in a dynamic tubular lysosomal network in vivo. eLife 2015, 4, e07366. [Google Scholar] [CrossRef]
- Jakobsdottir, J.; van der Lee, S.J.; Bis, J.C.; Chouraki, V.; Li-Kroeger, D.; Yamamoto, S.; Grove, M.L.; Naj, A.; Vronskaya, M.; Salazar, J.L.; et al. Rare Functional Variant in TM2D3 is Associated with Late-Onset Alzheimer’s Disease. PLoS Genet. 2016, 12, e1006327, Erratum in PLoS Genet. 2016, 12, e1006456. [Google Scholar] [CrossRef]
- Buechling, T.; Bartscherer, K.; Ohkawara, B.; Chaudhary, V.; Spirohn, K.; Niehrs, C.; Boutros, M. Wnt/Frizzled signaling requires dPRR, the Drosophila homolog of the prorenin receptor. Curr. Biol. 2010, 20, 1263–1268. [Google Scholar] [CrossRef]
- Ghosh, S.G.; Becker, K.; Huang, H.; Dixon-Salazar, T.; Chai, G.; Salpietro, V.; Al-Gazali, L.; Waisfisz, Q.; Wang, H.; Vaux, K.K.; et al. Biallelic Mutations in ADPRHL2, Encoding ADP-Ribosylhydrolase 3, Lead to a Degenerative Pediatric Stress-Induced Epileptic Ataxia Syndrome. Am. J. Hum. Genet. 2018, 103, 431–439. [Google Scholar] [CrossRef]
- Broeck, L.V.; Kleinberger, G.; Chapuis, J.; Gistelinck, M.; Amouyel, P.; Van Broeckhoven, C.; Lambert, J.-C.; Callaerts, P.; Dermaut, B. Functional complementation in Drosophila to predict the pathogenicity of TARDBP variants: Evidence for a loss-of-function mechanism. Neurobiol. Aging 2014, 36, 1121–1129. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Briere, L.C.; Kanca, O.; Marcogliese, P.; Walker, M.A.; High, F.A.; Vanderver, A.; Krier, J.; Carmichael, N.; Callahan, C.; et al. De novo mutations in TOMM70, a receptor of the mitochondrial import translocase, cause neurological impairment. Hum. Mol. Genet. 2020, 29, 1568–1579. [Google Scholar] [CrossRef] [PubMed]
- Sunderhaus, E.R.; Law, A.D.; Kretzschmar, D. Disease-Associated PNPLA6 Mutations Maintain Partial Functions When Analyzed in Drosophila. Front. Neurosci. 2019, 13, 1207. [Google Scholar] [CrossRef] [PubMed]
- Topaloglu, A.K.; Lomniczi, A.; Kretzschmar, D.; Dissen, G.A.; Kotan, L.D.; McArdle, C.A.; Koc, A.F.; Hamel, B.C.; Guclu, M.; Papatya, E.D.; et al. Loss-of-Function Mutations in PNPLA6 Encoding Neuropathy Target Esterase Underlie Pubertal Failure and Neurological Deficits in Gordon Holmes Syndrome. J. Clin. Endocrinol. Metab. 2014, 99, E2067–E2075. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wu, Z.; Kuo, Y.M.; Zhou, B. Dietary rescue of fumble–a Drosophila model for pantothenate-kinase-associated neurodegeneration. J. Inherit. Metab. Dis. 2005, 28, 1055–1064. [Google Scholar] [CrossRef]
- Ohno, M.; Hiraoka, Y.; Matsuoka, T.; Tomimoto, H.; Takao, K.; Miyakawa, T.; Oshima, N.; Kiyonari, H.; Kimura, T.; Kita, T.; et al. Nardilysin regulates axonal maturation and myelination in the central and peripheral nervous system. Nat. Neurosci. 2009, 12, 1506–1513. [Google Scholar] [CrossRef]
- Yoon, W.H.; Sandoval, H.; Nagarkar-Jaiswal, S.; Jaiswal, M.; Yamamoto, S.; Haelterman, N.A.; Putluri, N.; Putluri, V.; Sreekumar, A.; Tos, T.; et al. Loss of Nardilysin, a Mitochondrial Co-chaperone for α-Ketoglutarate Dehydrogenase, Promotes mTORC1 Activation and Neurodegeneration. Neuron 2016, 93, 115–131. [Google Scholar] [CrossRef]
- Vonk, J.J.; Yeshaw, W.M.; Pinto, F.; Faber, A.I.; Lahaye, L.L.; Kanon, B.; van der Zwaag, M.; Velayos-Baeza, A.; Freire, R.; van IJzendoorn, S.C.; et al. Drosophila Vps13 Is Required for Protein Homeostasis in the Brain. PLoS ONE 2017, 12, e0170106. [Google Scholar] [CrossRef]
- Yeshaw, W.M.; van der Zwaag, M.; Pinto, F.; Lahaye, L.L.; Faber, A.I.; Gómez-Sánchez, R.; Dolga, A.M.; Poland, C.; Monaco, A.P.; van IJzendoorn, S.C.; et al. Human VPS13A is associated with multiple organelles and influences mitochondrial morphology and lipid droplet motility. eLife 2019, 8, e43561. [Google Scholar] [CrossRef]
- Xiong, B.; Bayat, V.; Jaiswal, M.; Zhang, K.; Sandoval, H.; Charng, W.L.; Li, T.; David, G.; Duraine, L.; Lin, Y.Q.; et al. Crag Is a GEF for Rab11 required for rhodopsin trafficking and maintenance of adult photoreceptor cells. PLOS Biol. 2012, 10, e1001438. [Google Scholar] [CrossRef]
- Miyake, N.; Fukai, R.; Ohba, C.; Chihara, T.; Miura, M.; Shimizu, H.; Kakita, A.; Imagawa, E.; Shiina, M.; Ogata, K.; et al. Biallelic TBCD Mutations Cause Early-Onset Neurodegenerative Encephalopathy. Am. J. Hum. Genet. 2016, 99, 950–961. [Google Scholar] [CrossRef] [PubMed]
- Hekmat-Scafe, D.S.; Mercado, A.; Fajilan, A.A.; Lee, A.W.; Hsu, R.; Mount, D.B.; Tanouye, M.A. Seizure Sensitivity Is Ameliorated by Targeted Expression of K+–Cl− Cotransporter Function in the Mushroom Body of the Drosophila Brain. Genetics 2010, 184, 171–183. [Google Scholar] [CrossRef][Green Version]
- Praschberger, R.; Lowe, S.A.; Malintan, N.T.; Giachello, C.; Patel, N.; Houlden, H.; Kullmann, D.M.; Baines, R.A.; Usowicz, M.M.; Krishnakumar, S.S.; et al. Mutations in Membrin/GOSR2 Reveal Stringent Secretory Pathway Demands of Dendritic Growth and Synaptic Integrity. Cell Rep. 2017, 21, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Tea, J.S.; Luo, L. The chromatin remodeling factor Bap55 functions through the TIP60 complex to regulate olfactory projection neuron dendrite targeting. Neural Dev. 2011, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Kunduri, G.; Turner-Evans, D.; Konya, Y.; Izumi, Y.; Nagashima, K.; Lockett, S.; Holthuis, J.; Bamba, T.; Acharya, U.; Acharya, J.K. Defective cortex glia plasma membrane structure underlies light-induced epilepsy in cpes mutants. Proc. Natl. Acad. Sci. USA 2018, 115, E8919. [Google Scholar] [CrossRef]
- Tenedini, F.M.; Saez Gonzalez, M.; Hu, C.; Pedersen, L.H.; Petruzzi, M.M.; Spitzweck, B.; Wang, D.; Richter, M.; Petersen, M.; Szpotowicz, E.; et al. Maintenance of cell type-specific connectivity and circuit function requires Tao kinase. Nat. Commun. 2019, 10, 3506. [Google Scholar] [CrossRef]
- Hu, C.; Kanellopoulos, A.; Richter, M.; Petersen, M.; Konietzny, A.; Tenedini, F.M.; Hoyer, N.; Cheng, L.; Poon, C.L.; Harvey, K.F.; et al. Conserved Tao Kinase Activity Regulates Dendritic Arborization, Cytoskeletal Dynamics, and Sensory Function in Drosophila. J. Neurosci. 2020, 40, 1819–1833. [Google Scholar] [CrossRef]
- Hamilton, P.J.; Campbell, N.G.; Sharma, S.; Erreger, K.; Herborg Hansen, F.; Saunders, C.; Belovich, A.N.; NIH ARRA Autism Sequencing Consortium; Sahai, M.A.; Cook, E.H.; et al. De novo mutation in the dopamine transporter gene associates dopamine dysfunction with autism spectrum disorder. Mol. Psychiatry 2013, 18, 1315–1323. [Google Scholar] [CrossRef]
- Campbell, N.G.; Shekar, A.; Aguilar, J.I.; Peng, D.; Navratna, V.; Yang, D.; Morley, A.N.; Duran, A.M.; Galli, G.; O’Grady, B.; et al. Structural, functional, and behavioral insights of dopamine dysfunction revealed by a deletion in SLC6A3. Proc. Natl. Acad. Sci. USA 2019, 116, 3853–3862. [Google Scholar] [CrossRef]
- Zheng, J.C.; Tham, C.T.; Keatings, K.; Fan, S.; Liou, A.Y.-C.; Numata, Y.; Allan, D.; Numata, M. Secretory Carrier Membrane Protein (SCAMP) deficiency influences behavior of adult flies. Front. Cell Dev. Biol. 2014, 2, 64. [Google Scholar] [CrossRef]
- Volders, K.; Scholz, S.; Slabbaert, J.R.; Nagel, A.C.; Verstreken, P.; Creemers, J.W.; Callaerts, P.; Schwärzel, M. Drosophila rugose is a functional homolog of mammalian Neurobeachin and affects synaptic architecture, brain morphology, and associative learning. J. Neurosci. 2012, 32, 15193–15204. [Google Scholar] [CrossRef] [PubMed]
- Wang, V.Y.; Hassan, B.A.; Bellen, H.J.; Zoghbi, H.Y. Drosophila atonal fully rescues the phenotype of Math1 null mice: New functions evolve in new cellular contexts. Curr Biol 2002, 12, 1611–1616. [Google Scholar] [CrossRef]
- Züchner, S.; Mersiyanova, I.V.; Muglia, M.; Bissar-Tadmouri, N.; Rochelle, J.; Dadali, E.L.; Zappia, M.; Nelis, E.; Patitucci, A.; Senderek, J.; et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004, 36, 449–451. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, H.; Yao, C.K.; Chen, K.; Jaiswal, M.; Donti, T.; Lin, Y.Q.; Bayat, V.; Xiong, B.; Zhang, K.; David, G.; et al. Mitochondrial fusion but not fission regulates larval growth and synaptic development through steroid hormone production. eLife 2014, 3, e03558. [Google Scholar] [CrossRef] [PubMed]
- Del Amo, V.L.; Seco-Cervera, M.; García-Giménez, J.L.; Whitworth, A.J.; Pallardó, F.V.; Galindo, M.I. Mitochondrial defects and neuromuscular degeneration caused by altered expression of Drosophila Gdap1: Implications for the Charcot–Marie–Tooth neuropathy. Hum. Mol. Genet. 2014, 24, 21–36. [Google Scholar] [CrossRef]
