
Investigation of the Molecular Details of the Interactions of Selenoglycosides and Human Galectin-3

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Results and Discussions
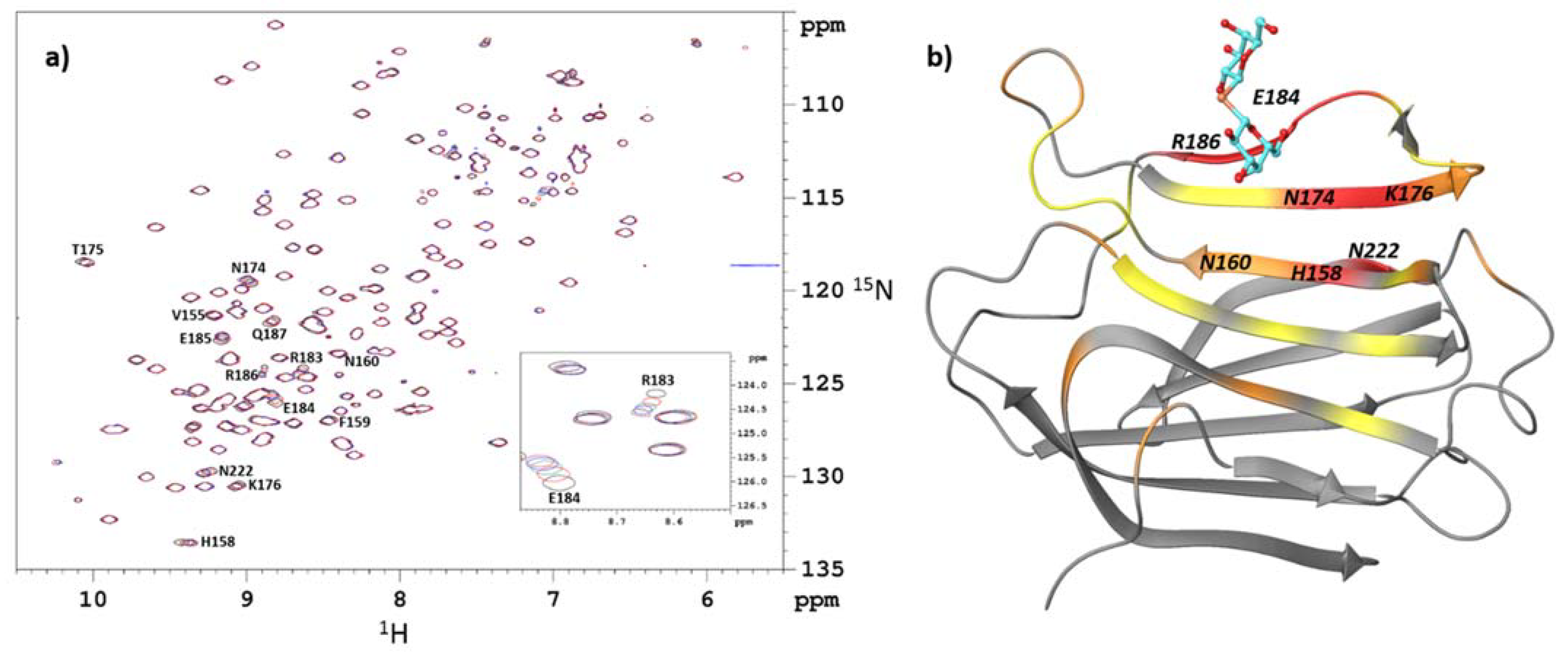
2.1. Identification of the Binding Site by NMR Chemical-Shift Mapping
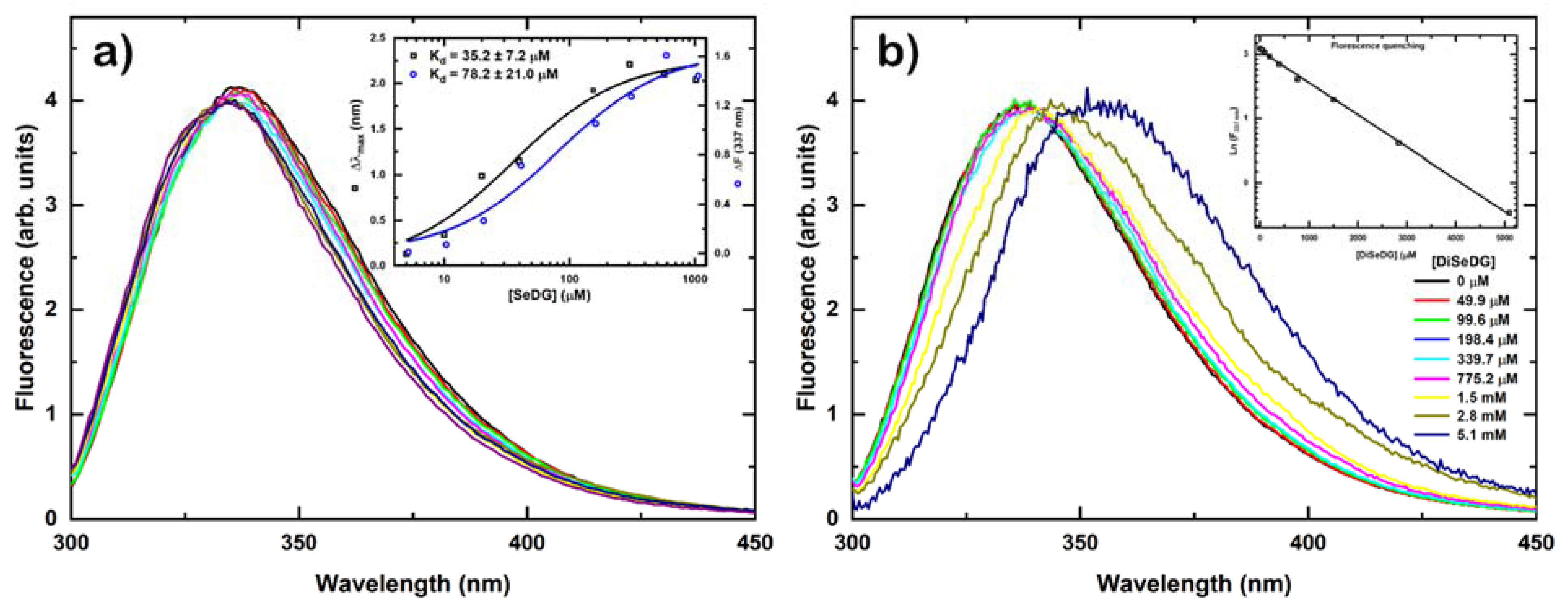
2.2. Determination of Binding Affinity by NMR Titrations
2.3. Determination of Binding Affinity by Fluorescence Anisotropy Titrations
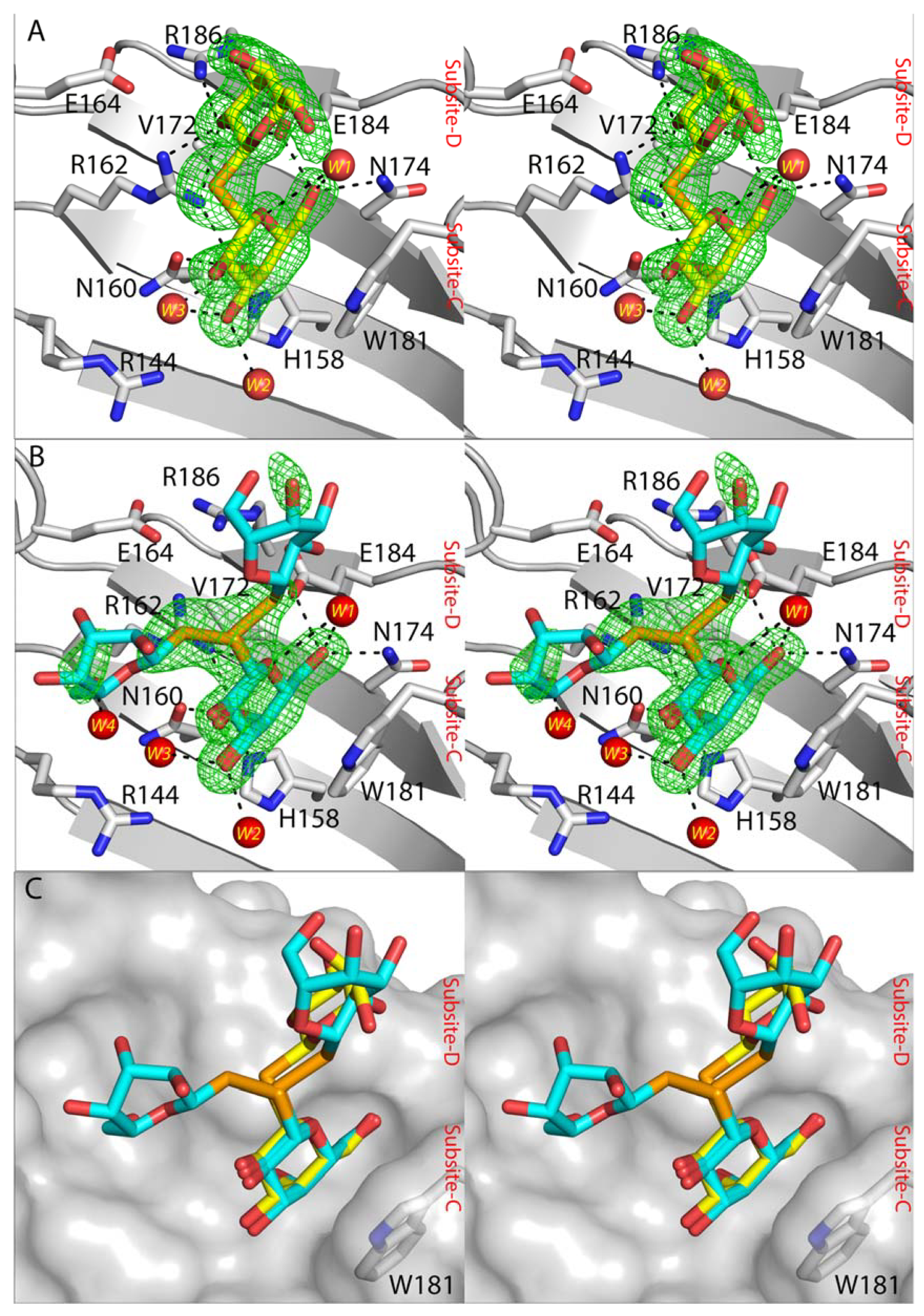
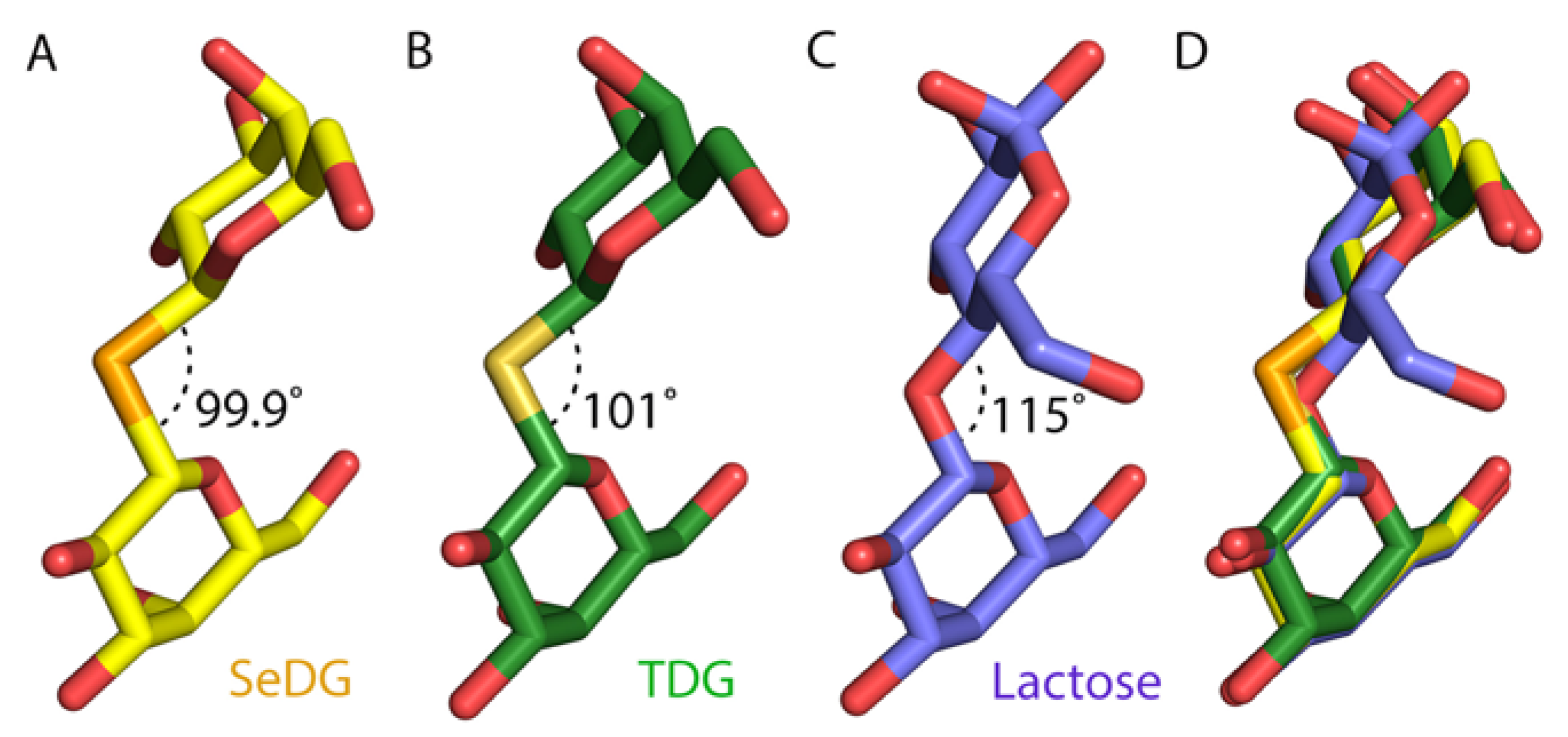
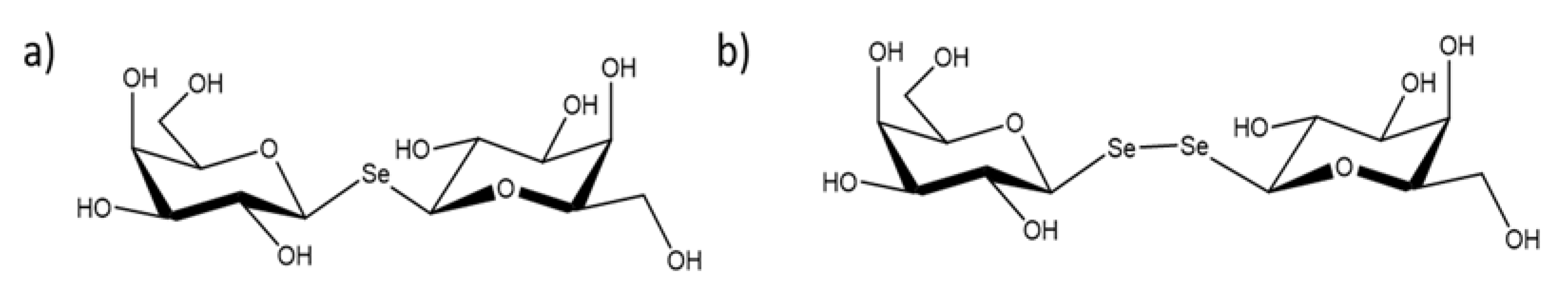
2.4. Crystal Structures of SeDG and DSeDG in Complex with hGal-3 CRD
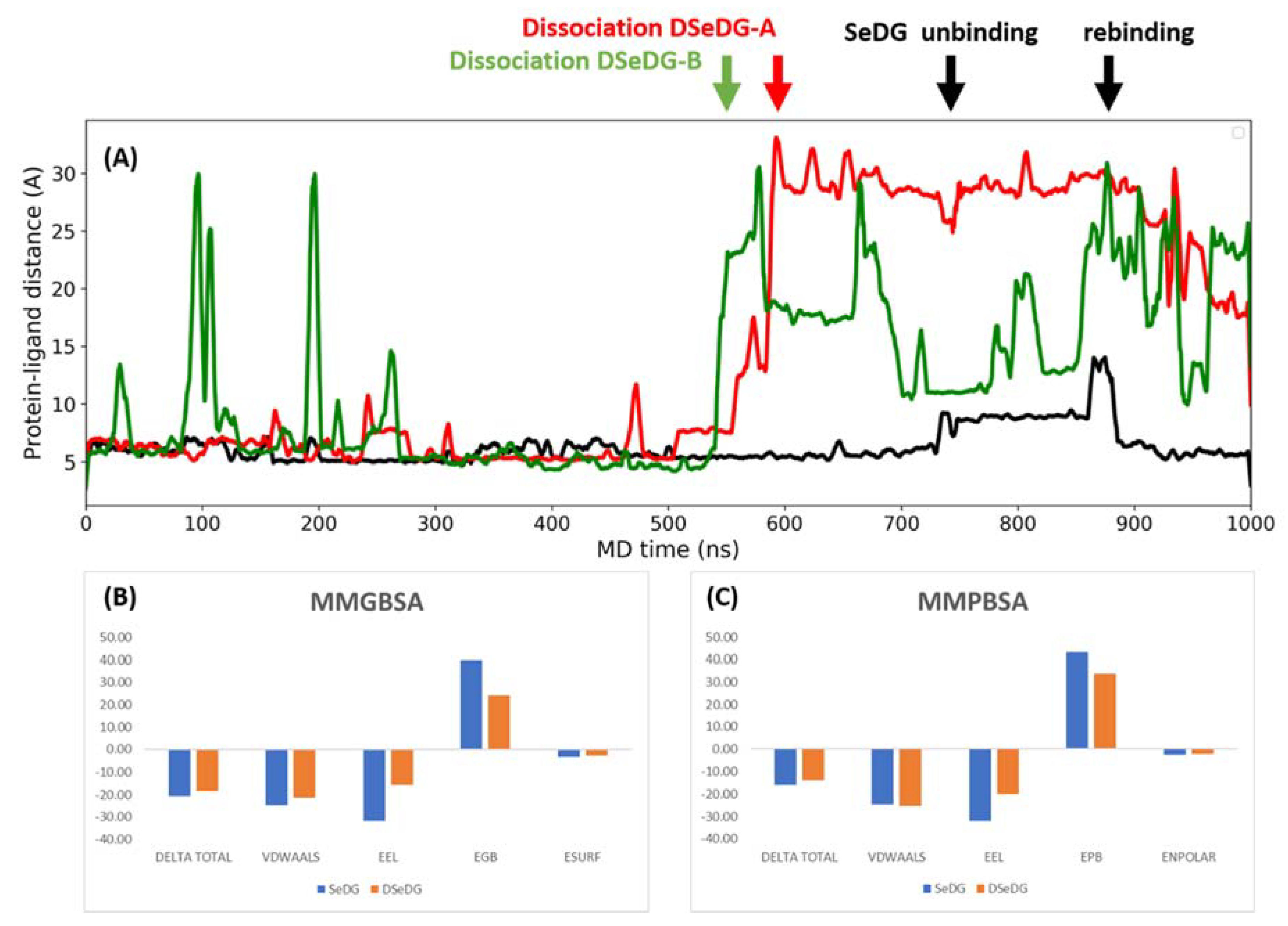
2.5. Dynamics and Energetics of the Ligand Binding by Molecular Dynamics (MD) Simulations
3. Materials and Methods
3.1. Expression and Purification of Human Galectin-3 CRD
3.2. Sample Preparation for NMR
3.3. NMR Measurements
3.4. Determination of Dissociation Constants from NMR Titration
3.5. Fluorescence Anisotropy
3.6. Crystallization and Structure Determination
3.7. Molecular Dynamics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, F.-T.; Rabinovich, G.A. Galectins: Regulators of acute and chronic inflammation. Ann. N. Y. Acad. Sci. 2010, 1183, 158–182. [Google Scholar] [CrossRef] [PubMed]
- Girotti, M.R.; Salatino, M.; Dalotto-Moreno, T.; Rabinovich, G.A. Sweetening the hallmarks of cancer: Galectins as multifunctional mediators of tumor progression. J. Exp. Med. 2019, 217, e20182041. [Google Scholar] [CrossRef] [PubMed]
- Salatino, M.; Girotti, M.R.; Rabinovich, G.A. Glycans Pave the Way for Immunotherapy in Triple-Negative Breast Cancer. Cancer Cell 2018, 33, 155–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hisrich, B.V.; Young, R.B.; Sansone, A.M.; Bowens, Z.; Green, L.J.; Lessey, B.A.; Blenda, A.V. Role of Human Galectins in Inflammation and Cancers Associated with Endometriosis. Biomolecules 2020, 10, 230. [Google Scholar] [CrossRef] [Green Version]
- Rabinovich, G.A.; Toscano, M.; Jackson, S.S.; Vasta, G.R. Functions of cell surface galectin-glycoprotein lattices. Curr. Opin. Struct. Biol. 2007, 17, 513–520. [Google Scholar] [CrossRef] [Green Version]
- Boscher, C.; Dennis, J.W.; Nabi, I.R. Glycosylation, galectins and cellular signaling. Curr. Opin. Cell Biol. 2011, 23, 383–392. [Google Scholar] [CrossRef]
