
Identification of Neoantigens in Cancer Cells as Targets for Immunotherapy

Abstract
:1. Introduction
Clinical Significance of Tumor Neoantigens
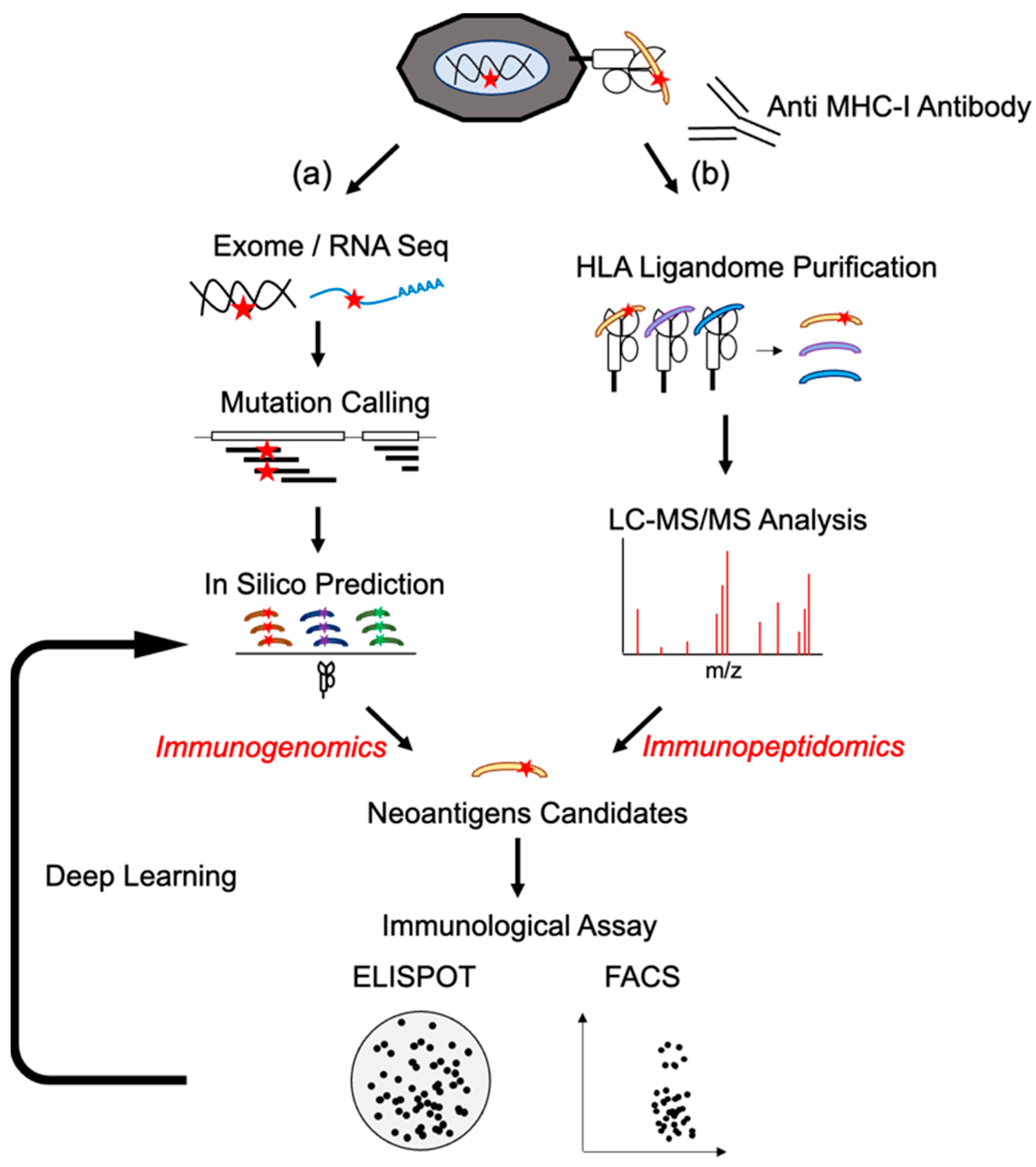
2. Neoantigen Identification Methodology Development
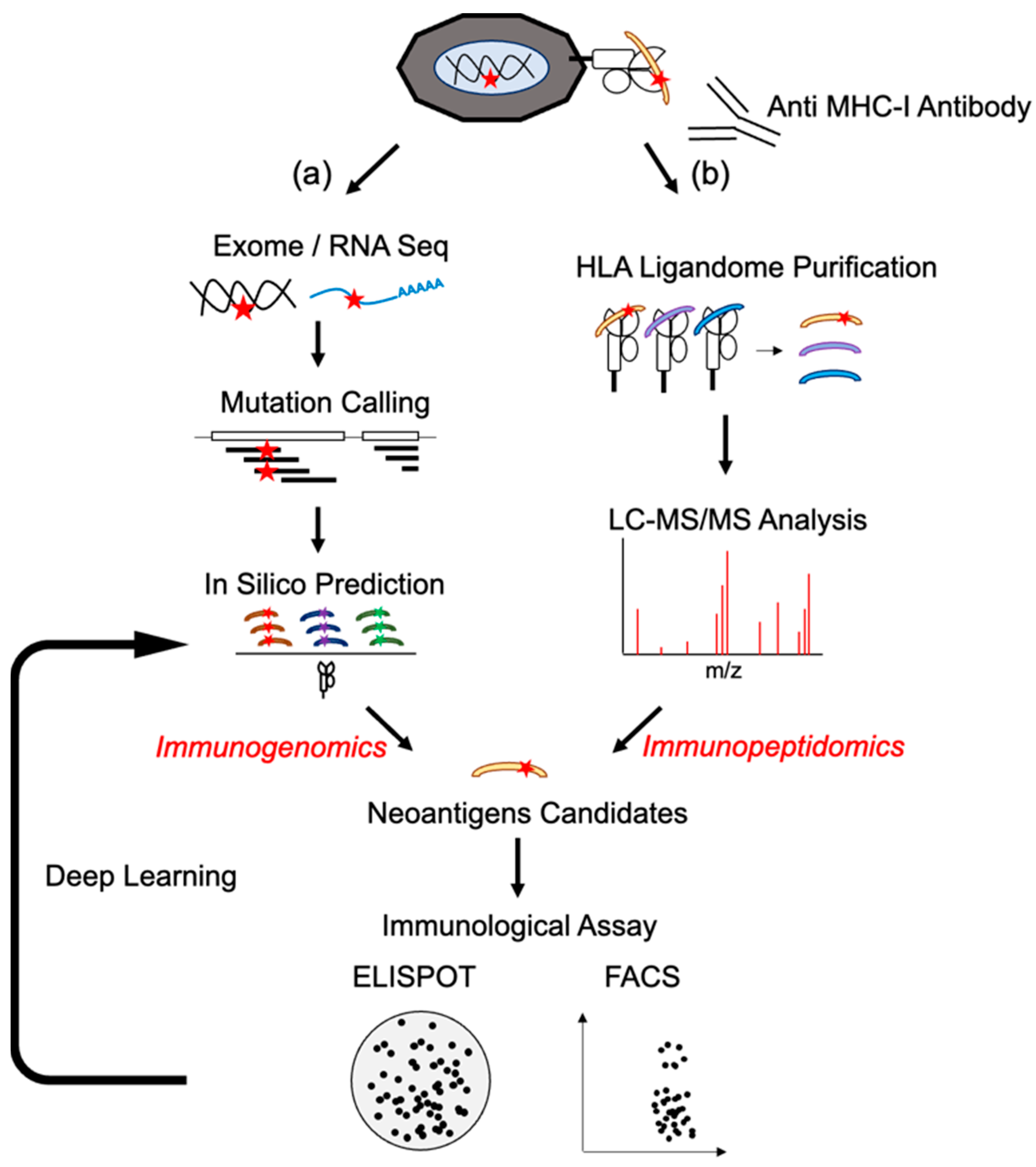
2.1. Next-Generation Sequences and Epitope Prediction as Immunogenomics Approach
2.2. HLA Typing for Neoantigen Detection
2.3. In Silico Prediction of Neoantigens
2.4. Mass Spectrometry Analysis (Immunopeptidomics)
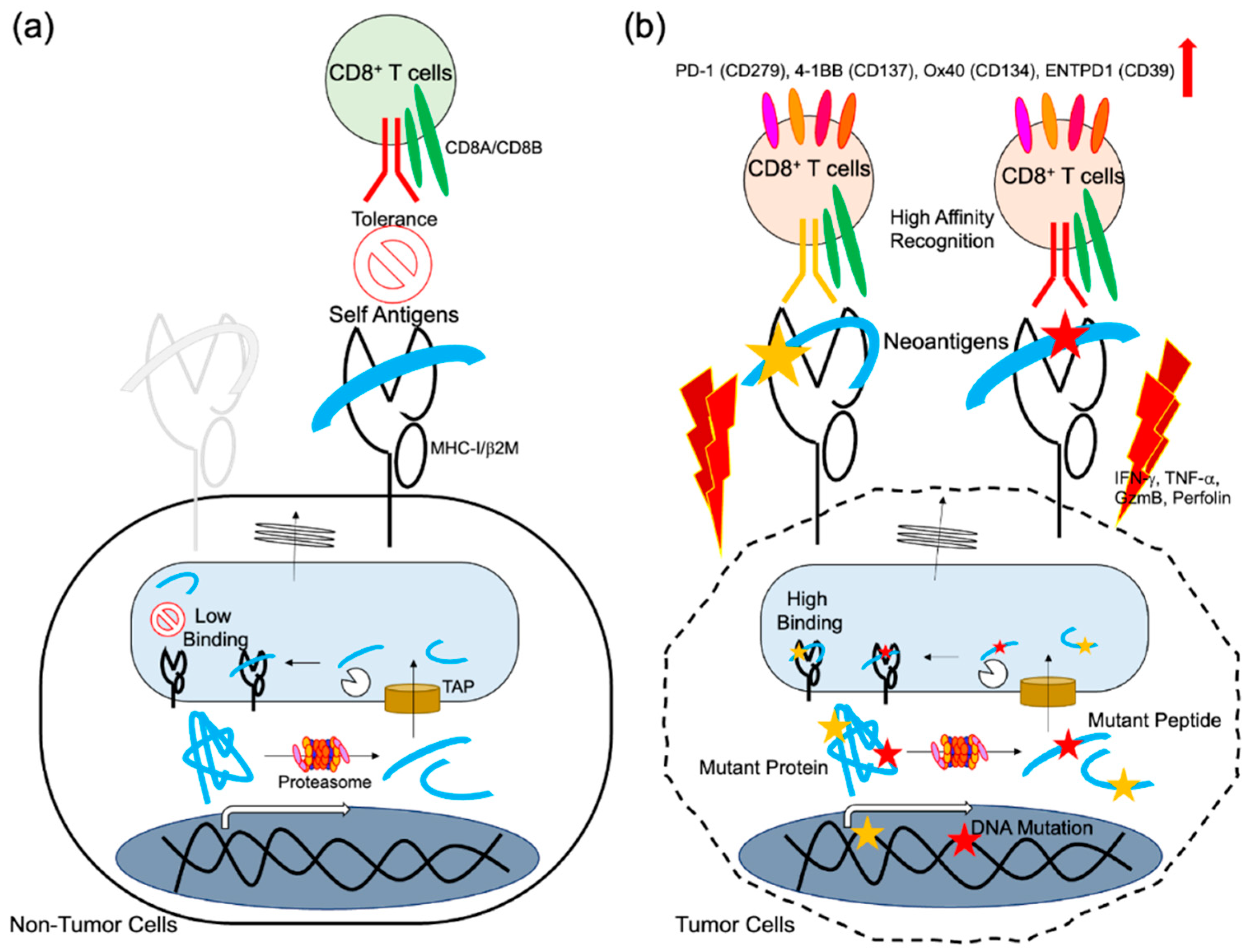
3. Neoantigen-Specific T Cell Responses
3.1. Reactivity of Neoantigen-Specific T Cells after Vaccines
3.2. Existence of Neoantigen-Specific T Cells in Cancer Patients without Vaccines Treatment
4. Neoantigen Candidates as Shared Antigens
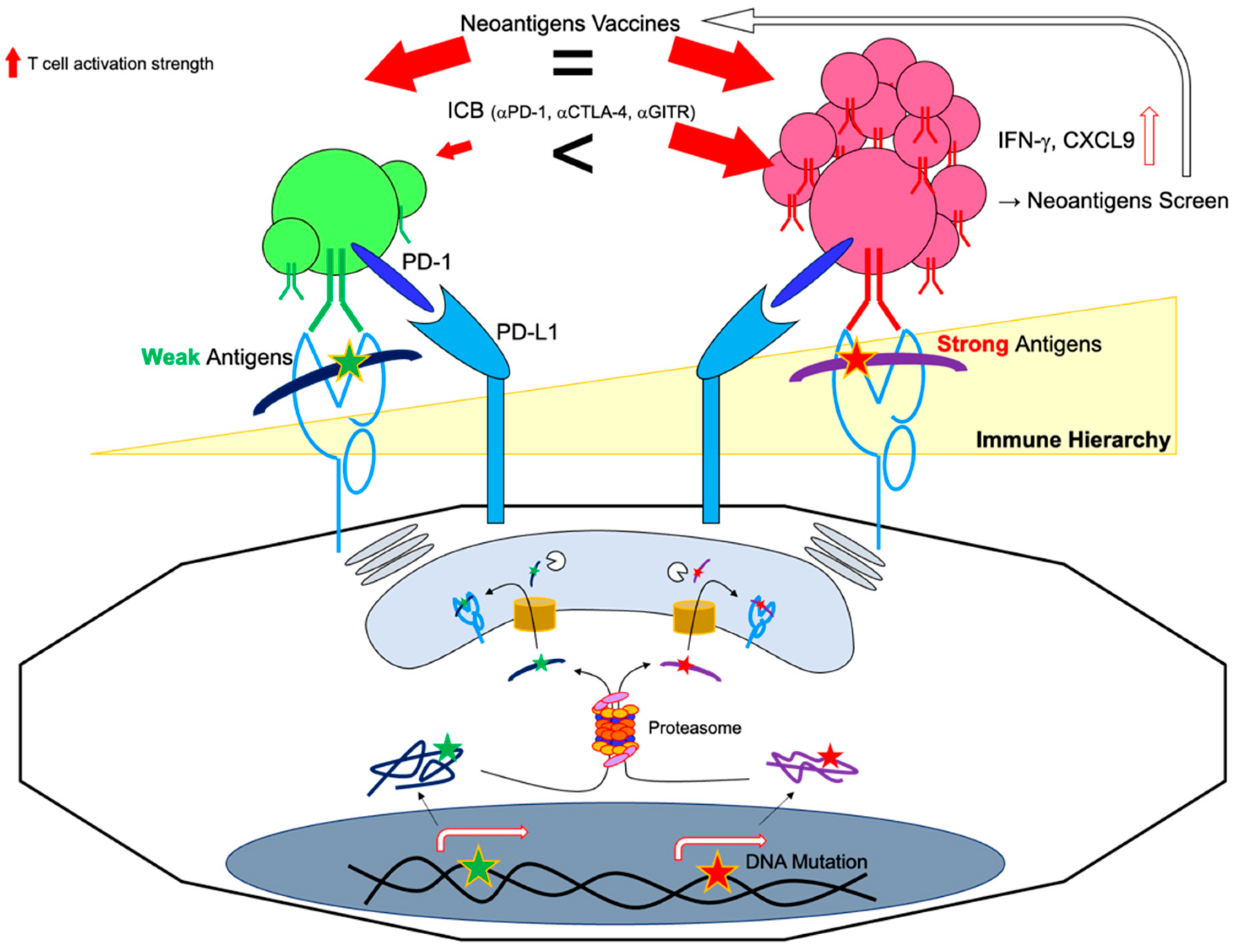
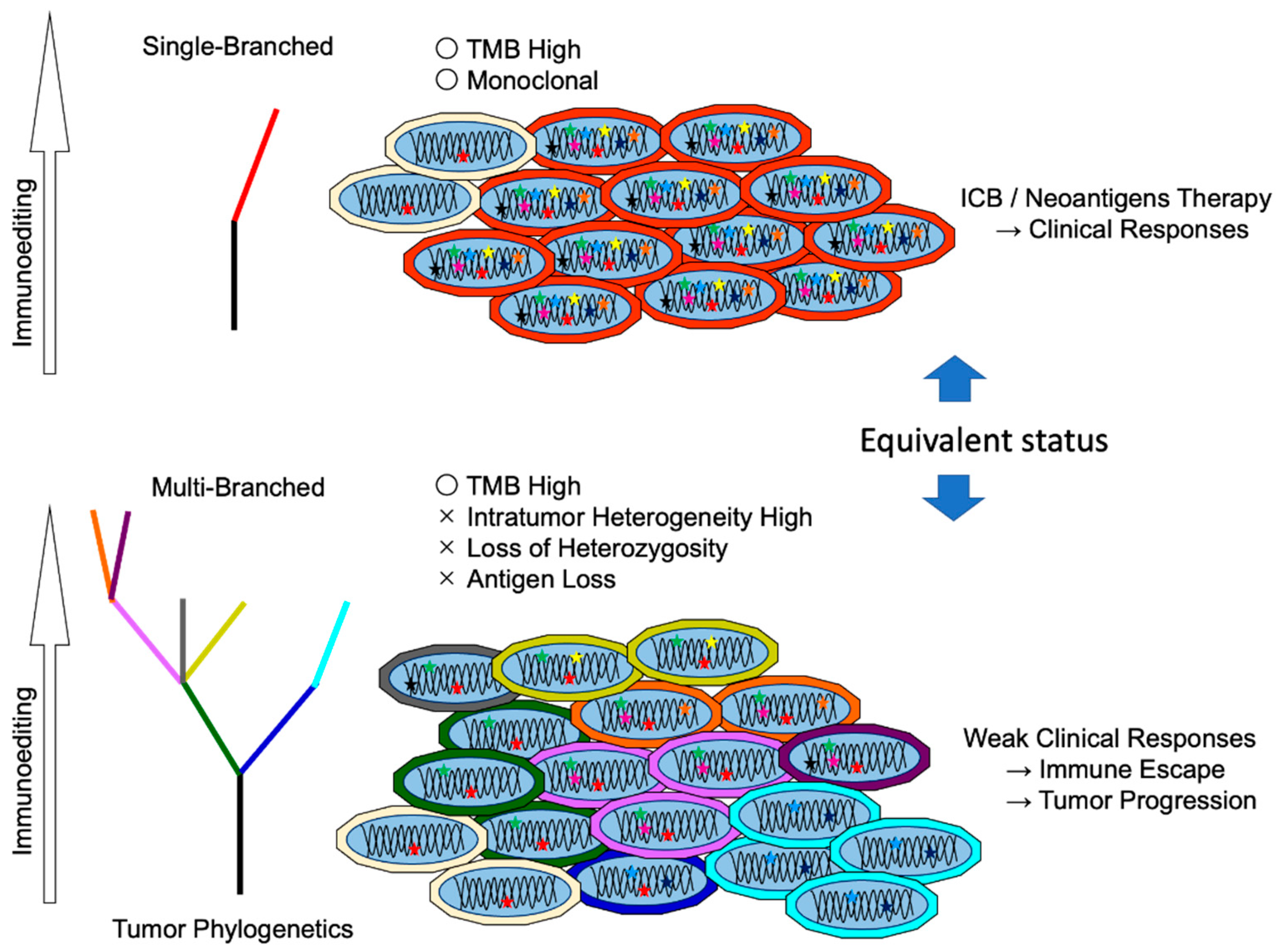
5. Neoantigen Responsiveness and Clonality
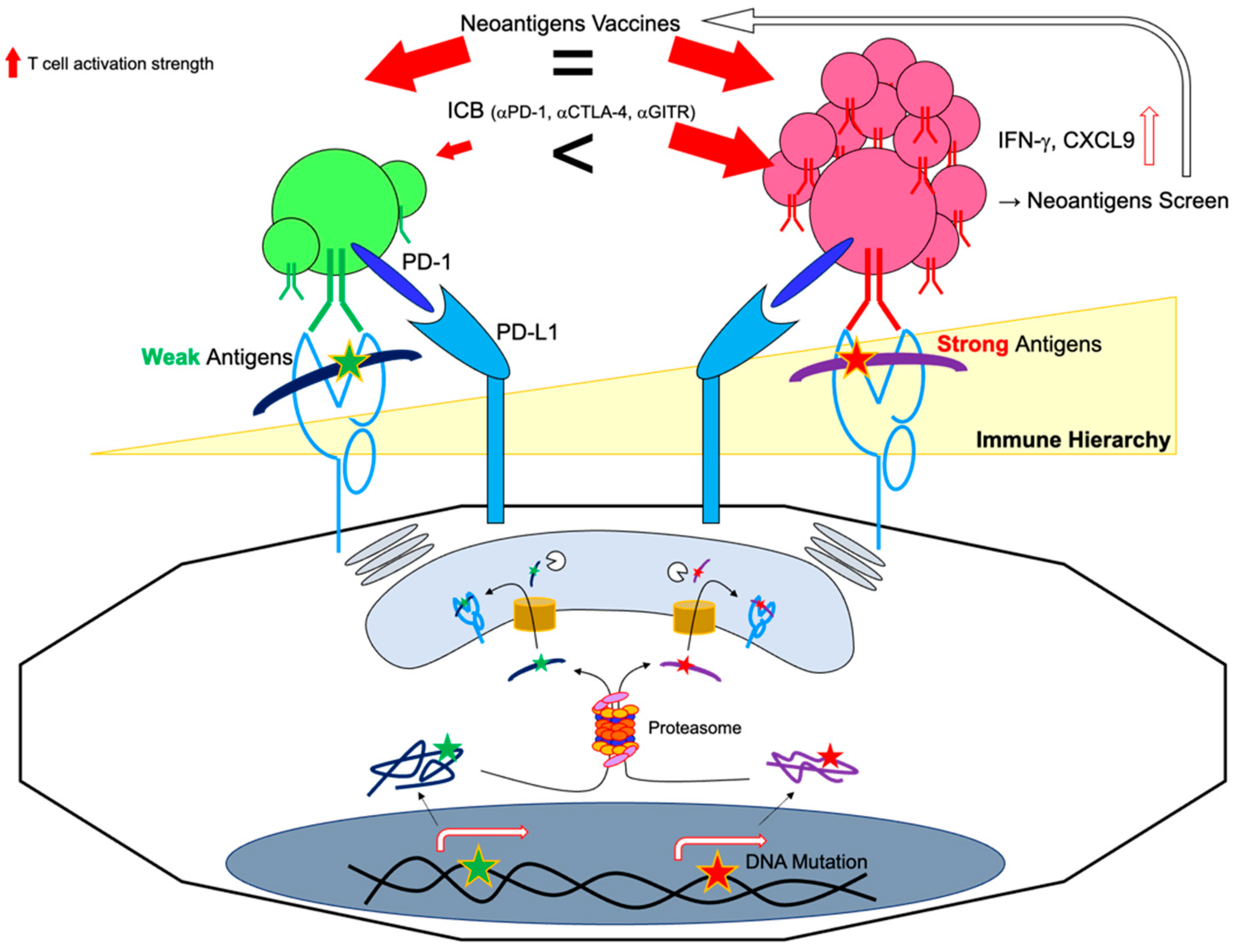
5.1. Immunodominant vs. Subdominant Neoantigens
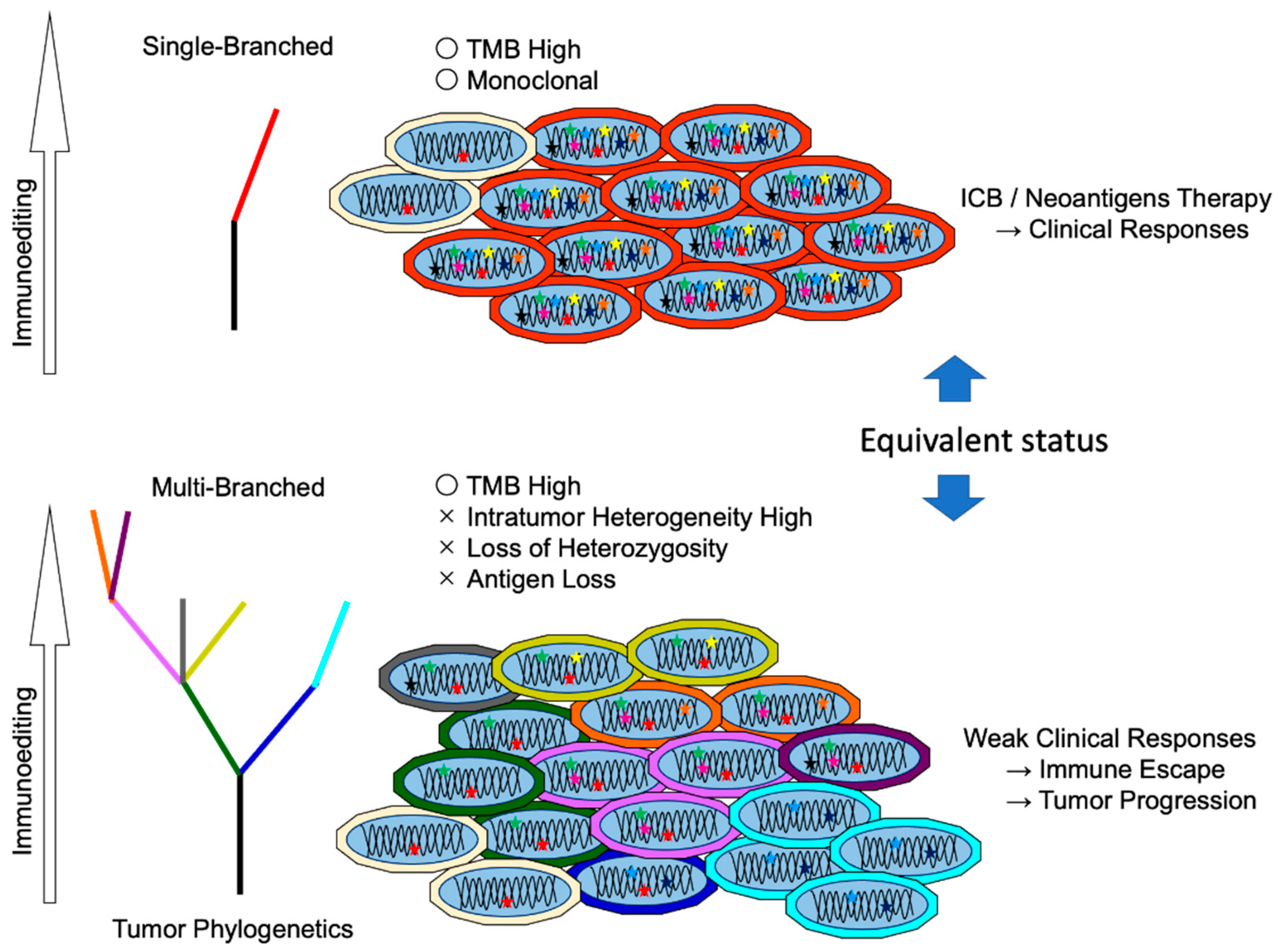
5.2. Difference between Clonal Neoantigen and Subclonal Neoantigens
6. Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in Cancer Immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Bobisse, S.; Foukas, P.G.; Coukos, G.; Harari, A. Neoantigen-Based Cancer Immunotherapy. Ann. Trans. Med. 2016, 4, 262. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.G.; Li, F.; Roszik, J.; Lizée, G. Exploiting Tumor Neoantigens to Target Cancer Evolution: Current Challenges and Promising Therapeutic Approaches. Cancer Discov. 2021, 11, 1024–1039. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef]
- Jhunjhunwala, S.; Hammer, C.; Delamarre, L. Antigen Presentation in Cancer: Insights into Tumour Immunogenicity and Immune Evasion. Nat. Rev. Cancer 2021, 21, 298–312. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of Mutational Processes in Human Cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer Immunology. Mutational Landscape Determines Sensitivity to PD-1 Blockade in Non-Small Cell Lung Cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.-J.; et al. Checkpoint Blockade Cancer Immunotherapy Targets Tumour-Specific Mutant Antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [Green Version]
- Germano, G.; Lamba, S.; Rospo, G.; Barault, L.; Magrì, A.; Maione, F.; Russo, M.; Crisafulli, G.; Bartolini, A.; Lerda, G.; et al. Inactivation of DNA Repair Triggers Neoantigen Generation and Impairs Tumour Growth. Nature 2017, 552, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [Green Version]
- Mandal, R.; Samstein, R.M.; Lee, K.-W.; Havel, J.J.; Wang, H.; Krishna, C.; Sabio, E.Y.; Makarov, V.; Kuo, F.; Blecua, P.; et al. Genetic Diversity of Tumors with Mismatch Repair Deficiency Influences Anti-PD-1 Immunotherapy Response. Science 2019, 364, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Deniger, D.C.; Pasetto, A.; Robbins, P.F.; Gartner, J.J.; Prickett, T.D.; Paria, B.C.; Malekzadeh, P.; Jia, L.; Yossef, R.; Langhan, M.M.; et al. T-Cell Responses to TP53 “Hotspot” Mutations and Unique Neoantigens Expressed by Human Ovarian Cancers. Clin. Cancer Res. 2018, 24, 5562–5573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, A.; Asmann, Y.; Cattaneo, L.; Braggio, E.; Keats, J.; Auclair, D.; Lonial, S.; MMRF CoMMpass Network; Russell, S.J.; Stewart, A.K. High Somatic Mutation and Neoantigen Burden Are Correlated with Decreased Progression-Free Survival in Multiple Myeloma. Blood Cancer J. 2017, 7, e612. [Google Scholar] [CrossRef] [Green Version]
- Robbins, P.F.; El-Gamil, M.; Li, Y.F.; Kawakami, Y.; Loftus, D.; Appella, E.; Rosenberg, S.A. A Mutated Beta-Catenin Gene Encodes a Melanoma-Specific Antigen Recognized by Tumor Infiltrating Lymphocytes. J. Exp. Med. 1996, 183, 1185–1192. [Google Scholar] [CrossRef] [Green Version]
- Wölfel, T.; Hauer, M.; Schneider, J.; Serrano, M.; Wölfel, C.; Klehmann-Hieb, E.; De Plaen, E.; Hankeln, T.; Meyer zum Büschenfelde, K.H.; Beach, D. A P16INK4a-Insensitive CDK4 Mutant Targeted by Cytolytic T Lymphocytes in a Human Melanoma. Science 1995, 269, 1281–1284. [Google Scholar] [CrossRef]
- Robinson, J.; Barker, D.J.; Georgiou, X.; Cooper, M.A.; Flicek, P.; Marsh, S.G.E. IPD-IMGT/HLA Database. Nucleic Acids Res. 2020, 48, D948–D955. [Google Scholar] [CrossRef]
- Boegel, S.; Löwer, M.; Schäfer, M.; Bukur, T.; de Graaf, J.; Boisguérin, V.; Türeci, Ö.; Diken, M.; Castle, J.C.; Sahin, U. HLA Typing from RNA-Seq Sequence Reads. Genome Med. 2012, 4, 102. [Google Scholar] [CrossRef] [Green Version]
- Szolek, A.; Schubert, B.; Mohr, C.; Sturm, M.; Feldhahn, M.; Kohlbacher, O. OptiType: Precision HLA Typing from next-Generation Sequencing Data. Bioinformatics 2014, 30, 3310–3316. [Google Scholar] [CrossRef] [Green Version]