- Storkebaum, E.; Leitão-Gonçalves, R.; Godenschwege, T.; Nangle, L.; Mejia, M.; Bosmans, I.; Ooms, T.; Jacobs, A.; Van Dijck, P.; Yang, X.L.; et al. Dominant mutations in the tyrosyl-tRNA synthetase gene recapitulate in Drosophila features of human Charcot-Marie-Tooth neuropathy. Proc. Natl. Acad. Sci. USA 2009, 106, 11782–11787. [Google Scholar] [CrossRef]
- Chihara, T.; Luginbuhl, D.; Luo, L. Cytoplasmic and mitochondrial protein translation in axonal and dendritic terminal arborization. Nat. Neurosci. 2007, 10, 828–837. [Google Scholar] [CrossRef]
- Duan, R.; Shi, Y.; Yu, L.; Zhang, G.; Li, J.; Lin, Y.; Guo, J.; Wang, J.; Shen, L.; Jiang, H.; et al. UBA5 Mutations Cause a New Form of Autosomal Recessive Cerebellar Ataxia. PLoS ONE 2016, 11, e0149039. [Google Scholar] [CrossRef]
- Chen, K.; Lin, G.; Haelterman, N.A.; Ho, T.S.-Y.; Li, T.; Li, Z.; DuRaine, L.; Graham, B.H.; Jaiswal, M.; Yamamoto, S.; et al. Loss of Frataxin induces iron toxicity, sphingolipid synthesis, and Pdk1/Mef2 activation, leading to neurodegeneration. eLife 2016, 5, e16043. [Google Scholar] [CrossRef]
- Kim, M.; Sandford, E.; Gatica, D.; Qiu, Y.; Liu, X.; Zheng, Y.; Schulman, B.A.; Xu, J.; Semple, I.; Ro, S.H.; et al. Mutation in ATG5 reduces autophagy and leads to ataxia with developmental delay. eLife 2016, 5, e12245. [Google Scholar] [CrossRef]
- Li, C.; Brazill, J.M.; Liu, S.; Bello, C.; Zhu, Y.; Morimoto, M.; Cascio, L.; Pauly, R.; Diaz-Perez, Z.; Malicdan, M.C.V.; et al. Spermine synthase deficiency causes lysosomal dysfunction and oxidative stress in models of Snyder-Robinson syndrome. Nat. Commun. 2017, 8, 1257. [Google Scholar] [CrossRef] [PubMed]
- Leiserson, W.M.; Forbush, B.; Keshishian, H. Drosophila glia use a conserved cotransporter mechanism to regulate extracellular volume. Glia 2010, 59, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Moussian, B.; Schaeffeler, E.; Schwab, M.; Nies, A.T. The fruit fly Drosophila melanogaster as an innovative preclinical ADME model for solute carrier membrane transporters, with consequences for pharmacology and drug therapy. Drug Discov. Today 2018, 23, 1746–1760. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Jaiswal, M.; Charng, W.L.; Gambin, T.; Karaca, E.; Mirzaa, G.; Wiszniewski, W.; Sandoval, H.; Haelterman, N.A.; Xiong, B.; et al. A Drosophila genetic resource of mutants to study mechanisms underlying human genetic diseases. Cell 2014, 159, 200–214. [Google Scholar] [CrossRef]
- Link, N.; Chung, H.; Jolly, A.; Withers, M.; Tepe, B.; Arenkiel, B.R.; Shah, P.S.; Krogan, N.J.; Aydin, H.; Geckinli, B.B.; et al. Mutations in ANKLE2, a ZIKA Virus Target, Disrupt an Asymmetric Cell Division Pathway in Drosophila Neuroblasts to Cause Microcephaly. Dev. Cell 2019, 51, 713–729.e6. [Google Scholar] [CrossRef]
- Curtin, K.D.; Meinertzhagen, I.A.; Wyman, R.J. Basigin (EMMPRIN/CD147) interacts with integrin to affect cellular architecture. J. Cell Sci. 2005, 118, 2649–2660. [Google Scholar] [CrossRef]
- Huang, Y.; Mao, X.; van Jaarsveld, R.; Shu, L.; Terhal, P.A.; Jia, Z.; Xi, H.; Peng, Y.; Yan, H.; Yuan, S.; et al. Variants in CAPZA2, a member of an F-actin capping complex, cause intellectual disability and developmental delay. Hum. Mol. Genet. 2020, 29, 1537–1546. [Google Scholar] [CrossRef]
- Kelly, S.M.; Leung, S.W.; Pak, C.; Banerjee, A.; Moberg, K.H.; Corbett, A.H. A conserved role for the zinc finger polyadenosine RNA binding protein, ZC3H14, in control of poly(A) tail length. RNA 2014, 20, 681–688. [Google Scholar] [CrossRef]
- Nahm, M.; Lee, M.-J.; Parkinson, W.; Lee, M.; Kim, H.; Kim, Y.-J.; Kim, S.; Cho, Y.S.; Min, B.-M.; Bae, Y.C.; et al. Spartin regulates synaptic growth and neuronal survival by inhibiting BMP-mediated microtubule stabilization. Neuron 2013, 77, 680–695. [Google Scholar] [CrossRef]
- Kim, S.; Kim, J.; Park, S.; Park, J.J.; Lee, S. Drosophila Graf regulates mushroom body β-axon extension and olfactory long-term memory. Mol. Brain 2021, 14, 73. [Google Scholar] [CrossRef]
- Malik, B.R.; Gillespie, J.M.; Hodge, J.J. CASK and CaMKII function in the mushroom body α’/β’ neurons during Drosophila memory formation. Front. Neural Circuits 2013, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Huang, Y.; Hu, W.; Huang, S.; Wang, Q.; Han, J.; Zhang, Y.Q. dAcsl, the Drosophila ortholog of acyl-CoA synthetase long-chain family member 3 and 4, inhibits synapse growth by attenuating bone morphogenetic protein signaling via endocytic recycling. J. Neurosci. 2014, 34, 2785–2796. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wang, Q.; Gong, N.N.; Kurolap, A.; Feldman, H.B.; Boy, N.; Brugger, M.; Grand, K.; McWalter, K.; Sacoto, M.J.; et al. Pathogenic variants in SMARCA5, a chromatin remodeler, cause a range of syndromic neurodevelopmental features. Sci. Adv. 2021, 7, eabf2066. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.T.; Davids, M.; Burke, E.; Pappas, J.G.; Rosenfeld, J.A.; McCarty, A.J.; Davis, T.; Wolfe, L.; Toro, C.; Tifft, C.; et al. A Syndromic Neurodevelopmental Disorder Caused by De Novo Variants in EBF3. Am. J. Hum. Genet. 2017, 100, 128–137. [Google Scholar] [CrossRef]
- Farhan, S.; Nixon, K.; Everest, M.; Edwards, T.N.; Long, S.; Segal, D.; Knip, M.J.; Arts, H.H.; Chakrabarti, R.; Wang, J.; et al. Identification of a novel synaptic protein, TMTC3, involved in periventricular nodular heterotopia with intellectual disability and epilepsy. Hum. Mol. Genet. 2017, 26, 4278–4289. [Google Scholar] [CrossRef]
- Ansar, M.; Chung, H.L.; Al-Otaibi, A.; Elagabani, M.N.; Ravenscroft, T.A.; Paracha, S.A.; Scholz, R.; Abdel Magid, T.; Sarwar, M.T.; Shah, S.F.; et al. Bi-allelic Variants in IQSEC1 Cause Intellectual Disability, Developmental Delay, and Short Stature. Am. J. Hum. Genet. 2019, 105, 907–920. [Google Scholar] [CrossRef]
- Yap, Z.Y.; Strucinska, K.; Matsuzaki, S.; Lee, S.; Si, Y.; Humphries, K.; Tarnopolsky, M.A.; Yoon, W.H. A biallelic pathogenic variant in the OGDH gene results in a neurological disorder with features of a mitochondrial disease. J. Inherit. Metab. Dis. 2020, 44, 388–400. [Google Scholar] [CrossRef]
- Chao, Y.-H.; Robak, L.A.; Xia, F.; Koenig, M.K.; Adesina, A.; Bacino, C.A.; Scaglia, F.; Bellen, H.; Wangler, M.F. Missense variants in the middle domain of DNM1L in cases of infantile encephalopathy alter peroxisomes and mitochondria when assayed in Drosophila. Hum. Mol. Genet. 2016, 25, 1846–1856. [Google Scholar] [CrossRef]
- Shao, L.; Shuai, Y.; Wang, J.; Feng, S.; Lu, B.; Li, Z.; Zhao, Y.; Wang, L.; Zhong, Y. Schizophrenia susceptibility gene dysbindin regulates glutamatergic and dopaminergic functions via distinctive mechanisms in Drosophila. Proc. Natl. Acad. Sci. USA 2011, 108, 18831–18836. [Google Scholar] [CrossRef]
- Tamberg, L.; Sepp, M.; Timmusk, T.; Palgi, M. Introducing Pitt-Hopkins syndrome-associated mutations of TCF4 to Drosophila daughterless. Biol. Open 2015, 4, 1762–1771. [Google Scholar] [CrossRef]
- Gavilan, H.S.; Kulikauskas, R.M.; Gutmann, D.H.; Fehon, R.G. In vivo functional analysis of the human NF2 tumor suppressor gene in Drosophila. PLoS ONE 2014, 9, e90853. [Google Scholar] [CrossRef] [PubMed]
- Bodmer, R. Heart development in Drosophila and its relationship to vertebrates. Trends Cardiovasc. Med. 1995, 5, 21–28. [Google Scholar] [CrossRef]
- Bodmer, R.; Venkatesh, T.V. Heart development in Drosophila and vertebrates: Conservation of molecular mechanisms. Dev. Genet. 1998, 22, 181–186. [Google Scholar] [CrossRef]
- Ahmad, S.M. Conserved signaling mechanisms in Drosophila heart development. Dev. Dyn. 2017, 246, 641–656. [Google Scholar] [CrossRef]
- Souidi, A.; Jagla, K. Drosophila Heart as a Model for Cardiac Development and Diseases. Cells 2021, 10, 3078. Available online: https://www.mdpi.com/2073-4409/10/11/3078 (accessed on 13 January 2022). [CrossRef] [PubMed]
- Jay, P.Y.; Harris, B.S.; Maguire, C.T.; Buerger, A.; Wakimoto, H.; Tanaka, M.; Kupershmidt, S.; Roden, D.M.; Schultheiss, T.M.; O’Brien, T.X.; et al. Nkx2-5 mutation causes anatomic hypoplasia of the cardiac conduction system. J. Clin. Investig. 2004, 113, 1130–1137. [Google Scholar] [CrossRef]
- Moskowitz, I.P.; Kim, J.B.; Moore, M.L.; Wolf, C.M.; Peterson, M.A.; Shendure, J.; Nobrega, M.A.; Yokota, Y.; Berul, C.; Izumo, S.; et al. A Molecular Pathway Including Id2, Tbx5, and Nkx2-5 Required for Cardiac Conduction System Development. Cell 2007, 129, 1365–1376. [Google Scholar] [CrossRef]
- Qian, L.; Mohapatra, B.; Akasaka, T.; Liu, J.; Ocorr, K.; Towbin, J.A.; Bodmer, R. Transcription factor neuromancer/TBX20 is required for cardiac function in Drosophila with implications for human heart disease. Proc. Natl. Acad. Sci. USA 2008, 105, 19833–19838. [Google Scholar] [CrossRef]
- Qian, L.; Bodmer, R. Partial loss of GATA factor Pannier impairs adult heart function in Drosophila. Hum. Mol. Genet. 2009, 18, 3153–3163. [Google Scholar] [CrossRef]