- Farhad, M.; Rolig, A.S.; Redmond, W.L. The role of Galectin-3 in modulating tumor growth and immunosuppression within the tumor microenvironment. Oncoimmunology 2018, 7, e1434467. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Tang, J.-W.; Owusu, L.; Sun, M.-Z.; Wu, J.; Zhang, J. Galectin-3 in cancer. Clin. Chim. Acta 2014, 431, 185–191. [Google Scholar] [CrossRef]
- Hara, A.; Niwa, M.; Noguchi, K.; Kanayama, T.; Niwa, A.; Matsuo, M.; Hatano, Y.; Tomita, H. Galectin-3 as a Next-Generation Biomarker for Detecting Early Stage of Various Diseases. Biomolecules 2020, 10, 389. [Google Scholar] [CrossRef] [Green Version]
- Li, L.-C.; Li, J.; Gao, J. Functions of Galectin-3 and Its Role in Fibrotic Diseases. J. Pharmacol. Exp. Ther. 2014, 351, 336–343. [Google Scholar] [CrossRef] [Green Version]
- Teichberg, V.I.; Silman, I.; Beitsch, D.D.; Resheff, G. A beta-D-galactoside binding protein from electric organ tissue of Electrophorus electricus. Proc. Natl. Acad. Sci. USA 1975, 72, 1383–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sparrow, C.P.; Leffler, H.; Barondes, S.H. Multiple soluble beta-galactoside-binding lectins from human lung. J. Biol. Chem. 1987, 262, 7383–7390. [Google Scholar] [CrossRef]
- Seetharaman, J.; Kanigsberg, A.; Slaaby, R.; Leffler, H.; Barondes, S.H.; Rini, J.M. X-ray Crystal Structure of the Human Galectin-3 Carbohydrate Recognition Domain at 2.1-Å Resolution. J. Biol. Chem. 1998, 273, 13047–13052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanchard, H.; Yu, X.; Collins, P.M.; Bum-Erdene, K. Galectin-3 inhibitors: A patent review (2008–present). Expert Opin. Ther. Patents 2014, 24, 1053–1065. [Google Scholar] [CrossRef]
- Öberg, C.T.; Leffler, H.; Nilsson, U.J. Inhibition of Galectins with Small Molecules. CHIMIA Int. J. Chem. 2011, 65, 18–23. [Google Scholar] [CrossRef]
- Szilagyi, L.; Varela, O. Non-Conventional Glycosidic Linkages: Syntheses and Structures of Thiooligosaccharides and Carbohydrates with Three-Bond Glycosidic Connections. Curr. Org. Chem. 2006, 10, 1745–1770. [Google Scholar] [CrossRef]
- Leffler, H.; Barondes, S.H. Specificity of binding of three soluble rat lung lectins to substituted and unsubstituted mammalian beta-galactosides. J. Biol. Chem. 1986, 261, 10119–10126. [Google Scholar] [CrossRef]
- Ito, K.; Scott, S.A.; Cutler, S.; Dong, L.; Neuzil, J.; Blanchard, H.; Ralph, S.J. Thiodigalactoside inhibits murine cancers by concurrently blocking effects of galectin-1 on immune dysregulation, angiogenesis and protection against oxidative stress. Angiogenesis 2011, 14, 293–307. [Google Scholar] [CrossRef] [Green Version]