- Buchkovich, M.L.; Brown, C.C.; Robasky, K.; Chai, S.; Westfall, S.; Vincent, B.G.; Weimer, E.T.; Powers, J.G. HLAProfiler Utilizes K-Mer Profiles to Improve HLA Calling Accuracy for Rare and Common Alleles in RNA-Seq Data. Genome Med. 2017, 9, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orenbuch, R.; Filip, I.; Comito, D.; Shaman, J.; Pe’er, I.; Rabadan, R. ArcasHLA: High-Resolution HLA Typing from RNAseq. Bioinformatics 2020, 36, 33–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yewdell, J.W.; Reits, E.; Neefjes, J. Making Sense of Mass Destruction: Quantitating MHC Class I Antigen Presentation. Nat. Rev. Immunol. 2003, 3, 952. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.; Lundegaard, C.; Lund, O.; Keşmir, C. The Role of the Proteasome in Generating Cytotoxic T-Cell Epitopes: Insights Obtained from Improved Predictions of Proteasomal Cleavage. Immunogenetics 2005, 57, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Stranzl, T.; Larsen, M.V.; Lundegaard, C.; Nielsen, M. NetCTLpan: Pan-Specific MHC Class I Pathway Epitope Predictions. Immunogenetics 2010, 62, 357–368. [Google Scholar] [CrossRef] [Green Version]
- Jurtz, V.; Paul, S.; Andreatta, M.; Marcatili, P.; Peters, B.; Nielsen, M. NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J. Immunol. 2017, 199, 3360–3368. [Google Scholar] [CrossRef] [PubMed]
- Reynisson, B.; Alvarez, B.; Paul, S.; Peters, B.; Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved Predictions of MHC Antigen Presentation by Concurrent Motif Deconvolution and Integration of MS MHC Eluted Ligand Data. Nucleic Acids Res. 2020, 48, W449–W454. [Google Scholar] [CrossRef]
- O’Donnell, T.J.; Rubinsteyn, A.; Bonsack, M.; Riemer, A.B.; Laserson, U.; Hammerbacher, J. MHCflurry: Open-Source Class I MHC Binding Affinity Prediction. Cell Syst. 2018, 7, 129–132.e4. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Sidney, J.; Pinilla, C.; Sette, A.; Peters, B. Derivation of an Amino Acid Similarity Matrix for Peptide: MHC Binding and Its Application as a Bayesian Prior. BMC Bioinform. 2009, 10, 394. [Google Scholar] [CrossRef] [Green Version]
- Abelin, J.G.; Keskin, D.B.; Sarkizova, S.; Hartigan, C.R.; Zhang, W.; Sidney, J.; Stevens, J.; Lane, W.; Zhang, G.L.; Eisenhaure, T.M.; et al. Mass Spectrometry Profiling of HLA-Associated Peptidomes in Mono-Allelic Cells Enables More Accurate Epitope Prediction. Immunity 2017, 46, 315–326. [Google Scholar] [CrossRef] [Green Version]
- Brennick, C.A.; George, M.M.; Moussa, M.M.; Hagymasi, A.T.; Seesi, S.A.; Shcheglova, T.V.; Englander, R.P.; Keller, G.L.; Balsbaugh, J.L.; Baker, B.M.; et al. An Unbiased Approach to Defining Bona Fide Cancer Neoepitopes That Elicit Immune-Mediated Cancer Rejection. J. Clin. Investig. 2021, 131, 142823. [Google Scholar] [CrossRef]
- Bulik-Sullivan, B.; Busby, J.; Palmer, C.D.; Davis, M.J.; Murphy, T.; Clark, A.; Busby, M.; Duke, F.; Yang, A.; Young, L.; et al. Deep Learning Using Tumor HLA Peptide Mass Spectrometry Datasets Improves Neoantigen Identification. Nat. Biotechnol. 2019, 37, 55–63. [Google Scholar] [CrossRef]
- Shi, Y.; Guo, Z.; Su, X.; Meng, L.; Zhang, M.; Sun, J.; Wu, C.; Zheng, M.; Shang, X.; Zou, X.; et al. DeepAntigen: A Novel Method for Neoantigen Prioritization via 3D Genome and Deep Sparse Learning. Bioinformatics 2020, 36, 4894–4901. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.H.; Qiao, R.; Xin, L.; Chen, X.; Shan, B.; Li, M. Personalized Deep Learning of Individual Immunopeptidomes to Identify Neoantigens for Cancer Vaccines. Nat. Mach. Intell. 2020, 2, 764–771. [Google Scholar] [CrossRef]
- Li, G.; Iyer, B.; Prasath, V.B.S.; Ni, Y.; Salomonis, N. DeepImmuno: Deep Learning-Empowered Prediction and Generation of Immunogenic Peptides for T-Cell Immunity. Brief. Bioinf. 2021, 22, bbab160. [Google Scholar] [CrossRef] [PubMed]
- Purcell, A.W.; Ramarathinam, S.H.; Ternette, N. Mass Spectrometry–Based Identification of MHC-Bound Peptides for Immunopeptidomics. Nat. Protoc. 2019, 14, 1687–1707. [Google Scholar] [CrossRef] [PubMed]
- Kote, S.; Pirog, A.; Bedran, G.; Alfaro, J.; Dapic, I. Mass Spectrometry-Based Identification of MHC-Associated Peptides. Cancers (Basel) 2020, 12, 535. [Google Scholar] [CrossRef] [Green Version]
- Yadav, M.; Jhunjhunwala, S.; Phung, Q.T.; Lupardus, P.; Tanguay, J.; Bumbaca, S.; Franci, C.; Cheung, T.K.; Fritsche, J.; Weinschenk, T.; et al. Predicting Immunogenic Tumour Mutations by Combining Mass Spectrometry and Exome Sequencing. Nature 2014, 515, 572–576. [Google Scholar] [CrossRef]
- Newey, A.; Griffiths, B.; Michaux, J.; Pak, H.S.; Stevenson, B.J.; Woolston, A.; Semiannikova, M.; Spain, G.; Barber, L.J.; Matthews, N.; et al. Immunopeptidomics of Colorectal Cancer Organoids Reveals a Sparse HLA Class I Neoantigen Landscape and No Increase in Neoantigens with Interferon or MEK-Inhibitor Treatment. J. Immunother Cancer 2019, 7, 309. [Google Scholar] [CrossRef]