- Qian, L.; Wythe, J.D.; Liu, J.; Cartry, J.; Vogler, G.; Mohapatra, B.; Otway, R.T.; Huang, Y.; King, I.N.; Maillet, M.; et al. Tinman/Nkx2-5 acts via miR-1 and upstream of Cdc42 to regulate heart function across species. J. Cell Biol. 2011, 193, 1181–1196. [Google Scholar] [CrossRef]
- Taghli-Lamallem, O.; Auxerre-Plantié, E.; Jagla, K. Drosophila in the heart of understanding cardiac diseases: Modeling channelopathies and cardiomyopathies in the fruitfly. J. Cardiovasc. Dev. Dis. 2016, 3, 7. [Google Scholar] [CrossRef] [PubMed]
- Reiter, L.T.; Potocki, L.; Chien, S.; Gribskov, M.; Bier, E. A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Res. 2001, 11, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, M.S.; Smith, A.G.C.; Sable, C.A.; Echko, M.M.; Wilner, L.B.; Olsen, H.E.; Atalay, H.T.; Awasthi, A.; Bhutta, Z.A.; Boucher, J.L.; et al. Global, regional, and national burden of congenital heart disease, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Child Adolesc. Health 2020, 4, 185–200. [Google Scholar] [CrossRef]
- Pediatric Cardiac Genomics, C.; Gelb, B.; Brueckner, M.; Chung, W.; Goldmuntz, E.; Kaltman, J.; Kaski, J.P.; Kim, R.; Kline, J.; Mercer-Rosa, L.; et al. The congenital heart disease genetic network study: Rationale, design, and early results. Circ Res. 2013, 112, 698–706. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.Y.; Fu, Y.; Nettleton, M.; Richman, A.; Han, Z. High throughput in vivo functional validation of candidate congenital heart disease genes in Drosophila. eLife 2017, 6, e22617. [Google Scholar] [CrossRef]
- McKenna, W.J.; Maron, B.J.; Thiene, G. Classification, epidemiology, and global burden of cardiomyopathies. Circ. Res. 2017, 121, 722–730. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Hedges, D.J.; Morales, A. Dilated cardiomyopathy: The complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 2013, 10, 531–547. [Google Scholar] [CrossRef]
- Jordan, E.; Peterson, L.; Ai, T.; Asatryan, B.; Bronicki, L.; Brown, E.; Celeghin, R.; Edwards, M.; Fan, J.; Ingles, J.; et al. Evidence-Based Assessment of Genes in Dilated Cardiomyopathy. Circulation 2021, 144, 7–19. [Google Scholar] [CrossRef]
- Semsarian, C.; Ingles, J.; Maron, M.S.; Maron, B.J. New perspectives on the prevalence of hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2015, 65, 1249–1254. [Google Scholar] [CrossRef]
- Geske, J.B.; Ommen, S.R.; Gersh, B.J. Hypertrophic Cardiomyopathy: Clinical Update. JACC Heart Fail. 2018, 6, 364–375. [Google Scholar] [CrossRef]
- Ingles, J.; Goldstein, J.; Thaxton, C.; Caleshu, C.; Corty, E.W.; Crowley, S.B.; Dougherty, K.; Harrison, S.M.; McGlaughon, J.; Milko, L.V.; et al. Evaluating the Clinical Validity of Hypertrophic Cardiomyopathy Genes. Circ. Genom. Precis. Med. 2019, 12, e002460. [Google Scholar] [CrossRef] [PubMed]
- Musunuru, K.; Hershberger, R.E.; Day, S.M.; Klinedinst, N.J.; Landstrom, A.P.; Parikh, V.N.; Prakash, S.; Semsarian, C.; Sturm, A.C.; American Heart Association Council on Genomic and Precision Medicine; et al. Genetic testing for inherited cardiovascular diseases: A scientific statement from the american heart association. Circ. Genom. Precis. Med. 2020, 13, e000067. [Google Scholar] [CrossRef] [PubMed]
- Manivannan, S.N.; Darouich, S.; Masmoudi, A.; Gordon, D.; Zender, G.; Han, Z.; Fitzgerald-Butt, S.; White, P.; McBride, K.L.; Kharrat, M.; et al. Novel frameshift variant in MYL2 reveals molecular differences between dominant and recessive forms of hypertrophic cardiomyopathy. PLoS Genet. 2020, 16, e1008639. [Google Scholar] [CrossRef]
- Moore, J.R.; Dickinson, M.H.; Vigoreaux, J.O.; Maughan, D.W. The effect of removing the N-terminal extension of the Drosophila myosin regulatory light chain upon flight ability and the contractile dynamics of indirect flight muscle. Biophys. J. 2000, 78, 1431–1440. [Google Scholar] [CrossRef]
- Campuzano, V.; Montermini, L.; Molto, M.D.; Pianese, L.; Cossee, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef]
- Weidemann, F.; Liu, D.; Hu, K.; Florescu, C.; Niemann, M.; Herrmann, S.; Kramer, B.; Klebe, S.; Doppler, K.; Uceyler, N.; et al. The cardiomyopathy in Friedreich’s ataxia—New biomarker for staging cardiac involvement. Int. J. Cardiol. 2015, 194, 50–57. [Google Scholar] [CrossRef]
- Tricoire, H.; Palandri, A.; Bourdais, A.; Camadro, J.-M.; Monnier, V. Methylene blue rescues heart defects in a Drosophila model of friedreich’s ataxia. Hum. Mol. Genet. 2014, 23, 968–979. [Google Scholar] [CrossRef]
- Gonçalves, S.; Patat, J.; Guida, M.C.; Lachaussee, N.; Arrondel, C.; Helmstädter, M.; Boyer, O.; Gribouval, O.; Gubler, M.C.; Mollet, G.; et al. A homozygous KAT2B variant modulates the clinical phenotype of ADD3 deficiency in humans and flies. PLoS Genet. 2018, 14, e1007386. [Google Scholar] [CrossRef] [PubMed]
- Carré, C.; Szymczak, D.; Pidoux, J.; Antoniewski, C. The histone H3 acetylase dGcn5 is a key player in Drosophila melanogaster metamorphosis. Mol. Cell. Biol. 2005, 25, 8228–8238. [Google Scholar] [CrossRef]
- Casas-Tintó, S.; Arnés, M.; Ferrús, A. Drosophila enhancer-Gal4 lines show ectopic expression during development. R. Soc. Open Sci. 2017, 4, 170039. [Google Scholar] [CrossRef]
- Ocorr, K.; Reeves, N.L.; Wessells, R.J.; Fink, M.; Chen, H.S.; Akasaka, T.; Yasuda, S.; Metzger, J.M.; Giles, W.; Posakony, J.W.; et al. KCNQ potassium channel mutations cause cardiac arrhythmias in Drosophila that mimic the effects of aging. Proc. Natl. Acad. Sci. USA 2007, 104, 3943–3948. [Google Scholar] [CrossRef]
- Zhang, D.; Ke, L.; Mackovicova, K.; Van Der Want, J.J.; Sibon, O.C.; Tanguay, R.M.; Morrow, G.; Henning, R.H.; Kampinga, H.H.; Brundel, B.J. Effects of different small HSPB members on contractile dysfunction and structural changes in a Drosophila melanogaster model for Atrial Fibrillation. J. Mol. Cell. Cardiol. 2011, 51, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Santalla, M.; Valverde, C.A.; Harnichar, E.; Lacunza, E.; Aguilar-Fuentes, J.; Mattiazzi, A.; Ferrero, P. Aging and CaMKII alter intracellular Ca2+ transients and heart rhythm in Drosophila melanogaster. PLoS ONE. 2014, 9, e101871. [Google Scholar] [CrossRef] [PubMed]
- Limpitikul, W.B.; Viswanathan, M.C.; O’Rourke, B.; Yue, D.T.; Cammarato, A. Conservation of cardiac L-type Ca2+ channels and their regulation in Drosophila: A novel genetically-pliable channelopathic model. J. Mol. Cell. Cardiol. 2018, 119, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Pineda, S.; Nikolova-Krstevski, V.; Leimena, C.; Atkinson, A.J.; Altekoester, A.K.; Cox, C.D.; Jacoby, A.; Huttner, I.G.; Ju, Y.K.; Soka, M.; et al. Conserved Role of the Large Conductance Calcium-Activated Potassium Channel, KCa1.1, in Sinus Node Function and Arrhythmia Risk. Circ. Genom. Precis Med. 2021, 14, e003144. [Google Scholar] [CrossRef]
- Yu, L.; Daniels, J.; Glaser, A.E.; Wolf, M.J. Raf-mediated cardiac hypertrophy in adult Drosophila. DMM Dis. Model. Mech. 2013, 6, 964–976. [Google Scholar]
- Yu, L.; Daniels, J.P.; Wu, H.; Wolf, M.J. Cardiac hypertrophy induced by active Raf depends on Yorkie-mediated transcription. Sci. Signal. 2015, 8, ra13. [Google Scholar] [CrossRef]
- Migunova, E.; Theophilopoulos, J.; Mercadante, M.; Men, J.; Zhou, C.; Dubrovsky, E.B. ELAC2/RNaseZ-linked cardiac hypertrophy in Drosophila melanogaster. DMM Dis. Model. Mech. 2021, 14, dmm048931. [Google Scholar] [CrossRef]
- Bloemink, M.J.; Melkani, G.C.; Dambacher, C.M.; Bernstein, S.I.; Geeves, M.A. Two Drosophila myosin transducer mutants with distinct cardiomyopathies have divergent ADP and actin affinities. J. Biol. Chem. 2011, 286, 28435–28443. [Google Scholar] [CrossRef]
- Achal, M.; Trujillo, A.S.; Melkani, G.C.; Farman, G.P.; Ocorr, K.; Viswanathan, M.C.; Kaushik, G.; Newhard, C.S.; Glasheen, B.M.; Melkani, A.; et al. A Restrictive Cardiomyopathy Mutation in an Invariant Proline at the Myosin Head/Rod Junction Enhances Head Flexibility and Function, Yielding Muscle Defects in Drosophila. J. Mol. Biol. 2016, 428, 2446–2461. [Google Scholar] [CrossRef]
- Taghli-Lamallem, O.; Akasaka, T.; Hogg, G.; Nudel, U.; Yaffe, D.; Chamberlain, J.S.; Ocorr, K.; Bodmer, R. Dystrophin deficiency in Drosophila reduces lifespan and causes a dilated cardiomyopathy phenotype. Aging Cell 2008, 7, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.M.; Wolf, M.J. Serial examination of an inducible and reversible dilated cardiomyopathy in individual adult Drosophila. PLoS ONE 2009, 4, e7132. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, M.C.; Kaushik, G.; Engler, A.J.; Lehman, W.; Cammarato, A. A Drosophila melanogaster model of diastolic dysfunction and cardiomyopathy based on impaired troponin-T function. Circ Res. 2014, 114, e6–e17. [Google Scholar]