- Cumpstey, I.; Sundin, A.; Leffler, H.; Nilsson, U.J. C2-Symmetrical Thiodigalactoside Bis-Benzamido Derivatives as High-Affinity Inhibitors of Galectin-3: Efficient Lectin Inhibition through Double Arginine–Arene Interactions. Angew. Chem. Int. Ed. 2005, 44, 5110–5112. [Google Scholar] [CrossRef]
- van Hattum, H.; Branderhorst, H.M.; Moret, E.E.; Nilsson, U.J.; Leffler, H.; Pieters, R.J. Tuning the Preference of Thiodigalactoside- and Lactosamine-Based Ligands to Galectin-3 over Galectin-1. J. Med. Chem. 2013, 56, 1350–1354. [Google Scholar] [CrossRef]
- Hsieh, T.-J.; Lin, H.-Y.; Tu, Z.; Lin, T.-C.; Wu, S.-C.; Tseng, Y.-Y.; Liu, F.-T.; Hsu, S.-T.D.; Lin, C.-H. Dual thio-digalactoside-binding modes of human galectins as the structural basis for the design of potent and selective inhibitors. Sci. Rep. 2016, 6, 29457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, K.; Kumar, R.; Stenström, O.; Verma, P.; Verma, P.R.; Håkansson, M.; Kahl-Knutsson, B.; Zetterberg, F.; Leffler, H.; Akke, M.; et al. Systematic Tuning of Fluoro-galectin-3 Interactions Provides Thiodigalactoside Derivatives with Single-Digit nM Affinity and High Selectivity. J. Med. Chem. 2018, 61, 1164–1175. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Hartz, R.A.; Beno, B.R.; Ghosh, K.; Shukla, J.K.; Kumar, A.; Patel, D.; Kalidindi, N.; Lemos, N.; Gautam, S.S.; et al. Synthesis, Structure–Activity Relationships, and In Vivo Evaluation of Novel Tetrahydropyran-Based Thiodisaccharide Mimics as Galectin-3 Inhibitors. J. Med. Chem. 2021, 64, 6634–6655. [Google Scholar] [CrossRef]
- Kostlánová, N.; Mitchell, E.P.; Lortat-Jacob, H.; Oscarson, S.; Lahmann, M.; Gilboa-Garber, N.; Chambat, G.; Wimmerová, M.; Imberty, A. The fucose-binding lectin from Ralstonia solanacearum. A new type of beta-propeller architecture formed by oligomerization and interacting with fucoside, fucosyllactose, and plant xyloglucan. J. Biol. Chem. 2005, 280, 27839–27849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimabukuro, J.; Makyio, H.; Suzuki, T.; Nishikawa, Y.; Kawasaki, M.; Imamura, A.; Ishida, H.; Ando, H.; Kato, R.; Kiso, M. Synthesis of seleno-fucose compounds and their application to the X-ray structural determination of carbohydrate-lectin complexes using single/multi-wavelength anomalous dispersion phasing. Bioorg. Med. Chem. 2016, 25, 1132–1142. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Makyio, H.; Ando, H.; Komura, N.; Menjo, M.; Yamada, Y.; Imamura, A.; Ishida, H.; Wakatsuki, S.; Kato, R.; et al. Expanded potential of seleno-carbohydrates as a molecular tool for X-ray structural determination of a carbohydrate–protein complex with single/multi-wavelength anomalous dispersion phasing. Bioorg. Med. Chem. 2014, 22, 2090–2101. [Google Scholar] [CrossRef]
- Uzawa, J.; Shimabukuro, J.; Suzuki, T.; Imamura, A.; Ishida, H.; Ando, H.; Yamaguchi, Y. J(77Se,1H) and J(77Se,13C) couplings of seleno-carbohydrates obtained by 77Se satellite 1D13C spectroscopy and 77Se selective HR-HMBC spectroscopy. Org. Magn. Reson. 2018, 56, 836–846. [Google Scholar] [CrossRef]
- Timári, I.; Balla, S.; Fehér, K.; Kövér, K.E.; Szilágyi, L. 77Se-Enriched Selenoglycoside Enables Significant Enhancement in NMR Spectroscopic Monitoring of Glycan–Protein Interactions. Pharmaceutics 2022, 14, 201. [Google Scholar] [CrossRef]
- Suzuki, T.; Hayashi, C.; Komura, N.; Tamai, R.; Uzawa, J.; Ogawa, J.; Tanaka, H.-N.; Imamura, A.; Ishida, H.; Kiso, M.; et al. Synthesis and Glycan–Protein Interaction Studies of Se-Sialosides by 77Se NMR. Org. Lett. 2019, 21, 6393–6396. [Google Scholar] [CrossRef]
- André, S.; Kövér, K.E.; Gabius, H.-J.; Szilágyi, L. Thio- and selenoglycosides as ligands for biomedically relevant lectins: Valency–activity correlations for benzene-based dithiogalactoside clusters and first assessment for (di)selenodigalactosides. Bioorg. Med. Chem. Lett. 2014, 25, 931–935. [Google Scholar] [CrossRef] [Green Version]
- Williamson, M.P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 73, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Bum-Erdene, K.; Gagarinov, I.A.; Collins, P.M.; Winger, M.; Pearson, A.G.; Wilson, J.C.; Leffler, H.; Nilsson, U.J.; Grice, I.D.; Blanchard, H. Investigation into the Feasibility of Thioditaloside as a Novel Scaffold for Galectin-3-Specific Inhibitors. ChemBioChem 2013, 14, 1331–1342. [Google Scholar] [CrossRef] [PubMed]
- Halimi, H.; Rigato, A.; Byrne, D.; Ferracci, G.; Sebban-Kreuzer, C.; ElAntak, L.; Guerlesquin, F. Glycan Dependence of Galectin-3 Self-Association Properties. PLoS ONE 2014, 9, e111836. [Google Scholar] [CrossRef] [PubMed]