- Ebrahimi-Nik, H.; Michaux, J.; Corwin, W.L.; Keller, G.L.J.; Shcheglova, T.; Pak, H.; Coukos, G.; Baker, B.M.; Mandoiu, I.I.; Bassani-Sternberg, M.; et al. Mass Spectrometry–Driven Exploration Reveals Nuances of Neoepitope-Driven Tumor Rejection. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, J.P.; Konda, P.; Kowalewski, D.J.; Schuster, H.; Clements, D.; Kim, Y.; Cohen, A.M.; Sharif, T.; Nielsen, M.; Stevanovic, S.; et al. MHC-I Ligand Discovery Using Targeted Database Searches of Mass Spectrometry Data: Implications for T-Cell Immunotherapies. J. Proteome Res. 2017, 16, 1806–1816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Fauteux, F.; Foote, S.; Stupak, J.; Tremblay, T.-L.; Gurnani, K.; Fulton, K.M.; Weeratna, R.D.; Twine, S.M.; Li, J. Chemical Derivatization Strategy for Extending the Identification of MHC Class I Immunopeptides. Anal. Chem. 2018, 90, 11409–11416. [Google Scholar] [CrossRef] [PubMed]
- Kochin, V.; Kanaseki, T.; Tokita, S.; Miyamoto, S.; Shionoya, Y.; Kikuchi, Y.; Morooka, D.; Hirohashi, Y.; Tsukahara, T.; Watanabe, K.; et al. HLA-A24 Ligandome Analysis of Colon and Lung Cancer Cells Identifies a Novel Cancer-Testis Antigen and a Neoantigen That Elicits Specific and Strong CTL Responses. Oncoimmunology 2017, 6, e1293214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassani-Sternberg, M.; Bräunlein, E.; Klar, R.; Engleitner, T.; Sinitcyn, P.; Audehm, S.; Straub, M.; Weber, J.; Slotta-Huspenina, J.; Specht, K.; et al. Direct Identification of Clinically Relevant Neoepitopes Presented on Native Human Melanoma Tissue by Mass Spectrometry. Nat. Commun. 2016, 7, 13404. [Google Scholar] [CrossRef] [Green Version]
- Laumont, C.M.; Vincent, K.; Hesnard, L.; Audemard, É.; Bonneil, É.; Laverdure, J.-P.; Gendron, P.; Courcelles, M.; Hardy, M.-P.; Côté, C.; et al. Noncoding Regions Are the Main Source of Targetable Tumor-Specific Antigens. Sci. Trans. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Castle, J.C.; Kreiter, S.; Diekmann, J.; Löwer, M.; van de Roemer, N.; de Graaf, J.; Selmi, A.; Diken, M.; Boegel, S.; Paret, C.; et al. Exploiting the Mutanome for Tumor Vaccination. Cancer Res. 2012, 72, 1081–1091. [Google Scholar] [CrossRef] [Green Version]
- Duan, F.; Duitama, J.; Al Seesi, S.; Ayres, C.M.; Corcelli, S.A.; Pawashe, A.P.; Blanchard, T.; McMahon, D.; Sidney, J.; Sette, A.; et al. Genomic and Bioinformatic Profiling of Mutational Neoepitopes Reveals New Rules to Predict Anticancer Immunogenicity. J. Exp. Med. 2014, 211, 2231–2248. [Google Scholar] [CrossRef]
- Kinkead, H.L.; Hopkins, A.; Lutz, E.; Wu, A.A.; Yarchoan, M.; Cruz, K.; Woolman, S.; Vithayathil, T.; Glickman, L.H.; Ndubaku, C.O.; et al. Combining STING-Based Neoantigen-Targeted Vaccine with Checkpoint Modulators Enhances Antitumor Immunity in Murine Pancreatic Cancer. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Hu, R.; Wan, Y.; Sun, F.; Wang, Z.; Yue, J.; Chen, J.; Han, G.; Wei, G.; Dong, Z. Comprehensive Mutanome Analysis of Lewis Lung Cancer Reveals Immunogenic Neoantigens for Therapeutic Vaccines. Biochem. Biophys. Res. Commun. 2020, 525, 607–613. [Google Scholar] [CrossRef]
- Schoenberger, S.P.; Toes, R.E.M.; van der Voort, E.I.H.; Offringa, R.; Melief, C.J.M. T-Cell Help for Cytotoxic T Lymphocytes Is Mediated by CD40–CD40L Interactions. Nature 1998, 393, 480–483. [Google Scholar] [CrossRef]
- Wong, S.B.J.; Bos, R.; Sherman, L.A. Tumor-Specific CD4+ T Cells Render the Tumor Environment Permissive for Infiltration by Low-Avidity CD8+ T Cells. J. Immunol. 2008, 180, 3122–3131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zander, R.; Schauder, D.; Xin, G.; Nguyen, C.; Wu, X.; Zajac, A.; Cui, W. CD4+ T Cell Help Is Required for the Formation of a Cytolytic CD8+ T Cell Subset That Protects against Chronic Infection and Cancer. Immunity 2019, 51, 1028–1042.e4. [Google Scholar] [CrossRef]
- Alspach, E.; Lussier, D.M.; Miceli, A.P.; Kizhvatov, I.; DuPage, M.; Luoma, A.M.; Meng, W.; Lichti, C.F.; Esaulova, E.; Vomund, A.N.; et al. MHC-II Neoantigens Shape Tumor Immunity and Response to Immunotherapy. Nature 2019, 574, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Quezada, S.A.; Simpson, T.R.; Peggs, K.S.; Merghoub, T.; Vider, J.; Fan, X.; Blasberg, R.; Yagita, H.; Muranski, P.; Antony, P.A.; et al. Tumor-Reactive CD4+ T Cells Develop Cytotoxic Activity and Eradicate Large Established Melanoma after Transfer into Lymphopenic Hosts. J. Exp. Med. 2010, 207, 637–650. [Google Scholar] [CrossRef] [Green Version]
- Sato, Y.; Bolzenius, J.K.; Eteleeb, A.M.; Su, X.; Maher, C.A.; Sehn, J.K.; Arora, V.K. CD4+ T Cells Induce Rejection of Urothelial Tumors after Immune Checkpoint Blockade. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Kreiter, S.; Vormehr, M.; van de Roemer, N.; Diken, M.; Löwer, M.; Diekmann, J.; Boegel, S.; Schrörs, B.; Vascotto, F.; Castle, J.C.; et al. Mutant MHC Class II Epitopes Drive Therapeutic Immune Responses to Cancer. Nature 2015, 520, 692–696. [Google Scholar] [CrossRef] [Green Version]
- Duperret, E.K.; Perales-Puchalt, A.; Stoltz, R.; G H, H.; Mandloi, N.; Barlow, J.; Chaudhuri, A.; Sardesai, N.Y.; Weiner, D.B. A Synthetic DNA, Multi-Neoantigen Vaccine Drives Predominately MHC Class I CD8+ T-Cell Responses, Impacting Tumor Challenge. Cancer Immunol. Res. 2019, 7, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.D.; Brown, S.D.; Wick, D.A.; Nielsen, J.S.; Kroeger, D.R.; Twumasi-Boateng, K.; Holt, R.A.; Nelson, B.H. Low Mutation Burden in Ovarian Cancer May Limit the Utility of Neoantigen-Targeted Vaccines. PLoS ONE 2016, 11, e0155189. [Google Scholar] [CrossRef]