- Selma-Soriano, E.; Casillas-Serra, C.; Artero, R.; Llamusi, B.; Navarro, J.A.; Redón, J. Rabphilin silencing causes dilated cardiomyopathy in a Drosophila model of nephrocyte damage. Sci. Rep. 2021, 11, 15287. [Google Scholar] [CrossRef]
- Allikian, M.J.; Bhabha, G.; Dospoy, P.; Heydemann, A.; Ryder, P.; Earley, J.U.; Wolf, M.J.; Rockman, H.A.; McNally, E.M. Reduced life span with heart and muscle dysfunction in Drosophila sarcoglycan mutants. Hum. Mol. Genet. 2007, 16, 2933–2943. [Google Scholar] [CrossRef]
- Taghli-Lamallem, O.; Jagla, K.; Chamberlain, J.S.; Bodmer, R. Mechanical and non-mechanical functions of Dystrophin can prevent cardiac abnormalities in Drosophila. Exp. Gerontol. 2014, 49, 26–34. [Google Scholar] [CrossRef]
- Gao, Q.Q.; Wyatt, E.; Goldstein, J.A.; LoPresti, P.; Castillo, L.M.; Gazda, A.; Petrossian, N.; Earley, J.U.; Hadhazy, M.; Barefield, D.Y.; et al. Reengineering a transmembrane protein to treat muscular dystrophy using exon skipping. J. Clin. Investig. 2015, 125, 4186–4195. [Google Scholar] [CrossRef]
- Tang, M.; Yuan, W.; Fan, X.; Liu, M.; Bodmer, R.; Ocorr, K.; Wu, X. Pygopus maintains heart function in aging Drosophila independently of canonical Wnt signaling. Circ. Cardiovasc. Genet. 2013, 6, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Yuan, W.; Bodmer, R.; Wu, X.; Ocorr, K. The role of pygopus in the differentiation of intracardiac valves in Drosophila. Genesis 2014, 52, 19–28. [Google Scholar] [CrossRef]
- Kramps, T.; Peter, O.; Brunner, E.; Nellen, D.; Froesch, B.; Chatterjee, S.; Murone, M.; Zullig, S.; Basler, K. Wnt/wingless signaling requires BCL9/legless-mediated recruitment of pygopus to the nuclear beta-catenin-TCF complex. Cell 2002, 109, 47–60. [Google Scholar] [CrossRef]
- Thomson, K.L.; Ormondroyd, E.; Harper, A.R.; Dent, T.; McGuire, K.; Baksi, J.; Blair, E.; Brennan, P.; Buchan, R.; Bueser, T.; et al. Analysis of 51 proposed hypertrophic cardiomyopathy genes from genome sequencing data in sarcomere negative cases has negligible diagnostic yield. Genet. Med. 2019, 21, 1576–1584. [Google Scholar] [CrossRef] [PubMed]
- Micheu, M.M.; Popa-Fotea, N.M.; Oprescu, N.; Dorobantu, M.; Ratiu, A.C.; Ecovoiu, A.A. NGS data validated by Sanger sequencing reveal a puzzling small deletion of MYBPC3 gene associated with hypertrophic cardiomyopathy. Rom. Biotechnol. Lett. 2019, 24, 91–99. [Google Scholar] [CrossRef]
- Micheu, M.M.; Popa-Fotea, N.M.; Oprescu, N.; Bogdan, S.; Dan, M.; Deaconu, A.; Dorobantu, L.; Gheorghe-Fronea, O.; Greavu, M.; Iorgulescu, C.; et al. Yield of Rare Variants Detected by Targeted Next-Generation Sequencing in a Cohort of Romanian Index Patients with Hypertrophic Cardiomyopathy. Diagnostics 2020, 10, 1061. [Google Scholar] [CrossRef] [PubMed]
- Alimohamed, M.Z.; Johansson, L.F.; Posafalvi, A.; Boven, L.G.; van Dijk, K.K.; Walters, L.; Vos, Y.J.; Westers, H.; Hoedemaekers, Y.M.; Sinke, R.J.; et al. Diagnostic yield of targeted next generation sequencing in 2002 Dutch cardiomyopathy patients. Int. J. Cardiol. 2021, 332, 90–104. [Google Scholar] [CrossRef]
- Kim, A.R.; Choi, K.W. TRiC/CCT chaperonins are essential for organ growth by interacting with insulin/TOR signaling in Drosophila. Oncogene 2019, 38, 4739–4754. [Google Scholar] [CrossRef]
- Jung, W.H.; Liu, C.C.; Yu, Y.L.; Chang, Y.C.; Lien, W.Y.; Chao, H.C.; Huang, S.Y.; Kuo, C.H.; Ho, H.C.; Chan, C.C. Lipophagy prevents activity-dependent neurodegeneration due to dihydroceramide accumulation in vivo. EMBO Rep. 2017, 18, 1150–1165. [Google Scholar] [CrossRef] [PubMed]
- Muyrers-Chen, I.; Rozovskaia, T.; Lee, N.; Kersey, J.H.; Nakamura, T.; Canaani, E.; Paro, R. Expression of leukemic MLL fusion proteins in Drosophila affects cell cycle control and chromosome morphology. Oncogene 2004, 23, 8639–8648. [Google Scholar] [CrossRef] [PubMed]
- Perkins, L.A.; Johnson, M.R.; Melnick, M.B.; Perrimon, N. The nonreceptor protein tyrosine phosphatase corkscrew functions in multiple receptor tyrosine kinase pathways in Drosophila. Dev. Biol. 1996, 180, 63–81. [Google Scholar] [CrossRef]
- Tan, K.L.; Haelterman, N.A.; Kwartler, C.S.; Regalado, E.S.; Lee, P.T.; Nagarkar-Jaiswal, S.; Guo, D.C.; Duraine, L.; Wangler, M.F.; University of Washington Center for Mendelian Genomics; et al. Ari-1 regulates myonuclear organization together with parkin and is associated with aortic aneurysms. Dev. Cell 2018, 45, 226–244. [Google Scholar] [CrossRef]
- Mirzoyan, Z.; Sollazzo, M.; Allocca, M.; Valenza, A.M.; Grifoni, D.; Bellosta, P. Drosophila melanogaster: A Model Organism to Study Cancer. Front. Genet. 2019, 10, 51. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Lechel, A.; Gunes, C. Telomerase: The devil inside. Genes 2016, 7, 43. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the intersections between metabolism and cancer biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Millburn, G.H.; Crosby, M.A.; Gramates, L.S.; Tweedie, S.; FlyBase, C. FlyBase portals to human disease re-search using Drosophila models. Dis. Model. Mech. 2016, 9, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Grifoni, D.; Garoia, F.; Schimanski, C.C.; Schmitz, G.; Laurenti, E.; Galle, P.R.; Pession, A.; Cavicchi, S.; Strand, D. The human protein Hugl-1 substitutes for Drosophila Lethal giant larvae tumour suppressor function in vivo. Oncogene 2004, 23, 8688–8694. [Google Scholar] [CrossRef]
- Tao, W.; Zhang, S.; Turenchalk, G.S.; Stewart, R.A.; St John, M.A.; Chen, W.; Xu, T. Human homologue of the Drosophila melanogaster lats tumour suppressor modulates CDC2 activity. Nat. Genet. 1999, 21, 177–181. [Google Scholar] [CrossRef]
- Dow, L.E.; Brumby, A.M.; Muratore, R.; Coombe, M.L.; Sedelies, K.A.; Trapani, J.A.; Russell, S.M.; Richardson, H.E.; Humbert, P.O. hScrib is a functional homologue of the Drosophila tumour suppressor Scribble. Oncogene 2003, 22, 9225–9230. [Google Scholar] [CrossRef]
- Benchabane, H.; Xin, N.; Tian, A.; Hafler, B.P.; Nguyen, K.; Ahmed, A.; Ahmed, Y. Jerky/Earthbound facilitates cell-specific Wnt/Wingless signalling by modulating β-catenin-TCF activity. EMBO J. 2011, 30, 1444–1458. [Google Scholar] [CrossRef]
- Drusenheimer, N.; Migdal, B.; Jäckel, S.; Tveriakhina, L.; Scheider, K.; Schulz, K.; Gröper, J.; Köhrer, K.; Klein, T. The Mam-malian Orthologs of Drosophila Lgd, CC2D1A and CC2D1B, Function in the Endocytic Pathway, but Their Individual Loss of Function Does Not Affect Notch Signalling. PLoS Genet. 2015, 11, e1005749. [Google Scholar] [CrossRef]
- D’Brot, A.; Kurtz, P.; Regan, E.; Jakubowski, B.; Abrams, J.M. A platform for interrogating cancer-associated p53 alleles. Oncogene 2016, 36, 286–291. [Google Scholar] [CrossRef]
- Bras, S.; Martin-Lanneree, S.; Gobert, V.; Auge, B.; Breig, O.; Sanial, M.; Yamaguchi, M.; Haenlin, M.; Plessis, A.; Waltzer, L. Myeloid leukemia factor is a conserved regulator of RUNX transcription factor activity involved in hematopoiesis. Proc. Natl. Acad. Sci. USA 2012, 109, 4986–4991. [Google Scholar] [CrossRef] [PubMed]
- Ludlow, C.; Choy, R.; Blochlinger, K. Functional analysis of Drosophila and mammalian cut proteins in files. Dev. Biol. 1996, 178, 149–159. [Google Scholar] [CrossRef]
- Brumby, A.M.; Richardson, H.E. scribble mutants cooperate with oncogenic Ras or Notch to cause neo-plastic overgrowth in Drosophila. EMBO J. 2003, 22, 5769–5779. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Luo, L. Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron 1999, 22, 451–461. [Google Scholar] [CrossRef]
- Pagliarini, R.A.; Xu, T. A genetic screen in Drosophila for metastatic behavior. Science 2003, 302, 1227–1231. [Google Scholar] [CrossRef] [PubMed]
- Tipping, M.; Perrimon, N. Drosophila as a model for context-dependent tumorigenesis. J. Cell. Physiol. 2014, 229, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhou, B.P. Inflammation: A driving force speeds cancer metastasis. Cell Cycle 2009, 8, 3267–3273. [Google Scholar] [CrossRef]
- Grzeschik, N.A.; Parsons, L.M.; Richardson, H.E. Lgl, the SWH pathway and tumorigenesis: It’s a matter of context & competition! Cell Cycle 2010, 9, 3222–3232. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Woodhouse, E.; Hersperger, E.; Shearn, A. Growth, metastasis, and invasiveness of Drosophila tumors caused by mutations in specific tumor suppressor genes. Dev. Genes Evol. 1998, 207, 542–550. [Google Scholar] [CrossRef]
- Woodhouse, E.; Hersperger, E.; Stetler-Stevenson, W.G.; Liotta, L.A.; Shearn, A. Increased type IV collagenase in lgl-induced invasive tumors of Drosophila. Cell Growth Differ. 1994, 5, 151–159. [Google Scholar] [PubMed]