- Ippel, H.; Miller, M.C.; Vértesy, S.; Zheng, Y.; Cañada, F.J.; Suylen, D.; Umemoto, K.; Romanò, C.; Hackeng, T.; Tai, G.; et al. Intra- and intermolecular interactions of human galectin-3: Assessment by full-assignment-based NMR. Glycobiology 2016, 26, 888–903. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Ippel, H.; Miller, M.C.; Wong, T.J.; Griffioen, A.W.; Mayo, K.H.; Pieters, R.J. Hybrid ligands with calixarene and thiodigalactoside groups: Galectin binding and cytotoxicity. Org. Chem. Front. 2019, 6, 2981–2990. [Google Scholar] [CrossRef]
- Miller, M.C.; Cai, C.; Wichapong, K.; Bhaduri, S.; Pohl, N.L.B.; Linhardt, R.J.; Gabius, H.-J.; Mayo, K.H. Structural insight into the binding of human galectins to corneal keratan sulfate, its desulfated form and related saccharides. Sci. Rep. 2020, 10, 15708. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Miller, M.C.; Xu, X.J.; Song, C.C.; Zhang, F.; Zheng, Y.; Zhou, Y.F.; Tai, G.H.; Mayo, K.H. NMR-based insight into galectin-3 binding to endothelial cell adhesion molecule CD146: Evidence for noncanonical interactions with the lectin’s CRD beta-sandwich F-face. Glycobiology 2019, 29, 608–618. [Google Scholar] [CrossRef]
- Miller, M.C.; Zheng, Y.; Suylen, D.; Ippel, H.; Cañada, F.J.; Berbís, M.A.; Jiménez-Barbero, J.; Tai, G.; Gabius, H.; Mayo, K.H. Targeting the CRD F-face of Human Galectin-3 and Allosterically Modulating Glycan Binding by Angiostatic PTX008 and a Structurally Optimized Derivative. ChemMedChem 2020, 16, 713–723. [Google Scholar] [CrossRef]
- Lima, C.D.L.; Coelho, H.; Gimeno, A.; Trovão, F.; Diniz, A.; Dias, J.S.; Jiménez-Barbero, J.; Corzana, F.; Carvalho, A.L.; Cabrita, E.J.; et al. Structural Insights into the Molecular Recognition Mechanism of the Cancer and Pathogenic Epitope, LacdiNAc by Immune-Related Lectins. Chem. Eur. J. 2021, 27, 7951–7958. [Google Scholar] [CrossRef]
- Diercks, T.; Medrano, F.J.; FitzGerald, F.G.; Beckwith, D.; Pedersen, M.J.; Reihill, M.; Ludwig, A.; Romero, A.; Oscarson, S.; Cudic, M.; et al. Galectin–Glycan Interactions: Guidelines for Monitoring by 77 Se NMR Spectroscopy, and Solvent (H2O/D2O) Impact on Binding. Chem. Eur. J. 2020, 27, 316–325. [Google Scholar] [CrossRef]
- Collins, P.M.; Hidari, K.I.P.J.; Blanchard, H. Slow diffusion of lactose out of galectin-3 crystals monitored by X-ray crystallography: Possible implications for ligand-exchange protocols. Acta Crystallogr. Sect. D Biol. Crystallogr. 2007, 63, 415–419. [Google Scholar] [CrossRef]
- Dussouy, C.; Kishor, C.; Lambert, A.; Lamoureux, C.; Blanchard, H.; Grandjean, C. Linear triazole-linked pseudo oligogalactosides as scaffolds for galectin inhibitor development. Chem. Biol. Drug Des. 2020, 96, 1123–1133. [Google Scholar] [CrossRef] [PubMed]