- Carreno, B.M.; Magrini, V.; Becker-Hapak, M.; Kaabinejadian, S.; Hundal, J.; Petti, A.A.; Ly, A.; Lie, W.-R.; Hildebrand, W.H.; Mardis, E.R.; et al. Cancer Immunotherapy. A Dendritic Cell Vaccine Increases the Breadth and Diversity of Melanoma Neoantigen-Specific T Cells. Science 2015, 348, 803–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An Immunogenic Personal Neoantigen Vaccine for Patients with Melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.-P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA Mutanome Vaccines Mobilize Poly-Specific Therapeutic Immunity against Cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Hilf, N.; Kuttruff-Coqui, S.; Frenzel, K.; Bukur, V.; Stevanović, S.; Gouttefangeas, C.; Platten, M.; Tabatabai, G.; Dutoit, V.; van der Burg, S.H.; et al. Actively Personalized Vaccination Trial for Newly Diagnosed Glioblastoma. Nature 2019, 565, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen Vaccine Generates Intratumoral T Cell Responses in Phase Ib Glioblastoma Trial. Nature 2019, 565, 234–239. [Google Scholar] [CrossRef]
- Johanns, T.M.; Miller, C.A.; Liu, C.J.; Perrin, R.J.; Bender, D.; Kobayashi, D.K.; Campian, J.L.; Chicoine, M.R.; Dacey, R.G.; Huang, J.; et al. Detection of Neoantigen-Specific T Cells Following a Personalized Vaccine in a Patient with Glioblastoma. Oncoimmunology 2019, 8, e1561106. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu-Lieskovan, S.; Chmielowski, B.; Govindan, R.; Naing, A.; Bhardwaj, N.; Margolin, K.; Awad, M.M.; Hellmann, M.D.; Lin, J.J.; et al. A Phase Ib Trial of Personalized Neoantigen Therapy Plus Anti-PD-1 in Patients with Advanced Melanoma, Non-Small Cell Lung Cancer, or Bladder Cancer. Cell 2020, 183, 347–362.e24. [Google Scholar] [CrossRef]
- Chen, F.; Zou, Z.; Du, J.; Su, S.; Shao, J.; Meng, F.; Yang, J.; Xu, Q.; Ding, N.; Yang, Y.; et al. Neoantigen Identification Strategies Enable Personalized Immunotherapy in Refractory Solid Tumors. J. Clin. Investig. 2019, 130. [Google Scholar] [CrossRef]
- Kloor, M.; Reuschenbach, M.; Pauligk, C.; Karbach, J.; Rafiyan, M.-R.; Al-Batran, S.-E.; Tariverdian, M.; Jäger, E.; von Knebel Doeberitz, M. A Frameshift Peptide Neoantigen-Based Vaccine for Mismatch Repair-Deficient Cancers: A Phase I/IIa Clinical Trial. Clin. Cancer Res. 2020, 26, 4503–4510. [Google Scholar] [CrossRef]
- Cafri, G.; Gartner, J.J.; Zaks, T.; Hopson, K.; Levin, N.; Paria, B.C.; Parkhurst, M.R.; Yossef, R.; Lowery, F.J.; Jafferji, M.S.; et al. MRNA Vaccine–Induced Neoantigen-Specific T Cell Immunity in Patients with Gastrointestinal Cancer. J. Clin. Investig. 2020, 130, 5976–5988. [Google Scholar] [CrossRef]
- Fang, Y.; Mo, F.; Shou, J.; Wang, H.; Luo, K.; Zhang, S.; Han, N.; Li, H.; Ye, S.; Zhou, Z.; et al. A Pan-Cancer Clinical Study of Personalized Neoantigen Vaccine Monotherapy in Treating Patients with Various Types of Advanced Solid Tumors. Clin. Cancer Res. 2020, 26, 4511–4520. [Google Scholar] [CrossRef]
- Hu, Z.; Leet, D.E.; Allesøe, R.L.; Oliveira, G.; Li, S.; Luoma, A.M.; Liu, J.; Forman, J.; Huang, T.; Iorgulescu, J.B.; et al. Personal Neoantigen Vaccines Induce Persistent Memory T Cell Responses and Epitope Spreading in Patients with Melanoma. Nat. Med. 2021. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhang, S.; Han, N.; Jiang, J.; Xu, Y.; Ma, D.; Lu, L.; Guo, X.; Qiu, M.; Huang, Q.; et al. A Neoantigen-Based Peptide Vaccine for Patients With Advanced Pancreatic Cancer Refractory to Standard Treatment. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Bunse, L.; Wick, A.; Bunse, T.; Le Cornet, L.; Harting, I.; Sahm, F.; Sanghvi, K.; Tan, C.L.; Poschke, I.; et al. A Vaccine Targeting Mutant IDH1 in Newly Diagnosed Glioma. Nature 2021, 592, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Scheper, W.; Kelderman, S.; Fanchi, L.F.; Linnemann, C.; Bendle, G.; de Rooij, M.A.J.; Hirt, C.; Mezzadra, R.; Slagter, M.; Dijkstra, K.; et al. Low and Variable Tumor Reactivity of the Intratumoral TCR Repertoire in Human Cancers. Nat. Med. 2019, 25, 89–94. [Google Scholar] [CrossRef]
- Strønen, E.; Toebes, M.; Kelderman, S.; van Buuren, M.M.; Yang, W.; van Rooij, N.; Donia, M.; Böschen, M.-L.; Lund-Johansen, F.; Olweus, J.; et al. Targeting of Cancer Neoantigens with Donor-Derived T Cell Receptor Repertoires. Science 2016, 352, 1337–1341. [Google Scholar] [CrossRef]
- Forde, P.M.; Chaft, J.E.; Smith, K.N.; Anagnostou, V.; Cottrell, T.R.; Hellmann, M.D.; Zahurak, M.; Yang, S.C.; Jones, D.R.; Broderick, S.; et al. Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. N. Engl. J. Med. 2018, 378, 1976–1986. [Google Scholar] [CrossRef]
- Zolkind, P.; Przybylski, D.; Marjanovic, N.; Nguyen, L.; Lin, T.; Johanns, T.; Alexandrov, A.; Zhou, L.; Allen, C.T.; Miceli, A.P.; et al. Cancer Immunogenomic Approach to Neoantigen Discovery in a Checkpoint Blockade Responsive Murine Model of Oral Cavity Squamous Cell Carcinoma. Oncotarget 2018, 9, 4109–4119. [Google Scholar] [CrossRef] [Green Version]