- Xu, J.; Liu, L.Z.; Deng, X.F.; Timmons, L.; Hersperger, E.; Steeg, P.S.; Veron, M.; Shearn, A. The Enzymatic Activity of Drosophila AWD/NDP Kinase Is Necessary but Not Suffi-cient for Its Biological Function. Dev. Biol. 1996, 177, 544–557. [Google Scholar] [CrossRef] [PubMed]
- Gateff, E. Malignant neoplasms of genetic origin in Drosophila melanogaster. Science 1978, 200, 1448–1459. [Google Scholar] [CrossRef]
- Stoker, M.G.; Shearer, M.; O’Neill, C. Growth inhibition of polyoma-transformed cells by contact with static normal fibroblasts. J. Cell Sci. 1966, 1, 297–310. [Google Scholar] [CrossRef]
- Baker, N.E.; Li, W. Cell competition and its possible relation to cancer. Cancer Res. 2008, 68, 5505–5507. [Google Scholar] [CrossRef] [PubMed]
- Moreno, E. Is cell competition relevant to cancer? Nat. Rev. Cancer 2008, 8, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.-P.; Fletcher, A.G.; Baena-Lopez, L.A. Mechanisms and mechanics of cell competition in epithelia. Nat. Rev. Mol. Cell Biol. 2013, 14, 581–591. [Google Scholar] [CrossRef]
- Vincent, J.-P.; Kolahgar, G.; Gagliardi, M.; Piddini, E. Steep differences in wingless signaling trigger Myc-independent competitive cell interactions. Dev. Cell 2011, 21, 366–374. [Google Scholar] [CrossRef]
- McCartney, B.M.; Price, M.H.; Webb, R.L.; Hayden, M.A.; Holot, L.M.; Zhou, M.; Bejsovec, A.; Peifer, M. Testing hypotheses for the functions of APC family proteins using null and truncation alleles in Drosophila. Development 2006, 133, 2407–2418. [Google Scholar] [CrossRef]
- Jiang, H.; Grenley, M.O.; Bravo, M.-J.; Blumhagen, R.Z.; Edgar, B.A. EGFR/Ras/MAPK signaling mediates adult midgut epithelial homeostasis and regeneration in Drosophila. Cell Stem Cell 2011, 8, 84–95. [Google Scholar] [CrossRef]
- Jiang, H.; Edgar, B.A. Intestinal stem cell function in Drosophila and mice. Curr. Opin. Genet. Dev. 2012, 22, 354–360. [Google Scholar] [CrossRef]
- Patel, P.H.; Edgar, B.A. Tissue design: How Drosophila tumors remodel their neighborhood. Semin. Cell Dev. Biol. 2014, 28, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Cordero, J.; Vidal, M.; Sansom, O. APC as a master regulator of intestinal homeostasis and transformation: From flies to vertebrates. Cell Cycle 2009, 8, 2926–2931. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.C.; Beebe, K.; Sudmeier, L.; Micchelli, C.A. Adenomatous polyposis coli regulates Drosophila intestinal stem cell proliferation. Development 2009, 136, 2255–2264. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Waltzer, L.; Bienz, M. A new Drosophila APC homologue associated with adhesive zones of epithelial cells. Nat. Cell Biol. 1999, 1, 144–151. [Google Scholar] [CrossRef]
- Tian, A.; Benchabane, H.; Wang, Z.; Zimmerman, C.; Xin, N.; Perochon, J.; Kalna, G.; Sansom, O.J.; Cheng, C.; Cordero, J.; et al. Intestinal stem cell overproliferation resulting from inactivation of the APC tumor suppressor requires the transcription cofactors Earthbound and Erect wing. PLoS Genet. 2017, 13, e1006870. [Google Scholar] [CrossRef]
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009, 457, 608–611. [Google Scholar] [CrossRef]
- Benchabane, H.; Hughes, E.G.; Takacs, C.M.; Baird, J.R.; Ahmed, Y. Adenomatous polyposis coli is present near the minimal level required for accurate graded responses to the Wingless morphogen. Development 2008, 135, 963–971. [Google Scholar] [CrossRef]
- Ahmed, Y.; Hayashi, S.; Levine, A.; Wieschaus, E. Regulation of armadillo by a Drosophila APC inhibits neuronal apoptosis during retinal development. Cell 1998, 93, 1171–1182. [Google Scholar] [CrossRef]
- Xin, N.; Benchabane, H.; Tian, A.; Nguyen, K.; Klofas, L.; Ahmed, Y. Erect Wing facilitates context-dependent Wnt/Wingless signaling by recruiting the cell-specific Armadillo-TCF adaptor Earthbound to chromatin. Development 2011, 138, 4955–4967. [Google Scholar] [CrossRef]
- Söderholm, S.; Cantù, C. The WNT/β-catenin dependent transcription: A tissue-specific business. WIREs Syst. Biol. Med. 2020, 13, e1511. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Lai, L.C.; Cheng, H.C.; Chen, K.R.; Syue, Y.Z.; Lu, H.C.; Lin, W.Y.; Chen, S.H.; Huang, H.S.; Shiau, A.L.; et al. TBK1-associated protein in endolysosomes (TAPE) is an innate immune regulator modulating the TLR3 and TLR4 signaling pathways. J. Biol. Chem. 2011, 286, 7043–7051. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.R.; Chang, C.H.; Huang, C.Y.; Lin, C.Y.; Lin, W.Y.; Lo, Y.C.; Yang, C.Y.; Hsing, E.W.; Chen, L.F.; Shih, S.R.; et al. TBK1-associated protein in endolysosomes (TAPE)/CC2D1A is a key regulator linking RIG-I-like receptors to antiviral immunity. J. Biol. Chem. 2012, 287, 32216–32221. [Google Scholar] [CrossRef] [PubMed]
- Rothenberg, E.V.; Taghon, T. Molecular genetics of T cell development. Annu. Rev. Immunol. 2005, 23, 601–649. [Google Scholar] [CrossRef] [PubMed]
- Schweisguth, F. Regulation of notch signaling activity. Curr. Biol. 2004, 14, R129–R138. [Google Scholar] [CrossRef]
- Mumm, J.S.; Schroeter, E.H.; Saxena, M.T.; Griesemer, A.; Tian, X.; Pan, D.J.; Ray, W.J.; Kopan, R. A ligand-induced extracellular cleavage regulates gamma-secretase-like proteolytic activation of Notch1. Mol. Cell 2000, 5, 197–206. [Google Scholar] [CrossRef]
- Bryant, P.J.; Schubiger, G. Giant and duplicated imaginal discs in a new lethal mutant of Drosophila melanogaster. Dev. Biol. 1971, 24, 233–263. [Google Scholar] [CrossRef]
- Jaekel, R.; Klein, T. The Drosophila Notch inhibitor and tumor suppressor gene lethal (2) giant discs encodes a conserved regulator of endosomal trafficking. Dev. Cell 2006, 11, 655–669. [Google Scholar] [CrossRef]
- Klein, T. The tumour suppressor gene l(2)giant discs is required to restrict the activity of Notch to the dorsoventral boundary during Drosophila wing development. Dev. Biol. 2003, 255, 313–333. [Google Scholar] [CrossRef]
- Parr, C.; Watkins, G.; Jiang, W.G. The possible correlation of Notch-1 and Notch-2 with clinical outcome and tumour clinicopathological parameters in human breast cancer. Int. J. Mol. Med. 2004, 14, 779–786. [Google Scholar] [CrossRef]
- Jin, M.M.; Ye, Y.Z.; Qian, Z.D.; Zhang, Y.B. Notch signaling molecules as prognostic biomarkers for non-small cell lung cancer. Oncol. Lett. 2015, 10, 3252–3260. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Wen, J.; Ning, Y.; Li, Y. Higher notch expression implies poor survival in pancreatic ductal adenocarcinoma: A systematic review and meta-analysis. Pancreatology 2018, 18, 954–961. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Wu, H.; Xu, H.; Han, N.; Chu, Q.; Yu, S.; Chen, Y.; Wu, K. Meta-analysis reveals the correlation of Notch signaling with non-small cell lung cancer progression and prognosis. Sci. Rep. 2015, 5, 10338. [Google Scholar] [CrossRef] [PubMed]
- Harnish, J.M.; Link, N.; Yamamoto, S. Drosophila as a Model for Infectious Diseases. Int. J. Mol. Sci. 2021, 22, 2724. [Google Scholar] [CrossRef]
- Evans, A.S. Limitations of the Henle—Koch postulates. In Causation and Disease; Springer: Boston, MA, USA, 1993. [Google Scholar] [CrossRef]
- Florescu, S.A.; Cotar, A.I.; Popescu, C.P.; Ceianu, C.S.; Zaharia, M.; Vancea, G.; Codreanu, D.; Badescu, D.; Ceausu, E. First Two Imported Cases of Zika Virus Infections in Romania. Vector Borne Zoonotic Dis. 2017, 17, 354–357. [Google Scholar] [CrossRef] [PubMed]
- Oehler, E.; Watrin, L.; Larre, P.; Leparc-Goffart, I.; Lastère, S.; Valour, F.; Baudouin, L.; Mallet, H.P.; Musso, D.; Ghawche, F. Zika Virus Infection Complicated by Guillain-Barré Syndrome Acase Report, French Polynesia, December 2013. Eurosurveillance 2014, 19, 20720. [Google Scholar] [CrossRef]
- Mlakar, J.; Korva, M.; Tul, N.; Popović, M.; Poljšak-Prijatelj, M.; Mraz, J.; Kolenc, M.; Resman Rus, K.; Vesnaver Vipotnik, T.; Fabjan Vodušek, V.; et al. Zika Virus Associated with Microcephaly. N. Engl. J. Med. 2016, 374, 951–958. [Google Scholar] [CrossRef]
- Klaitong, P.; Smith, D.R. Roles of non-structural protein 4A in flavivirus infection. Viruses 2021, 13, 2077. [Google Scholar] [CrossRef]
- Shah, P.S.; Link, N.; Jang, G.M.; Sharp, P.P.; Zhu, T.; Swaney, D.L.; Johnson, J.R.; Von Dollen, J.; Ramage, H.R.; Sat-kamp, L.; et al. Comparative Flavivirus-host protein interaction mapping reveals mechanisms of dengue and Zika virus pathogenesis. Cell 2018, 175, 1931–1945. [Google Scholar] [CrossRef]
- Shaheen, R.; Maddirevula, S.; Ewida, N.; Alsahli, S.; Abdel-Salam, G.M.H.; Zaki, M.S.; Al Tala, S.; Alhashem, A.; Softah, A.; Al-Owain, M.; et al. Genomic and phenotypic delineation of congenital microcephaly. Genet. Med. 2018, 21, 545–552. [Google Scholar] [CrossRef]
- Link, N.; Bellen, H.J.; Dunwoodie, S.; Wallingford, J. Using Drosophila to drive the diagnosis and understand the mechanisms of rare human diseases. Development 2020, 147, dev191411. [Google Scholar] [CrossRef]