- Atmanene, C.; Ronin, C.; Téletchéa, S.; Gautier, F.-M.; Djedaïni-Pilard, F.; Ciesielski, F.; Vivat, V.; Grandjean, C. Biophysical and structural characterization of mono/di-arylated lactosamine derivatives interaction with human galectin-3. Biochem. Biophys. Res. Commun. 2017, 489, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Fehér, K.; Matthews, R.P.; Kövér, K.E.; Naidoo, K.J.; Szilágyi, L. Conformational preferences in diglycosyl disulfides: NMR and molecular modeling studies. Carbohydr. Res. 2011, 346, 2612–2621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartok, A.; Fehér, K.; Bodor, A.; Rákosi, K.; Tóth, G.K.; Kövér, K.E.; Panyi, G.; Varga, Z. An engineered scorpion toxin analogue with improved Kv1.3 selectivity displays reduced conformational flexibility. Sci. Rep. 2015, 5, 18397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fehér, K.; Timári, I.; Rákosi, K.; Szolomájer, J.; Illyés, T.Z.; Bartok, A.; Varga, Z.; Panyi, G.; Tóth, G.K.; Kövér, K.E. Probing pattern and dynamics of disulfide bridges using synthesis and NMR of an ion channel blocker peptide toxin with multiple diselenide bonds. Chem. Sci. 2015, 7, 2666–2673. [Google Scholar] [CrossRef] [Green Version]
- Zetterberg, F.R.; Peterson, K.; Johnsson, R.E.; Brimert, T.; Håkansson, M.; Logan, D.; Leffler, H.; Nilsson, U.J. Monosaccharide Derivatives with Low-Nanomolar Lectin Affinity and High Selectivity Based on Combined Fluorine-Amide, Phenyl-Arginine, Sulfur-π, and Halogen Bond Interactions. ChemMedChem 2018, 13, 133–137. [Google Scholar] [CrossRef] [Green Version]
- Balogh, G.; Gyöngyösi, T.; Timári, I.; Herczeg, M.; Borbás, A.; Sadiq, S.K.; Fehér, K.; Kövér, K.E. Conformational Analysis of Heparin-Analogue Pentasaccharides by Nuclear Magnetic Resonance Spectroscopy and Molecular Dynamics Simulations. J. Chem. Inf. Model. 2021, 61, 2926–2936. [Google Scholar] [CrossRef]
- Srinivasan, J.; Cheatham, T.E.; Cieplak, P.; Kollman, P.A.; Case, D.A. Continuum Solvent Studies of the Stability of DNA, RNA, and Phosphoramidate—DNA Helices. J. Am. Chem. Soc. 1998, 120, 9401–9409. [Google Scholar] [CrossRef]
- Umemoto, K.; Leffler, H. Letter to the Editor: Assignment of H-1, N-15 and C-13 resonances of the carbohydrate recognition domain of human galectin-3. J. Biomol. NMR 2001, 20, 91–92. [Google Scholar] [CrossRef]
- Mertens, M.L.; Kägi, J.H.R. A graphical correction procedure for inner filter effect in fluorescence quenching titrations. Anal. Biochem. 1979, 96, 448–455. [Google Scholar] [CrossRef]