- Veatch, J.R.; Singhi, N.; Jesernig, B.; Paulson, K.G.; Zalevsky, J.; Iacucci, E.; Tykodi, S.S.; Riddell, S.R. Mobilization of Pre-Existing Polyclonal T Cells Specific to Neoantigens but Not Self-Antigens during Treatment of a Patient with Melanoma with Bempegaldesleukin and Nivolumab. J. Immunother Cancer 2020, 8. [Google Scholar] [CrossRef]
- Fujii, K.; Miyahara, Y.; Harada, N.; Muraoka, D.; Komura, M.; Yamaguchi, R.; Yagita, H.; Nakamura, J.; Sugino, S.; Okumura, S.; et al. Identification of an Immunogenic Neo-Epitope Encoded by Mouse Sarcoma Using CXCR3 Ligand MRNAs as Sensors. Oncoimmunology 2017, 6, e1306617. [Google Scholar] [CrossRef]
- Okada, M.; Shimizu, K.; Iyoda, T.; Ueda, S.; Shinga, J.; Mochizuki, Y.; Watanabe, T.; Ohara, O.; Fujii, S. PD-L1 Expression Affects Neoantigen Presentation. iScience 2020, 23, 101238. [Google Scholar] [CrossRef]
- Gros, A.; Parkhurst, M.R.; Tran, E.; Pasetto, A.; Robbins, P.F.; Ilyas, S.; Prickett, T.D.; Gartner, J.J.; Crystal, J.S.; Roberts, I.M.; et al. Prospective Identification of Neoantigen-Specific Lymphocytes in the Peripheral Blood of Melanoma Patients. Nat. Med. 2016, 22, 433–438. [Google Scholar] [CrossRef]
- Simoni, Y.; Becht, E.; Fehlings, M.; Loh, C.Y.; Koo, S.-L.; Teng, K.W.W.; Yeong, J.P.S.; Nahar, R.; Zhang, T.; Kared, H.; et al. Bystander CD8+ T Cells Are Abundant and Phenotypically Distinct in Human Tumour Infiltrates. Nature 2018, 557, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Yossef, R.; Tran, E.; Deniger, D.C.; Gros, A.; Pasetto, A.; Parkhurst, M.R.; Gartner, J.J.; Prickett, T.D.; Cafri, G.; Robbins, P.F.; et al. Enhanced Detection of Neoantigen-Reactive T Cells Targeting Unique and Shared Oncogenes for Personalized Cancer Immunotherapy. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.-C.; Yao, X.; Crystal, J.S.; Li, Y.F.; El-Gamil, M.; Gross, C.; Davis, L.; Dudley, M.E.; Yang, J.C.; Samuels, Y.; et al. Efficient Identification of Mutated Cancer Antigens Recognized by T Cells Associated with Durable Tumor Regressions. Clin. Cancer Res. 2014, 20, 3401–3410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.-C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer Immunotherapy Based on Mutation-Specific CD4+ T Cells in a Patient with Epithelial Cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef]
- Cohen, C.J.; Gartner, J.J.; Horovitz-Fried, M.; Shamalov, K.; Trebska-McGowan, K.; Bliskovsky, V.V.; Parkhurst, M.R.; Ankri, C.; Prickett, T.D.; Crystal, J.S.; et al. Isolation of Neoantigen-Specific T Cells from Tumor and Peripheral Lymphocytes. J. Clin. Investig. 2015, 125, 3981–3991. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, T.; Leisegang, M.; Park, J.-H.; Ren, L.; Kato, T.; Ikeda, Y.; Harada, M.; Kiyotani, K.; Lengyel, E.; Fleming, G.F.; et al. Induction of Neoantigen-Specific Cytotoxic T Cells and Construction of T-Cell Receptor-Engineered T Cells for Ovarian Cancer. Clin. Cancer Res. 2018, 24, 5357–5367. [Google Scholar] [CrossRef] [Green Version]
- Ahmadzadeh, M.; Pasetto, A.; Jia, L.; Deniger, D.C.; Stevanović, S.; Robbins, P.F.; Rosenberg, S.A. Tumor-Infiltrating Human CD4+ Regulatory T Cells Display a Distinct TCR Repertoire and Exhibit Tumor and Neoantigen Reactivity. Sci Immunol 2019, 4, eaao4310. [Google Scholar] [CrossRef]
- Schumacher, T.; Bunse, L.; Pusch, S.; Sahm, F.; Wiestler, B.; Quandt, J.; Menn, O.; Osswald, M.; Oezen, I.; Ott, M.; et al. A Vaccine Targeting Mutant IDH1 Induces Antitumour Immunity. Nature 2014, 512, 324–327. [Google Scholar] [CrossRef]
- Khodadoust, M.S.; Olsson, N.; Wagar, L.E.; Haabeth, O.A.W.; Chen, B.; Swaminathan, K.; Rawson, K.; Liu, C.L.; Steiner, D.; Lund, P.; et al. Antigen Presentation Profiling Reveals Recognition of Lymphoma Immunoglobulin Neoantigens. Nature 2017, 543, 723–727. [Google Scholar] [CrossRef] [Green Version]
- Chheda, Z.S.; Kohanbash, G.; Okada, K.; Jahan, N.; Sidney, J.; Pecoraro, M.; Yang, X.; Carrera, D.A.; Downey, K.M.; Shrivastav, S.; et al. Novel and Shared Neoantigen Derived from Histone 3 Variant H3.3K27M Mutation for Glioma T Cell Therapy. J. Exp. Med. 2018, 215, 141–157. [Google Scholar] [CrossRef] [PubMed]
- van der Lee, D.I.; Reijmers, R.M.; Honders, M.W.; Hagedoorn, R.S.; de Jong, R.C.; Kester, M.G.; van der Steen, D.M.; de Ru, A.H.; Kweekel, C.; Bijen, H.M.; et al. Mutated Nucleophosmin 1 as Immunotherapy Target in Acute Myeloid Leukemia. J. Clin. Investig. 2019, 129, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Malekzadeh, P.; Pasetto, A.; Robbins, P.F.; Parkhurst, M.R.; Paria, B.C.; Jia, L.; Gartner, J.J.; Hill, V.; Yu, Z.; Restifo, N.P.; et al. Neoantigen Screening Identifies Broad TP53 Mutant Immunogenicity in Patients with Epithelial Cancers. J. Clin. Investig. 2019, 129, 1109–1114. [Google Scholar] [CrossRef] [PubMed]
- Malekzadeh, P.; Yossef, R.; Cafri, G.; Paria, B.C.; Lowery, F.J.; Jafferji, M.; Good, M.L.; Sachs, A.; Copeland, A.R.; Kim, S.P.; et al. Antigen Experienced T Cells from Peripheral Blood Recognize P53 Neoantigens. Clin. Cancer Res. 2020, 26, 1267–1276. [Google Scholar] [CrossRef] [Green Version]