- Link, N.; Chung, H.; Jolly, A.; Withers, M.; Tepe, B.; Arenkiel, B.R.; Shah, P.S.; Krogan, N.J.; Aydin, H.; Geckinli, B.B.; et al. Ankle2, a Target of Zika Virus, Controls Asymmetric Cell Division of Neuroblasts and Uncovers a Novel Microcephaly Pathway. bioRxiv 2019, 611384. [Google Scholar] [CrossRef]
- Almagor, L.; Ufimtsev, I.S.; Ayer, A.; Li, J.; Weis, W.I. Structural insights into the aPKC regulatory switch mechanism of the human cell polarity protein lethal giant larvae. Proc. Natl. Acad. Sci. USA 2019, 116, 10804–10812. [Google Scholar] [CrossRef]
- Gonzaga-Jauregui, C.; Lotze, T.; Jamal, L.; Penney, S.; Campbell, I.M.; Pehlivan, D.; Hunter, J.V.; Woodbury, S.L.; Raymond, G.; Adesina, A.M.; et al. Mutations in VRK1 associated with complex motor and sensory axonal neuropathy plus microcephaly. JAMA Neurol. 2013, 70, 1491–1498. [Google Scholar] [PubMed]
- Yakulov, T.; Günesdogan, U.; Jäckle, H.; Herzig, A. Bällchen participates in proliferation control and prevents the differentiation of Drosophila melanogaster neuronal stem cells. Biol. Open 2014, 3, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Nelson, A.; Lopez, A.L.; Sack, D. global burden of cholera in endemic countries. PLoS Neglected Trop. Dis. 2015, 9, e0003832. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4455997/ (accessed on 15 January 2022). [CrossRef] [PubMed]
- Israil, A.; Balotescu, C.; Damian, M.; Dinu, C.; Bucurenci, N. Comparative study of different methods for detection of toxic and other enzymatic factors in Vibrio cholerae strains. Rom. Arch. Microbiol. Immunol. 2005, 63, 63–77. [Google Scholar]
- Bhuin, T.; Roy, J.K. Rab11 in disease progression. Int. J. Mol. Cell. Med. 2015, 4, 1–8. [Google Scholar] [PubMed]
- Guichard, A.; Cruz-Moreno, B.; Aguilar, B.; van Sorge, N.; Kuang, J.; Kurkciyan, A.A.; Wang, Z.; Hang, S.; de Chambrun, G.P.P.; McCole, D.F.; et al. Cholera Toxin Disrupts Barrier Function by Inhibiting Exocyst-Mediated Trafficking of Host Proteins to Intestinal Cell Junctions. Cell Host Microbe 2013, 14, 294–305. [Google Scholar] [CrossRef]
- Ferlay, J.; Bray, P.; Parkin, D.M. Globocan 2000: Cancer Incidence, Mortality and Prevalence Worldwide, Version 1.0; IARC Cancer Base No. 5.; IARC Press: Lyon, France, 2001. [Google Scholar]
- Chen, Y.; Segers, S.; Blaser, M.J. Association between Helicobacter pylori and mortality in the NHANES III study. Gut 2013, 62, 1262–1269. [Google Scholar] [CrossRef]
- Ilie, M.; Dascalu, L.; Macovei, R.A. Helicobacter Pylori Cag A Antibodies and Their Clinical Implications: Correlation of Helicobacter Pylori CagA Antibodies with Treatment Resistance, Bleeding Ulcer and Gastric Cance; LAP LAMBERT Academic Publishing: Saarbrucken, Germany, 2014; ISBN1 103659526630. ISBN2 13978-3659526633. [Google Scholar]
- Hatakeyama, M.; Higashi, H. Helicobacter Pylori CagA: A New Paradigm for Bacterial Carcinogenesis. Cancer Sci. 2005, 96, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Butti, R.; Das, S.; Gunasekaran, V.P.; Yadav, A.S.; Kumar, D.; Kundu, G.C. Receptor tyrosine kinases (RTKs) in breast cancer: Signaling, therapeutic implications and challenges. Mol. Cancer 2018, 17, 34. [Google Scholar] [CrossRef]
- Saadat, I.; Higashi, H.; Obuse, C.; Umeda, M.; Murata-Kamiya, N.; Saito, Y.; Lu, H.; Ohnishi, N.; Azuma, T.; Suzuki, A.; et al. Helicobacter pylori CagA targets PAR1/MARK kinase to disrupt epithelial cell polarity. Nature 2007, 447, 330–333. [Google Scholar] [CrossRef]
- Hatakeyama, M. Structure and function of Helicobacter pylori CagA, the first-identified bacterial protein involved in human cancer. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 93, 196–219. [Google Scholar] [CrossRef] [PubMed]
- Tateno, M.; Nishida, Y.; Adachi-Yamada, T. Regulation of JNK by Src during Drosophila Development. Science 2000, 287, 324–327. [Google Scholar] [CrossRef] [PubMed]
- Yong, X.; Tang, B.; Li, B.S.; Xie, R.; Hu, C.J.; Luo, G.; Qin, Y.; Dong, H.; Yang, S.M. Helicobacter pylori virulence factor CagA promotes tumorigenesis of gastric cancer via multiple signaling pathways. Cell Commun. Signal. 2015, 13, 30. [Google Scholar] [CrossRef]
- Wandler, A.M.; Guillemin, K. Transgenic expression of the Helicobacter pylori virulence factor CagA promotes apoptosis or tumorigenesis through JNK activation in Drosophila. PLOS Pathog. 2012, 8, e1002939. [Google Scholar] [CrossRef]
- Igaki, T.; Pagliarini, R.A.; Xu, T. Loss of Cell Polarity Drives Tumor Growth and Invasion through JNK Activation in Drosophila. Curr. Biol. 2006, 16, 1139–1146. [Google Scholar] [CrossRef]
- Wu, M.; Pastor-Pareja, J.C.; Xu, T. Interaction between RasV12 and Scribbled Clones Induces Tumour Growth and Invasion. Nature 2010, 463, 545–548. [Google Scholar] [CrossRef]
- D’Souza, J.; Cheah, P.Y.; Gros, P.; Chia, W.; Rodrigues, V. Functional complementation of the malvolio mutation in the taste pathway of Drosophila melanogaster by the human natural resistance-associated macrophage protein 1 (Nramp-1). J. Exp. Biol. 1999, 202, 1909–1915, Printed in Great Britain © The Company of Biologists Limited 1999 JEB1976S. [Google Scholar] [CrossRef]
- Cellier, M.; Belouchi, A.; Gros, P. Resistance to intracellular infections: Comparative genomic analysis of Nramp. Trends Genet. 1996, 12, 201–204. [Google Scholar] [CrossRef]
- Rodrigues, V.; Cheah, P.Y.K.; Chia, W. Malvolio, the Drosophila homologue of mouse NRAMP-1( Bcg), is expressed in macrophages and in the nervous system and is required for normal taste behavior. EMBO J. 1995, 14, 3007–3020. [Google Scholar] [CrossRef] [PubMed]
- Orgad, S.; Nelson, H.; Segal, D.; Nelson, N. Metal ions suppress the abnormal taste behavior of the Drosophila mutant malvolio. J. Exp. Biol. 1998, 201, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Abel, L.; Sanchez, F.O.; Oberti, T.N.V.; Hoa, L.V.; Lap, V.D.; Skamene, E.; Lagrange, P.H.; Schurr, E. Susceptibility to leprosy is linked to the human NRAMP1 gene. J. Infect. Dis. 1998, 177, 133–145. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bellamy, R.; Ruwende, C.; Corrah, T.; McAdam, K.P.; Whittle, H.C.; Hill, A.V. Variations in the NRAMP1 gene and susceptibility to tuberculosis in West Africans. N. Engl. J. Med. 1998, 338, 640–644. [Google Scholar] [CrossRef]
- Gertler, F.B.; Comer, A.R.; Juang, J.-L.; Ahern, S.M.; Clark, M.J.; Liebl, E.C.; Hoffmann, F.M. Enabled, a dosage-sensitive suppressor of mutations in the Drosophila Abl tyrosine kinase, encodes an Abl substrate with SH3-binding properties. Genes Dev. 1995, 9, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Symons, M.; Derry, J.D.J.; Karlak, B.; Jiang, S.; Lemahieu, V.; McCormick, F.F.U.; Abo, A. Wiskott-Aldrich syndrome protein, a novel effector for the GTPase CDC42Hs, is implicated in actin polymerization. Cell 1996, 84, 723–734. [Google Scholar] [CrossRef]
- Ahern-Djamali, S.M.; Comer, A.R.; Bachmann, C.; Kastenmeier, A.S.; Reddy, S.K.; Beckerle, M.C.; Walter, U.; Hoffmann, F.M. Mutations in Drosophila enabled and rescue by human vasodilator-stimulated phosphoprotein (VASP) indicate important functional roles for Ena/VASP homology domain 1 (EVH1) and EVH2 domains. Mol. Biol. Cell 1998, 9, 2157–2171. [Google Scholar] [CrossRef]
- Muñoz-Alarcón, A.; Pavlovic, M.; Wismar, J.; Schmitt, B.; Eriksson, M.; Kylsten, P.; Dushay, M.S. Characterization of lamin mutation phenotypes in Drosophila and comparison to human laminopathies. PLoS ONE 2007, 2, e532. [Google Scholar] [CrossRef]
- Beard, G.S.; Bridger, J.M.; Kill, I.R.; Tree, D.R. Towards a Drosophila model of Hutchinson-Gilford progeria syndrome. Biochem. Soc. Trans. 2008, 36 Pt 6, 1389–1392. [Google Scholar] [CrossRef]
- Tsurumi, A.; Li, W.X. Aging mechanisms—A perspective mostly from Drosophila. Adv. Genet. 2020, 1, e10026. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Clinical Impact | Vertebrate Gene | Fly Gene | Mutant Phenotype (Fly) | Heterologous Rescue | HR References |
|---|---|---|---|---|---|
| motor neuron diseases | (h)VAPB | Vap33 | loss of Vapp33 determines larval lethality, with few adult escapers | expression of (h)VAPB alleviates the lethal phenotype determined by loss of Vap33 | [68] |
| Huntington’s disease | (h)UCP2 | UCP5 | expression of mutant Huntingtin protein in glia determines altered locomotor performances and uncommon vulnerability to mechanical stress | co-expression of (h)UCP2 | [69] |
| (h)HTT | htt | htt-null flies have severe thorax muscle loss and accelerated deterioration in mobility and lifespan | (h)HTT rescues the htt loss associated phenotypes | [70] | |