- Cowieson, N.P.; Aragao, D.; Clift, M.; Ericsson, D.; Gee, C.; Harrop, S.J.; Mudie, N.; Panjikar, S.; Price, J.; Riboldi-Tunnicliffe, A.; et al. MX1: A bending-magnet crystallography beamline serving both chemical and macromolecular crystallography communities at the Australian Synchrotron. J. Synchrotron Radiat. 2015, 22, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D Struct. Biol. 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Crystallogr. Sect. D Biol. Crystallogr. 1997, 53, 240–255. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D-Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Schuttelkopf, A.W.; van Aalten, D.M.F. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. Sect. D-Struct. Biol. 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Darden, T.A.; Cheatham, T.E.; Simmerling, C.L.I.; Wang, J.; Duke, R.E.; Luo, R.; Walker, R.C.; Zhang, W.; Merz, K.M.; et al. AMBER 12; University of California: San Francisco, CA, USA, 2012. [Google Scholar]
- Le Grand, S.; Götz, A.W.; Walker, R.C. SPFP: Speed without compromise—A mixed precision model for GPU accelerated molecular dynamics simulations. Comput. Phys. Commun. 2013, 184, 374–380. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Gotz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Vanquelef, E.; Simon, S.; Marquant, G.; Garcia, E.; Klimerak, G.; Delepine, J.C.; Cieplak, P.; Dupradeau, F.-Y.R.E.D. Server: A web service for deriving RESP and ESP charges and building force field libraries for new molecules and molecular fragments. Nucleic Acids Res. 2011, 39, W511–W517. [Google Scholar] [CrossRef] [Green Version]
- Dupradeau, F.-Y.; Pigache, A.; Zaffran, T.; Savineau, C.; Lelong, R.; Grivel, N.; Lelong, D.; Rosanski, W.; Cieplak, P. The R.E.D. tools: Advances in RESP and ESP charge derivation and force field library building. Phys. Chem. Chem. Phys. 2010, 12, 7821–7839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. Model. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Miller, B.R., 3rd; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Schroedinger, L. The PyMOL Molecular Graphics System; Version 1.2r3pre; Schrödinger Inc., LLC.: New York, NY, USA, 2011. [Google Scholar]
- Umemoto, K.; Leffler, H.; Venot, A.; Valafar, A.H.; Prestegard, J.H. Conformational Differences in Liganded and Unliganded States of Galectin-3. Biochemistry 2003, 42, 3688–3695. [Google Scholar] [CrossRef]