- Lo, W.; Parkhurst, M.R.; Robbins, P.F.; Tran, E.; Lu, Y.-C.; Jia, L.; Gartner, J.J.; Pasetto, A.; Deniger, D.C.; Malekzadeh, P.; et al. Immunologic Recognition of a Shared P53 Mutated Neoantigen in a Patient with Metastatic Colorectal Cancer. Cancer Immunol. Res. 2019, 7, 534–543. [Google Scholar] [CrossRef]
- Tran, E.; Robbins, P.F.; Lu, Y.-C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.J.; Yu, Z.; Griffith, K.; Hanada, K.; Restifo, N.P.; Yang, J.C. Identification of T-Cell Receptors Targeting KRAS-Mutated Human Tumors. Cancer Immunol. Res. 2016, 4, 204–214. [Google Scholar] [CrossRef] [Green Version]
- Veatch, J.R.; Jesernig, B.L.; Kargl, J.; Fitzgibbon, M.; Lee, S.M.; Baik, C.; Martins, R.; Houghton, A.M.; Riddell, S.R. Endogenous CD4+ T Cells Recognize Neoantigens in Lung Cancer Patients, Including Recurrent Oncogenic KRAS and ERBB2 (Her2) Driver Mutations. Cancer Immunol. Res. 2019, 7, 910–922. [Google Scholar] [CrossRef] [Green Version]
- Iiizumi, S.; Ohtake, J.; Murakami, N.; Kouro, T.; Kawahara, M.; Isoda, F.; Hamana, H.; Kishi, H.; Nakamura, N.; Sasada, T. Identification of Novel HLA Class II-Restricted Neoantigens Derived from Driver Mutations. Cancers (Basel) 2019, 11, 266. [Google Scholar] [CrossRef] [Green Version]
- Makohon-Moore, A.P.; Zhang, M.; Reiter, J.G.; Bozic, I.; Allen, B.; Kundu, D.; Chatterjee, K.; Wong, F.; Jiao, Y.; Kohutek, Z.A.; et al. Limited Heterogeneity of Known Driver Gene Mutations among the Metastases of Individual Patients with Pancreatic Cancer. Nat. Genet. 2017, 49, 358–366. [Google Scholar] [CrossRef]
- Yang, W.; Lee, K.-W.; Srivastava, R.M.; Kuo, F.; Krishna, C.; Chowell, D.; Makarov, V.; Hoen, D.; Dalin, M.G.; Wexler, L.; et al. Immunogenic Neoantigens Derived from Gene Fusions Stimulate T Cell Responses. Nat. Med. 2019, 25, 767. [Google Scholar] [CrossRef] [PubMed]
- Biernacki, M.A.; Foster, K.A.; Woodward, K.B.; Coon, M.E.; Cummings, C.; Cunningham, T.M.; Dossa, R.G.; Brault, M.; Stokke, J.; Olsen, T.M.; et al. CBFB-MYH11 Fusion Neoantigen Enables T Cell Recognition and Killing of Acute Myeloid Leukemia. J. Clin. Investig. 2020, 130, 5127–5141. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Zhou, C.; Zhang, Z.; Guan, M.; Zhang, C.; Liu, Z.; Liu, Q. The Landscape of Tumor Fusion Neoantigens: A Pan-Cancer Analysis. iScience 2019, 21, 249–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cukalac, T.; Chadderton, J.; Zeng, W.; Cullen, J.G.; Kan, W.T.; Doherty, P.C.; Jackson, D.C.; Turner, S.J.; La Gruta, N.L. The Influenza Virus-Specific CTL Immunodominance Hierarchy in Mice Is Determined by the Relative Frequency of High-Avidity T Cells. J. Immunol. 2014, 192, 4061–4068. [Google Scholar] [CrossRef] [Green Version]
- Burger, M.L.; Cruz, A.M.; Crossland, G.E.; Gaglia, G.; Ritch, C.C.; Blatt, S.E.; Bhutkar, A.; Canner, D.; Kienka, T.; Tavana, S.Z.; et al. Antigen Dominance Hierarchies Shape TCF1+ Progenitor CD8 T Cell Phenotypes in Tumors. Cell 2021, 184, 4996–5014.e26. [Google Scholar] [CrossRef]
- Verdegaal, E.M.E.; de Miranda, N.F.C.C.; Visser, M.; Harryvan, T.; van Buuren, M.M.; Andersen, R.S.; Hadrup, S.R.; van der Minne, C.E.; Schotte, R.; Spits, H.; et al. Neoantigen Landscape Dynamics during Human Melanoma-T Cell Interactions. Nature 2016, 536, 91–95. [Google Scholar] [CrossRef]
- Zhang, A.W.; McPherson, A.; Milne, K.; Kroeger, D.R.; Hamilton, P.T.; Miranda, A.; Funnell, T.; Little, N.; de Souza, C.P.E.; Laan, S.; et al. Interfaces of Malignant and Immunologic Clonal Dynamics in Ovarian Cancer. Cell 2018, 173, 1755–1769.e22. [Google Scholar] [CrossRef] [Green Version]
- Jiménez-Sánchez, A.; Memon, D.; Pourpe, S.; Veeraraghavan, H.; Li, Y.; Vargas, H.A.; Gill, M.B.; Park, K.J.; Zivanovic, O.; Konner, J.; et al. Heterogeneous Tumor-Immune Microenvironments among Differentially Growing Metastases in an Ovarian Cancer Patient. Cell 2017, 170, 927–938.e20. [Google Scholar] [CrossRef] [Green Version]
- Rosenthal, R.; Cadieux, E.L.; Salgado, R.; Bakir, M.A.; Moore, D.A.; Hiley, C.T.; Lund, T.; Tanić, M.; Reading, J.L.; Joshi, K.; et al. Neoantigen-Directed Immune Escape in Lung Cancer Evolution. Nature 2019, 567, 479–485. [Google Scholar] [CrossRef]
- Jia, Q.; Wu, W.; Wang, Y.; Alexander, P.B.; Sun, C.; Gong, Z.; Cheng, J.-N.; Sun, H.; Guan, Y.; Xia, X.; et al. Local Mutational Diversity Drives Intratumoral Immune Heterogeneity in Non-Small Cell Lung Cancer. Nat. Commun. 2018, 9, 5361. [Google Scholar] [CrossRef]