| PD | (h)MIC60 | MIC60 | MIC60mut-null allele determines pupal lethality in homozygous individuals; MIC60mut/+ flies are normal | expression of (h)MIC60 in MIC60mut/+ flies provides a normal phenotype, while expression of mutant (h)MIC60A4V, T11A or C17F leads to severe adult lethality and reduced larval crawling | [71] |
| (h)PRKN | park | park-null flies exhibit reduced lifespan, locomotor and fly defects, infertility, lower cell size and number, progressive degeneration of certain DA neurons | co-expression of (h)PRKN rescues the neurotoxicity; muscle-specific expression of (h)PRKN rescues the flight ability | [72,73] | |
| (h)LRRK2 | Lrrk | Lrrk-null mutants elicit autophagy defects and DA degeneration | overexpression of (h)LRRK2 rescues the mutant phenotype | [74,75] | |
| (h)VPS35 | Vps35 | downregulation of Vps35 in brain determines supernumerous neuroblast phenotype | expression of (h)VPS35 fully rescues the brain tumor phenotype exhibited by Vps35 mutants | [76] | |
| PD; frontotemporal dementia | (h)MAPT | tau | loss of tau determines lethality; deletion of tau in neurons determines neurodegeneration | expression of (h)MAPT partially rescues the neurodegenerative phenotype | [77] |
| ALS | (h)PFN1 | chic | RNAi-mediated downregulation of chic in motor neurons determines pupal lethality | the chic mutant phenotype is rescued by expressing (h)PFN1 in motor neurons | [78] |
| ALS and other neurodegenerative diseases | (h)VCP | TER94 | TER94 mutations determine tubular lysosome dysfunction | expression of (h)VCP rescues the phenotype determined by mutant TER94 | [79] |
| late-onset AD | (h)TM2D3 | amx | strong neurogenic phenotype when amx is maternally mutated | (h)TM2D3 is able to partially rescue the neurogenic phenotype and embryonal lethality | [80] |
| Parkinsonism with spasticity, X-linked; intellectual developmental disorder, X-linked, syndromic, Hedera type | (h)ATP6AP2 | ATP6AP2 | ATP6AP2 depletion is lethal; RNAi knockdown of ATP6AP2 in wing pouch leads to abnormal wing development and growth defects | expression of (h)ATP6AP2 in ATP6AP2 RNA1 background rescues the specific mutant phenotype | [81] |
| pediatric-onset neurodegenerative disorder | (h)ADPRHL2 | Parg | Parg LOF determines decreased survival in response to oxidative challenge | lethality is rescued by expressing (h)ADPRHL2 | [82] |
| neurodegeneration | (h)TARDBP | TBPH | TBPH-null mutants experience loss of the ventral nerve cord neurons (bursicon neurons) | expression of (h)TARDBP rescues the bursicon neurons | [83] |
| neurodegeneration; cancer; metabolic disorder | (h)TOMM70 | Tom70 | Tom70-null mutation conducted to pupal lethality | the lethality is rescued by the expression of (h)TOMM70 | [84] |
| neurodegeneration; Boucher–Neuhäuser, Gordon Holmes, Laurence–Moon and Oliver McFarlane syndromes | (h)PNPLA6 | sws | the sws1-null mutation causes locomotion deficits and neurodegeneration | (h)PNPLA6 rescues the mutant sws phenotype | [85] |
| the sws1 mutants showed characteristic vacuoles in central brain and optic lobes | (h)PNPLA6 partially rescues the vacuolization of mutant sws | [86] | |||
| pantothenate kinase-associated neurodegeneration | (h)PanK2 | fbl | a hypomorphic mutation in fumble results in flies that have brain lesions, defective neurological functions and severe motor impairment | the paralysis and impaired climbing activity are rescued by expressing (h)PanK2 | [87] |
| in mice, neonatal lethality, slow progressive neurodegeneration, enhanced limb-clasping reflexes, impaired motor activity, cognitive deficits and hypomyelination [88] | (h)NRD1 | Nrd1 | LOF allele causes neurodegeneration | expression of (h)NRD1 rescues the pupal lethality and electroretinogram defects | [89] |
| chorea-acanthocytosis, neurodegeneration, progressive loss of cognitive and locomotor functions | (h)VPS13A | Vps13 | mutant flies have age-linked neurodegeneration and reduced lifespan | overexpression of (h)VPS13A in mutant flies rescues the characteristic phenotype | [90,91] |
| Alkuraya-Kucinskas and Oliver Mcfarlane syndromes | (h)DENND4A | Crab | flies lacking Crab activity experience age-dependent decline in photoreceptor function and structural integrity | expression of (h)DENND4A rescues the eye defects exhibited by the mutant flies | [92] |
| neurodegenerative encephalopathy | (h)TBCD | TBCD | projection neurons expressing TBCD1 mutant allele have affected axonal branches | overexpression of (h)TBCD extensively suppresses the axonal mutant phenotype | [93] |
| neuronal K+–Cl− cotransporter; epilepsy | (h)SLC12A5 | kcc | kccDH1 hypomorphic allele acts as a seizure-enhancer mutation and exacerbates the bang-sensitive paralytic behavior | (h)KCC2 rescues the mutant phenotype induced by kccDH1 | [94] |
| progressive myoclonus epilepsy | (h)GOSR2 | membrin | homozygosity for membrin-null allele causes larval lethality | expressing (h)GOSR2 fully rescues the larval lethality, but the adults, although normal looking, display severe motor impairments | [95] |
| early infantile epileptic encephalopathy (EIEE) | (h)ACTL6B (BAF53B) | Bap55 | mutations in Bap55 affect the synaptic connections in olfactory neurons | (h)ACTL6B rescues the mutant phenotype of Bap55-null individuals | [96] |
| photosensitive epilepsy (PSE) | (h)SGMS1 | - | cpes-null mutants show compromised ceramide phosphoethanolamine synthase and fail to complete neuronal cell body encapsulation in the neuronal cortex | expression of (h)SGMS1 rescues the PSE and cortex glial aberrations | [97] |
| ASD | (h)TaoK2 | Tao | loss of Tao determines overgrowth of dendritic branching and behavioral defects | (h)TaoK2 restores the aberrant dendritic branches to control levels | [98,99] |
| (h)DAT (SLC6A3) | DAT | DAT KO flies are hyperactive | DAT KO flies expressing (h)DAT have reduced locomotion | [100,101] | |
| (h)SCAMP1, (h)SCAMP5 | Scamp | Scamp-null flies exhibit shortened lifespan, compromised climbing, heat-induced seizures and compromised learning and long-term memory | both (h)SCAMP1 and (h)SCAMP5 rescue the climbing mutant phenotype; (h)SCAMP1 significantly improves the learning index of Scamp-null flies | [102] | |
| autism; multiple myeloma | (m)Nbea | rg | rg-null mutants exhibit aberrant associative odor learning, modification of gross brain morphology and of synaptic architecture | the transgene (m)Nbea is able to rescue only aversive odor learning and synaptic architecture | [103] |
| affected development of distinct cell types in the central nervous system and in sensory systems | (m)Math1 | ato | mutant ato embryos lack precursor cell selection and chordotonal organ specification | expression of (m)Math1 under the control of the ato embryonic enhancer | [104] |
| in mouse, (m)Math1-null animals do not succeed to initiate respiration and die soon after birth | replacing (m)Math1 coding region with ato allowed the animals to survive to adulthood | ||||
| CMT type 2A, axon degeneration [105] | (h)MFN1, (h)MFN2 | Marf | mutant flies have affected mitochondria and, as a consequence, their nerves cannot send out signals to muscles; in addition, Marf is lost in the ring gland affecting the production of a hormone required for larva transition to adult, the mutants dying in their larval stage | expression of both (h)MFN1 and (h)MFN2 is necessary for hormone production and the rescue of all phenotypes | [106] |
| CMT neuropathy | (h)GDAP1 | Gdap1 | knockdown mutants experience retina and muscle degeneration | the mutant phenotype is rescued by (h)GDAP1 | [107] |
| dominant-intermediate CMT neuropathy | (h)YARS | TyrRS | RNAi-silenced TyrRS determines specific bristle phenotypes | expressing (h)YARS rescues the abnormal bristle phenotype | [108] |
| CMT neuropathy type 2D | (h)GARS | GlyRS | GlyRS-null flies lack dendritic and axonal terminal arborization | (h)GARS rescues the arborization defects in GlyRS-null flies | [109] |
| autosomal recessive cerebellar ataxia | (h)UBA5 | Uba5 | Uba5-null mutants have reduced lifespan and locomotor activity as well as neuromuscular junction (NMJ) defects | (h)UBA5 expression significantly rescues the NMJ mutant phenotype | [110] |
| FA | (h)FXN | fh | fh mutants have altered mitochondrial functions and exhibit age-dependent neurodegeneration | expression of (h)FXN rescues the neurodegeneration | [111] |
| ataxia determined by defects of autophagy | (h)ATG5 | Atg5 | flies lacking Atg5 activity are unable to walk and fly properly | (h)ATG5 restores the mutant flies’ normal movements; (h)ATG5E122D slightly improves the defective mobility | [112] |
| X-linked Snyder–Robinson syndrome | (h)SMS | Sms | Sms mutants have critically lowered transcript levels that reduce viability | (h)SMS rescues the viability of mutant flies | [113] |