- Wolf, Y.; Bartok, O.; Patkar, S.; Eli, G.B.; Cohen, S.; Litchfield, K.; Levy, R.; Jiménez-Sánchez, A.; Trabish, S.; Lee, J.S.; et al. UVB-Induced Tumor Heterogeneity Diminishes Immune Response in Melanoma. Cell 2019, 179, 219–235.e21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litchfield, K.; Reading, J.L.; Puttick, C.; Thakkar, K.; Abbosh, C.; Bentham, R.; Watkins, T.B.K.; Rosenthal, R.; Biswas, D.; Rowan, A.; et al. Meta-Analysis of Tumor- and T Cell-Intrinsic Mechanisms of Sensitization to Checkpoint Inhibition. Cell 2021, 184, 596–614.e14. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Rosenthal, R.; Hiley, C.T.; Rowan, A.J.; Watkins, T.B.K.; Wilson, G.A.; Birkbak, N.J.; Veeriah, S.; Van Loo, P.; Herrero, J.; et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 2017, 171, 1259–1271.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowell, D.; Morris, L.G.T.; Grigg, C.M.; Weber, J.K.; Samstein, R.M.; Makarov, V.; Kuo, F.; Kendall, S.M.; Requena, D.; Riaz, N.; et al. Patient HLA Class I Genotype Influences Cancer Response to Checkpoint Blockade Immunotherapy. Science 2018, 359, 582–587. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Shi, T.; Zhang, H.; Hu, J.; Song, Y.; Wei, J.; Ren, S.; Zhou, C. Tumor Neoantigens: From Basic Research to Clinical Applications. J. Hematol. Oncol. 2019, 12, 93. [Google Scholar] [CrossRef] [Green Version]
- Blass, E.; Ott, P.A. Advances in the Development of Personalized Neoantigen-Based Therapeutic Cancer Vaccines. Nat. Rev. Clin. Oncol. 2021, 18, 215–229. [Google Scholar] [CrossRef]
- Zhu, Y.; Liu, J. The Role of Neoantigens in Cancer Immunotherapy. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef]
- Lu, S.X.; De Neef, E.; Thomas, J.D.; Sabio, E.; Rousseau, B.; Gigoux, M.; Knorr, D.A.; Greenbaum, B.; Elhanati, Y.; Hogg, S.J.; et al. Pharmacologic Modulation of RNA Splicing Enhances Anti-Tumor Immunity. Cell 2021, 184, 4032–4047.e31. [Google Scholar] [CrossRef]
- Zhang, Z.; Lu, M.; Qin, Y.; Gao, W.; Tao, L.; Su, W.; Zhong, J. Neoantigen: A New Breakthrough in Tumor Immunotherapy. Front. Immunol. 2021, 12, 672356. [Google Scholar] [CrossRef]
- Zhao, X.; Pan, X.; Wang, Y.; Zhang, Y. Targeting Neoantigens for Cancer Immunotherapy. Biomark. Res. 2021, 9, 61. [Google Scholar] [CrossRef]
- Iyoda, T.; Yamasaki, S.; Kawamura, M.; Ueda, M.; Son, K.; Ito, Y.; Shimizu, K.; Fujii, S.-I. Optimal Therapeutic Strategy Using Antigen-Containing Liposomes Selectively Delivered to Antigen-Presenting Cells. Cancer Sci. 2019, 110, 875–887. [Google Scholar] [CrossRef] [PubMed]
- Kuai, R.; Ochyl, L.J.; Bahjat, K.S.; Schwendeman, A.; Moon, J.J. Designer Vaccine Nanodiscs for Personalized Cancer Immunotherapy. Nat. Mater. 2017, 16, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Lynn, G.M.; Jacobson, O.; Chen, K.; Liu, Y.; Zhang, H.; Ma, Y.; Zhang, F.; Tian, R.; Ni, Q.; et al. Albumin/Vaccine Nanocomplexes That Assemble in Vivo for Combination Cancer Immunotherapy. Nat. Commun. 2017, 8, 1954. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Yamasaki, S.; Shinga, J.; Sato, Y.; Watanabe, T.; Ohara, O.; Kuzushima, K.; Yagita, H.; Komuro, Y.; Asakura, M.; et al. Systemic DC Activation Modulates the Tumor Microenvironment and Shapes the Long-Lived Tumor-Specific Memory Mediated by CD8+ T Cells. Cancer Res. 2016, 76, 3756–3766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamasaki, S.; Shimizu, K.; Kometani, K.; Sakurai, M.; Kawamura, M.; Fujii, S.-I. In Vivo Dendritic Cell Targeting Cellular Vaccine Induces CD4+ Tfh Cell-Dependent Antibody against Influenza Virus. Sci. Rep. 2016, 6, 35173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, S.; Goto, A.; Shimizu, K. Antigen MRNA-Transfected, Allogeneic Fibroblasts Loaded with NKT-Cell Ligand Confer Antitumor Immunity. Blood 2009, 113, 4262–4272. [Google Scholar] [CrossRef]
- Lhuillier, C.; Rudqvist, N.-P.; Yamazaki, T.; Zhang, T.; Charpentier, M.; Galluzzi, L.; Dephoure, N.; Clement, C.C.; Santambrogio, L.; Zhou, X.K.; et al. Radiotherapy-Exposed CD8+ and CD4+ Neoantigens Enhance Tumor Control. J. Clin. Investig. 2021, 131, 138740. [Google Scholar] [CrossRef]
- Robbins, P.F.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; Dudley, M.E.; Wunderlich, J.R.; Nahvi, A.V.; Helman, L.J.; Mackall, C.L.; et al. Tumor Regression in Patients with Metastatic Synovial Cell Sarcoma and Melanoma Using Genetically Engineered Lymphocytes Reactive with NY-ESO-1. J. Clin. Oncol. 2011, 29, 917–924. [Google Scholar] [CrossRef]
- Rapoport, A.P.; Stadtmauer, E.A.; Binder-Scholl, G.K.; Goloubeva, O.; Vogl, D.T.; Lacey, S.F.; Badros, A.Z.; Garfall, A.; Weiss, B.; Finklestein, J.; et al. NY-ESO-1 Specific TCR Engineered T-Cells Mediate Sustained Antigen-Specific Antitumor Effects in Myeloma. Nat. Med. 2015, 21, 914–921. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.-C.; Parker, L.L.; Lu, T.; Zheng, Z.; Toomey, M.A.; White, D.E.; Yao, X.; Li, Y.F.; Robbins, P.F.; Feldman, S.A.; et al. Treatment of Patients With Metastatic Cancer Using a Major Histocompatibility Complex Class II-Restricted T-Cell Receptor Targeting the Cancer Germline Antigen MAGE-A3. J. Clin. Oncol. 2017, 35, 3322–3329. [Google Scholar] [CrossRef]
- Minagawa, A.; Yoshikawa, T.; Yasukawa, M.; Hotta, A.; Kunitomo, M.; Iriguchi, S.; Takiguchi, M.; Kassai, Y.; Imai, E.; Yasui, Y.; et al. Enhancing T Cell Receptor Stability in Rejuvenated IPSC-Derived T Cells Improves Their Use in Cancer Immunotherapy. Cell Stem. Cell 2018, 23, 850–858.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, M.; Chikuma, S.; Kondo, T.; Hibino, S.; Machiyama, H.; Yokosuka, T.; Nakano, M.; Yoshimura, A. Blockage of Core Fucosylation Reduces Cell-Surface Expression of PD-1 and Promotes Anti-Tumor Immune Responses of T Cells. Cell Rep. 2017, 20, 1017–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, C.T.; Hassan, M.; Morris, A.B.; Jeffery, J.; Lee, K.; Jagirdar, N.; Staton, A.D.; Raikar, S.S.; Spencer, H.T.; Sulchek, T.; et al. Improving T-Cell Expansion and Function for Adoptive T-Cell Therapy Using Ex Vivo Treatment with PI3Kδ Inhibitors and VIP Antagonists. Blood Adv. 2018, 2, 210–223. [Google Scholar] [CrossRef] [Green Version]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-Engineered T Cells in Patients with Refractory Cancer. Science 2020. [Google Scholar] [CrossRef] [PubMed]
- Wittman, V.P.; Woodburn, D.; Nguyen, T.; Neethling, F.A.; Wright, S.; Weidanz, J.A. Antibody Targeting to a Class I MHC-Peptide Epitope Promotes Tumor Cell Death. J. Immunol. 2006, 177, 4187–4195. [Google Scholar] [CrossRef] [Green Version]
- Ataie, N.; Xiang, J.; Cheng, N.; Brea, E.J.; Lu, W.; Scheinberg, D.A.; Liu, C.; Ng, H.L. Structure of a TCR Mimic Antibody with Target Predicts Pharmacogenetics. J. Mol. Biol 2016, 428, 194–205. [Google Scholar] [CrossRef] [Green Version]
- Skora, A.D.; Douglass, J.; Hwang, M.S.; Tam, A.J.; Blosser, R.L.; Gabelli, S.B.; Cao, J.; Diaz, L.A.; Papadopoulos, N.; Kinzler, K.W.; et al. Generation of MANAbodies Specific to HLA-Restricted Epitopes Encoded by Somatically Mutated Genes. PNAS 2015, 112, 9967–9972. [Google Scholar] [CrossRef] [Green Version]
- Kurosawa, N.; Wakata, Y.; Ida, K.; Midorikawa, A.; Isobe, M. High Throughput Development of TCR-Mimic Antibody That Targets Survivin-2B 80-88 /HLA-A*A24 and Its Application in a Bispecific T-Cell Engager. Sci. Rep. 2019, 9, 9827. [Google Scholar] [CrossRef] [Green Version]
- Low, L.; Goh, A.; Koh, J.; Lim, S.; Wang, C.-I. Targeting Mutant P53-Expressing Tumours with a T Cell Receptor-like Antibody Specific for a Wild-Type Antigen. Nat. Commun. 2019, 10, 5382. [Google Scholar] [CrossRef] [Green Version]
- Akahori, Y.; Wang, L.; Yoneyama, M.; Seo, N.; Okumura, S.; Miyahara, Y.; Amaishi, Y.; Okamoto, S.; Mineno, J.; Ikeda, H.; et al. Antitumor Activity of CAR-T Cells Targeting the Intracellular Oncoprotein WT1 Can Be Enhanced by Vaccination. Blood 2018, 132, 1134–1145. [Google Scholar] [CrossRef]
- Douglass, J.; Hsiue, E.H.-C.; Mog, B.J.; Hwang, M.S.; DiNapoli, S.R.; Pearlman, A.H.; Miller, M.S.; Wright, K.M.; Azurmendi, P.A.; Wang, Q.; et al. Bispecific Antibodies Targeting Mutant RAS Neoantigens. Sci. Immunol. 2021, 6. [Google Scholar] [CrossRef] [PubMed]
- Kather, J.N.; Pearson, A.T.; Halama, N.; Jäger, D.; Krause, J.; Loosen, S.H.; Marx, A.; Boor, P.; Tacke, F.; Neumann, U.P.; et al. Deep Learning Can Predict Microsatellite Instability Directly from Histology in Gastrointestinal Cancer. Nat. Med. 2019. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Trials | Tumor | Vaccines | Reference |
|---|---|---|---|
| NCT00683670 | Melanoma | Dendritic Cell | Carreno et al., 2015 [60] |
| NCT01970358 | Melanoma | Peptide | Ott et al., 2017 [61] |
| NCT02035956 | Melanoma | mRNA | Sahin et al., 2017 [62] |
| NCT02149225 | Glioblastoma | Peptide | Hilf et al., 2019 [63] |
| NCT02287428 | Glioblastoma | Peptide | Keskin et al., 2019 [64] |
| NCT02510950 | Glioblastoma | Peptide | Johanns et al., 2019 [65] |
| NCT02897765 | Advanced melanoma/Non-small cell lung cancer/Bladder cancer | Peptide + aPD-1 | Ott et al., 2020 [66] |
| NCT03171220 | Thymoma/Pancreatic cancer | Dendritic Cell | Chen et al., 2019 [67] |
| NCT01461148 | Colorectal cancer | Peptide | Kloor et al., 2020 [68] |
| NCT03480152 | Gastrointestinal cancer | mRNA | Cafri et al., 2020 [69] |
| NCT03662815 | Melanoma/Colon Cancer/Non-small cell lung cancer/Pancreatic Cancer/Biliary Tract Cancer/Ovarian Cancer | Peptide | Fang et al., 2020 [70] |
| NCT01970358 | Melanoma | Peptide | Hu et al., 2021 [71] |
| NCT03645148 | Advanced pancreatic cancer | Peptide | Chen et al., 2021 [72] |
| NCT02454634 | Gliomas | Peptide | Platten et al., 2021 [73] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okada, M.; Shimizu, K.; Fujii, S.-i. Identification of Neoantigens in Cancer Cells as Targets for Immunotherapy. Int. J. Mol. Sci. 2022, 23, 2594. https://doi.org/10.3390/ijms23052594
Okada M, Shimizu K, Fujii S-i. Identification of Neoantigens in Cancer Cells as Targets for Immunotherapy. International Journal of Molecular Sciences. 2022; 23(5):2594. https://doi.org/10.3390/ijms23052594
Chicago/Turabian StyleOkada, Masahiro, Kanako Shimizu, and Shin-ichiro Fujii. 2022. "Identification of Neoantigens in Cancer Cells as Targets for Immunotherapy" International Journal of Molecular Sciences 23, no. 5: 2594. https://doi.org/10.3390/ijms23052594
APA StyleOkada, M., Shimizu, K., & Fujii, S.-i. (2022). Identification of Neoantigens in Cancer Cells as Targets for Immunotherapy. International Journal of Molecular Sciences, 23(5), 2594. https://doi.org/10.3390/ijms23052594