| Delpire–Mcneill syndrome | (h)SCL12A2 (NKCC1) | Ncc69 | Ncc69 mutants reach adulthood but their abdominal nerves are swelled and form bulges | this neuropathy is rescued by (h)SCL12A2 | [114,115] |
| microcephaly; Zika virus target | (h)ANKLE2 | Ankle2 | mutations in Ankle2 can lead to loss of peripheral nervous system organs in adults and severely reduced brain size in hemizygous third instar larvae | expression of (h)ANKLE2 rescues the mutant phenotype | [116,117] |
| neural network formation; tumor progression | (m)Bsg | Bsg | mutations in Bsg alter the cell architecture and can lead to high embryo or larval lethality | Bsg LOF in adults’ eyes determines mislocalization of photoreceptor nuclei, a phenotype rescued by expressing (m)Bsg | [118] |
| global developmental disorders, intellectual disability | (h)CAPZA2 | cpa | cpa-null allele determines first instar lethality | (h)CAPZA2 rescues the lethal phenotype of cpa-null individuals | [119] |
| autosomal recessive, nonsyndromic intellectual disability | (h)ZC3H14 | Nab2 | Nab2-null flies experience developmental and locomotor defects | (h)ZC3H14 expressed in neurons rescues the Nab2-null phenotype | [120] |
| Troyer syndrome | (h)SPG20 | spartin | loss of spartin is associated with motor dysfunctions and brain neurodegeneration | synaptic overgrowth in spartin-null flies is rescued by presynaptic expression of Myc-tagged (h)ZC3H14 | [121] |
| intellectual disability | (h)OPHN1 | Graf | loss of Graf affects the mushroom body (MB) development | expression of (h)OPHN1 significantly ameliorates the MB mutant phenotype | [122] |
| intellectual disability, X-linked | (h)CASK | CASK | affected expression of CASK negatively impacts middle-term and long-term memory | overexpression of (h)CASK in neurons of CASK mutants fully rescues the memory | [123] |
| intellectual disability, X-linked | (h)ACSL4 | Acsl | Acsl mutants exhibit neuromuscular junction overgrowth | expression of (h)ACSL4 rescues the mutant phenotype particular to Acsl mutants | [124] |
| intellectual disability | (h)SMARCA5 | Iswi | Iswi LOF is related to decreased body size and movement in larvae and decreased brain size and locomotor dysfunctions in adults | (h)SMARCA5 expression rescues the Iswi specific mutations | [125] |
| nervous system developmental defects | (h)EBF3 | kn | homozygous kn-null genotype is embryo lethal | (h)EBF3 rescues the lethality | [126] |
| autosomal recessive neurologic disorder | (h)TMTC3 | Tmtc3 | neuron-specific knockdown of Tmtc3 rises the incidence of mechanically induced seizures | neuron-specific expression of (h)TMTC3 | [127] |
| intellectual developmental disorders | (h)IQSEC1 | siz | loss of siz affects the growth cones and causes embryonal lethality | overexpression of (h)IQSEC1 in WT fly background is toxic; lowered expression of (h)IQSEC1 in siz-null mutants partially rescues the embryonal lethality | [128] |
| developmental delay, movement disorders and metabolic decompensation | (h)OGDH | Ogdh | LOF allele is associated with early developmental lethality | the expression of (h)OGDH rescues the mutant phenotype | [129] |
| infantile encephalopathy (lethal) | (h)DNM1L | Drp1 | Drp1 mutants have altered mitochondrial trafficking and die as larvae | ubiquitous expression of (h)DNM1L rescues the lethality | [130] |
| schizophrenia | (h)DTNBP1 | Dysb | Dysb mutants have compromised memory, elevated climbing activity, abnormal male-male courtship behavior, hypoglutamatergic and hyperdopaminergic activities | pan-neuronal or glial expression of (h)DTNBP1 rescues various Dysb mutant phenotypes | [131] |
| Pitt–Hopkins syndrome | (h)TCF4-A, (h)TCF4-B | da | da-null allele severely impacts the embryonic nervous system development | both (h)TCF4-A and (h)TCF4-B rescue the mutant embryo phenotype | [132] |
| neurofibromatosis, type 2 | (h)NF2 | Mer | Mer-null mutations determine lethality | isoform 1 of (h)NF2 is able to rescue the lethality of Mer-null mutants | [133] |
| Clinical Impact | Vertebrate Gene | Fly Gene | Mutant Phenotype (Fly) | Heterologous Rescue | HR References |
|---|---|---|---|---|---|
| cardiac dysfunction (postulated), TRiC/CCT complex | (h)CCT4 | CCT4 | RNAi-silenced CCT4 determines pupal lethality and growth defects | overexpression of (h)CCT4 rescues the mutant phenotype | [187] |
| lipotoxic cardiomyopathy, ceramide/sphingolipid-related | (h)DEGS1 | ifc | knockout of ifc results in larval lethality | (h)DEGS1 rescues the lethal phenotype of ifc null individuals | [188] |
| congenital heart defect (postulated), KMT2-related | (h)KMT2A (MLL) | trx | LOF mutations determines larval to pupal lethality associated with aberrant cuticular patterns | expression of (h)MLL partially rescues the cuticular phenotype | [189] |
| dilated cardiomyopathy 3B | (m)Dmd | Dys | loss of Dys function leads to reduced lifespan, significantly increased heart rate, age-dependent myofibrillar disorganization, cardiac chamber enlargement and impaired systolic function | the mutant phenotype was partially reversed by expression of a truncated (m)Dmd which restores the cardiac diameters and function | [173,178] |
| Noonan syndrome | (h)PTPN11 (SHP-2) | csw | csw mutations determine zygotic lethality | expression of (h)SHP-2 rescues the zygotic lethality | [190] |
| muscle and aortic defects, ARIH1-related | (h)ARIH1 | ari-1 | ari-1-null allele is associated with affected larval muscle, lethality or reduced lifespan in adults | (h)ARIH1 rescues ari-1-related lethality. | [191] |
| Clinical Impact | Vertebrate Gene | Fly Gene | Mutant Phenotype (Fly) | Heterologous Rescue | HR References |
|---|---|---|---|---|---|
| epithelial cancer | (h)LLGL1 and (h)LLGL2 | l(2)gl | l(2)gl/l(2)gl genotype determines lethality | (h)LLGL1 partially rescues the homozygous l(2)gl lethal phenotype; imaginal tissues do not show any neoplastic features, with Dlg and Scrib exhibiting the correct localization; animals undergo a complete metamorphosis and hatch as viable adults | [197] |
| (h)HUGL-1 | lgl | mutations in lgl determine structural defects in larvae | (h)HUGL-1 expression in the homozygous lgl mutants leads to a partial development of rudimental eyes and larval structures comparable to wild type | ||
| (h)LATS1 and (h)LATS2 | Wts | developmental defects, lethality in flies | (h)LATS1 rescues all developmental defects including embryonic lethality in flies | [198] | |
| (h)Scrib | scrib | polarity and neoplastic overgrowth defects | (h)Scrib rescues the polarity and neoplastic overgrowth defects of scrib mutants | [199] | |
| (h)JRK/JH8 | ebd1 | muscle defects in ebd1 and ebd1/ebd2 double mutants | (h)JRK/JH8 has rescued the flight muscle defects in ebd1 as well as in ebd1/ebd2 double mutants | [200] | |
| (h)CC2D1A and (h)CC2D1B | Lgd | tissue hyperplasia in Lgd mutant phenotype | (h)CC2D1A and (h)CC2D1B rescue the Lgd mutant phenotype | [201] | |
| various cancers | (h)TP53 | p53 | p53-null embryos have high sensitivity to genotoxic stressors such as irradiation | (h)TP53 partially rescues the embryo liability to irradiation | [202] |
| acute myeloid leukemia | (h)MLF1 and (h)MLF2 | Mlf | Mlf LOF phenotypes include the decrease in embryonic crystal cell numbers and adult bristle and wing phenotypes | expression of (h)MLF1 and (h)MLF2 rescues several Mlf LOF phenotypes | [203] |
| (h)CUX1 | ct | ct deficient flies exhibit abnormal wing phenotypes | (h)CUX1 rescues the ct mutant phenotypes | [204] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ecovoiu, A.A.; Ratiu, A.C.; Micheu, M.M.; Chifiriuc, M.C. Inter-Species Rescue of Mutant Phenotype—The Standard for Genetic Analysis of Human Genetic Disorders in Drosophila melanogaster Model. Int. J. Mol. Sci. 2022, 23, 2613. https://doi.org/10.3390/ijms23052613
Ecovoiu AA, Ratiu AC, Micheu MM, Chifiriuc MC. Inter-Species Rescue of Mutant Phenotype—The Standard for Genetic Analysis of Human Genetic Disorders in Drosophila melanogaster Model. International Journal of Molecular Sciences. 2022; 23(5):2613. https://doi.org/10.3390/ijms23052613
Chicago/Turabian StyleEcovoiu, Alexandru Al., Attila Cristian Ratiu, Miruna Mihaela Micheu, and Mariana Carmen Chifiriuc. 2022. "Inter-Species Rescue of Mutant Phenotype—The Standard for Genetic Analysis of Human Genetic Disorders in Drosophila melanogaster Model" International Journal of Molecular Sciences 23, no. 5: 2613. https://doi.org/10.3390/ijms23052613
APA StyleEcovoiu, A. A., Ratiu, A. C., Micheu, M. M., & Chifiriuc, M. C. (2022). Inter-Species Rescue of Mutant Phenotype—The Standard for Genetic Analysis of Human Genetic Disorders in Drosophila melanogaster Model. International Journal of Molecular Sciences, 23(5), 2613. https://doi.org/10.3390/ijms23052613