- Rauthu, S.R.; Shiao, T.C.; André, S.; Miller, M.C.; Madej, É.; Mayo, K.H.; Gabius, H.-J.; Roy, R. Defining the Potential of Aglycone Modifications for Affinity/Selectivity Enhancement against Medically Relevant Lectins: Synthesis, Activity Screening, and HSQC-Based NMR Analysis. ChemBioChem 2014, 16, 126–139. [Google Scholar] [CrossRef]
- Bianchet, M.A.; Ahmed, H.; Vasta, G.R.; Amzel, L.M. Soluble beta-galactosyl-binding lectin (galectin) from toad ovary: Crystallographic studies of two protein-sugar complexes. Proteins 2000, 40, 378–388. [Google Scholar] [CrossRef]
- Stannard, K.A.; Collins, P.M.; Ito, K.; Sullivan, E.M.; Scott, S.A.; Gabutero, E.; Grice, I.D.; Low, P.; Nilsson, U.J.; Leffler, H.; et al. Galectin inhibitory disaccharides promote tumour immunity in a breast cancer model. Cancer Lett. 2010, 299, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Raics, M.; Timári, I.; Diercks, T.; Szilágyi, L.; Gabius, H.; Kövér, K.E. Selenoglycosides as Lectin Ligands: 77 Se-Edited CPMG-HSQMBC NMR Spectroscopy To Monitor Biomedically Relevant Interactions. ChemBioChem 2019, 20, 1688–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raics, M.; Timári, I.; Szilágyi, L.; Gabius, H.-J.; Kövér, K.E. Introducing 77Se NMR Spectroscopy to Analyzing Galectin–Ligand Interaction. In Galectins: Methods and Protocols, Methods in Molecular Biology; Stowell, S.R., Arthur, C.M., Cummings, R.D., Eds.; Springer Science+Business Media, LLC.: Berlin/Heidelberg, Germany, 2022; Volume 2442, p. 22. [Google Scholar]
- Martínez, J.D.; Manzano, A.I.; Calviño, E.; de Diego, A.; de Francisco, B.R.; Romanò, C.; Oscarson, S.; Millet, O.; Gabius, H.-J.; Jiménez-Barbero, J.; et al. Fluorinated Carbohydrates as Lectin Ligands: Simultaneous Screening of a Monosaccharide Library and Chemical Mapping by 19F NMR Spectroscopy. J. Org. Chem. 2020, 85, 16072–16081. [Google Scholar] [CrossRef]
- Martínez, J.D.; Infantino, A.S.; Valverde, P.; Diercks, T.; Delgado, S.; Reichardt, N.-C.; Ardá, A.; Cañada, F.J.; Oscarson, S.; Jiménez-Barbero, J. The Interaction of Fluorinated Glycomimetics with DC-SIGN: Multiple Binding Modes Disentangled by the Combination of NMR Methods and MD Simulations. Pharmaceuticals 2020, 13, 179. [Google Scholar] [CrossRef]
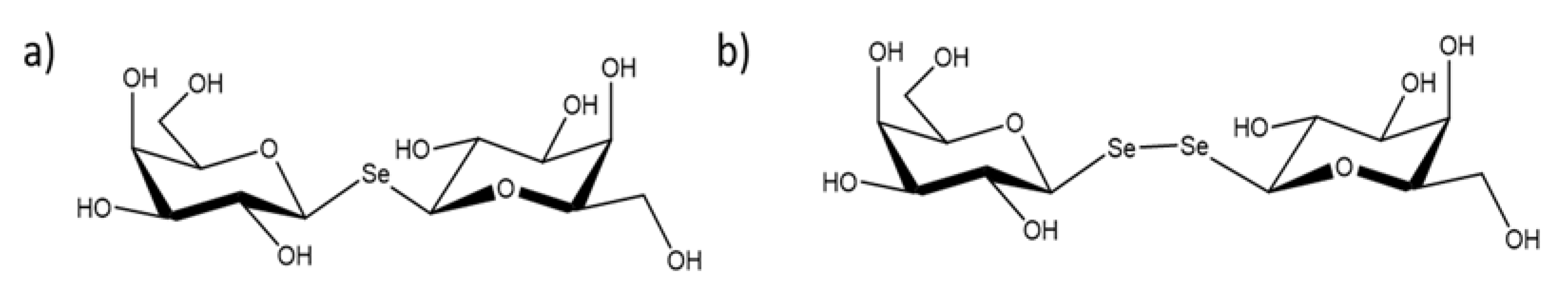
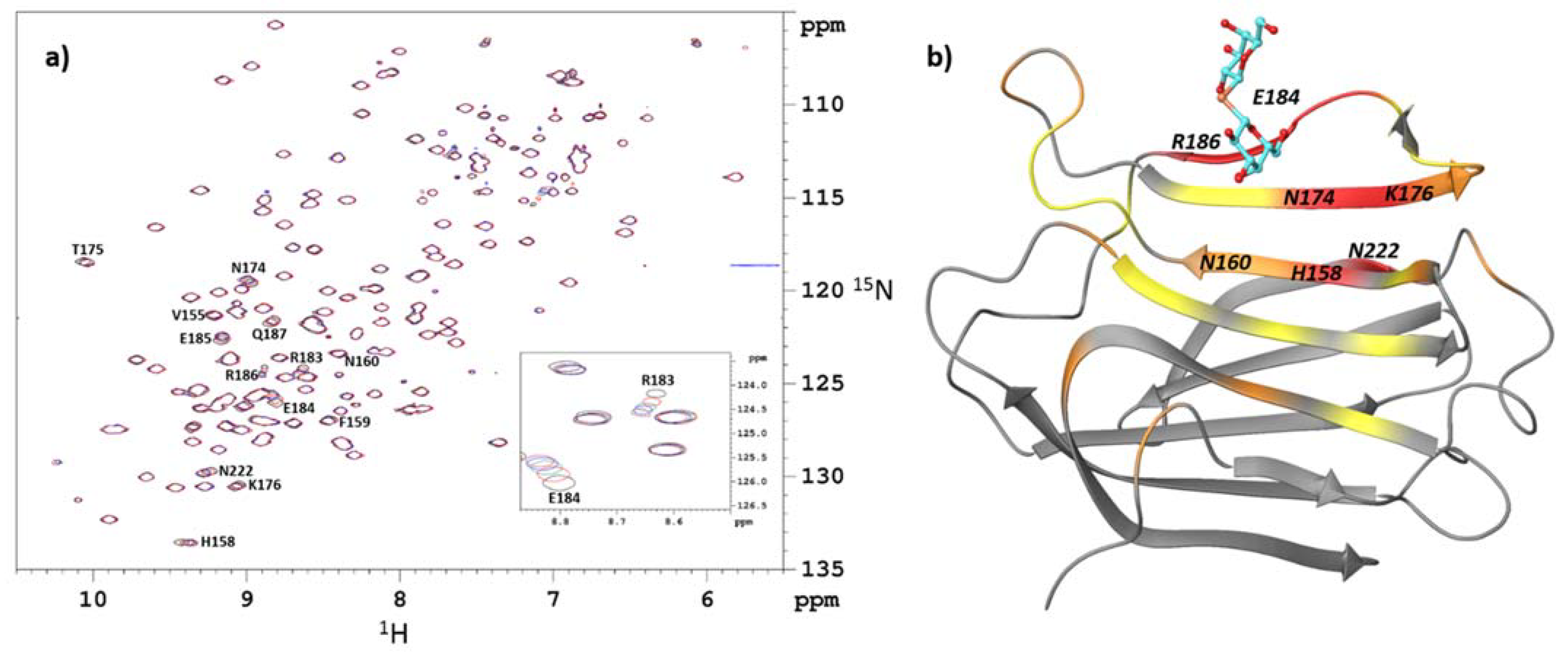
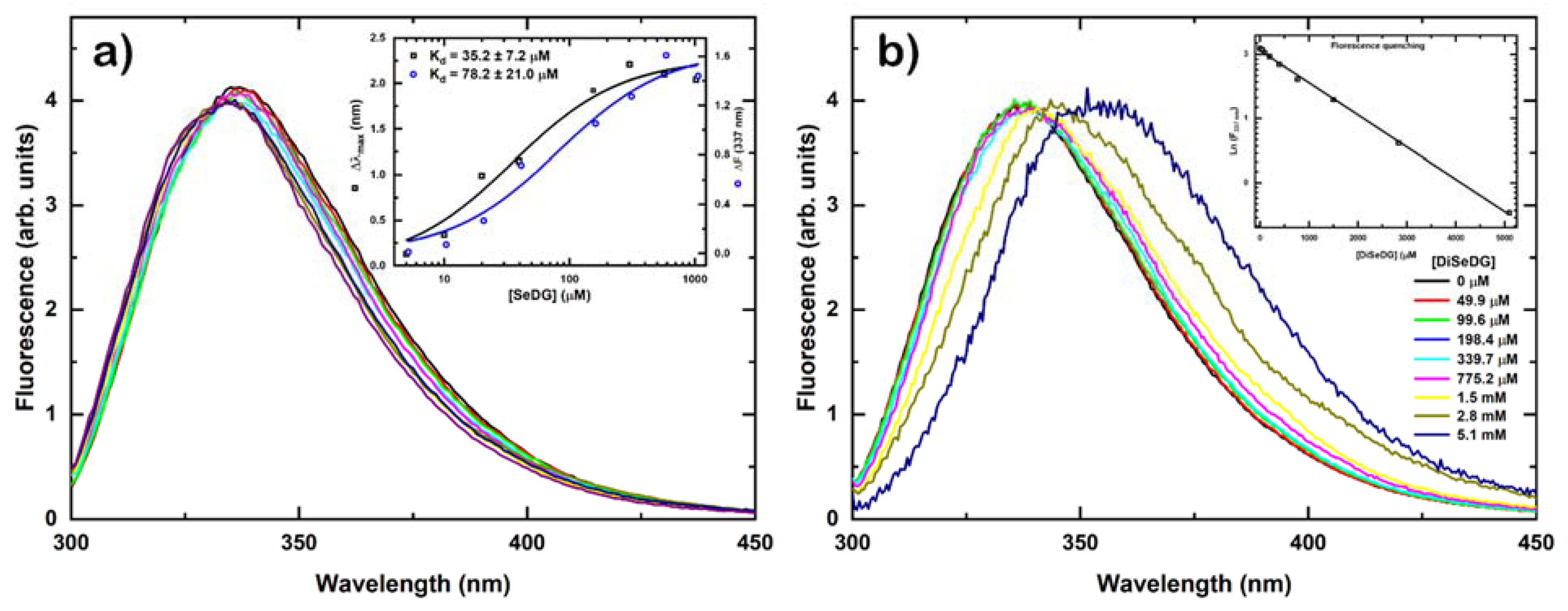

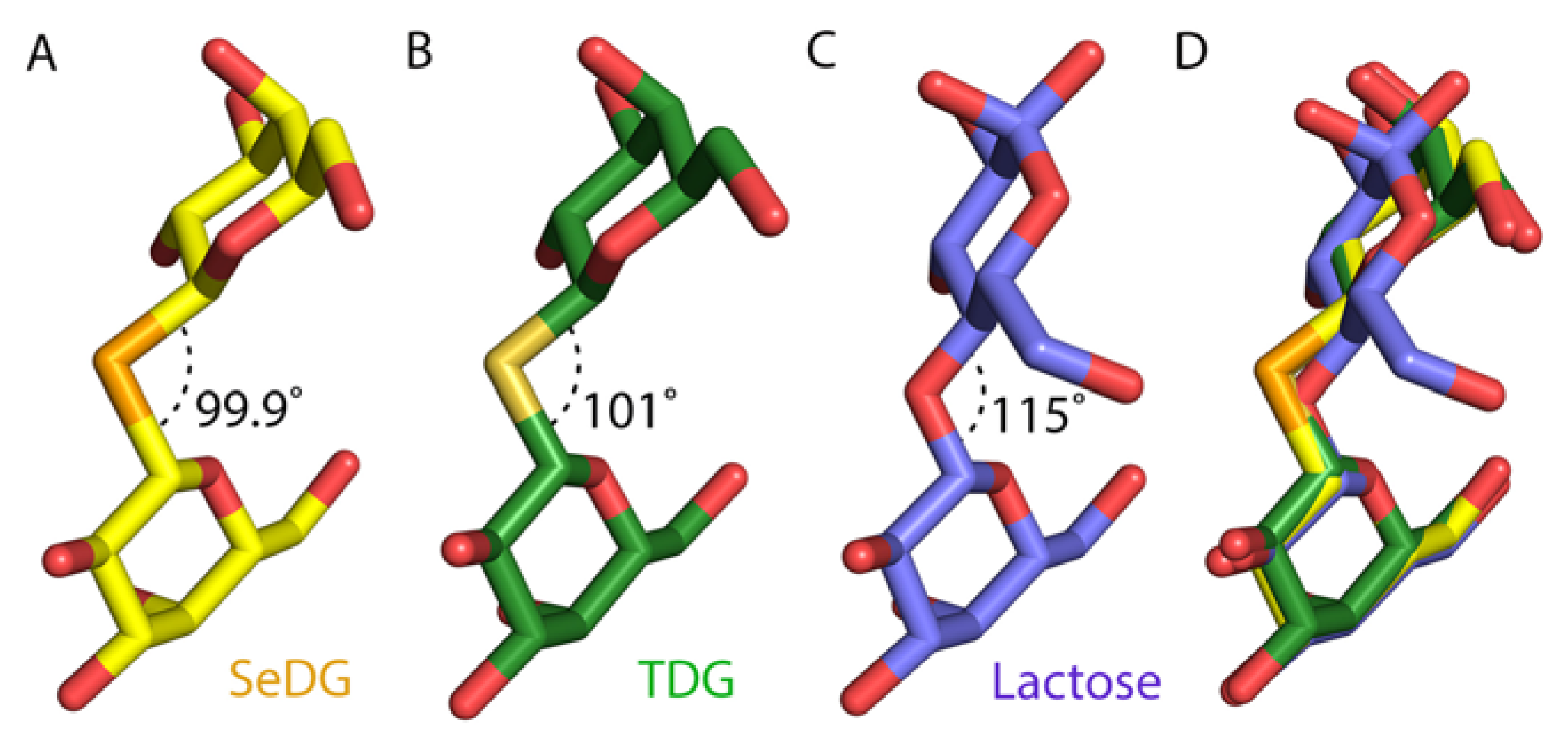
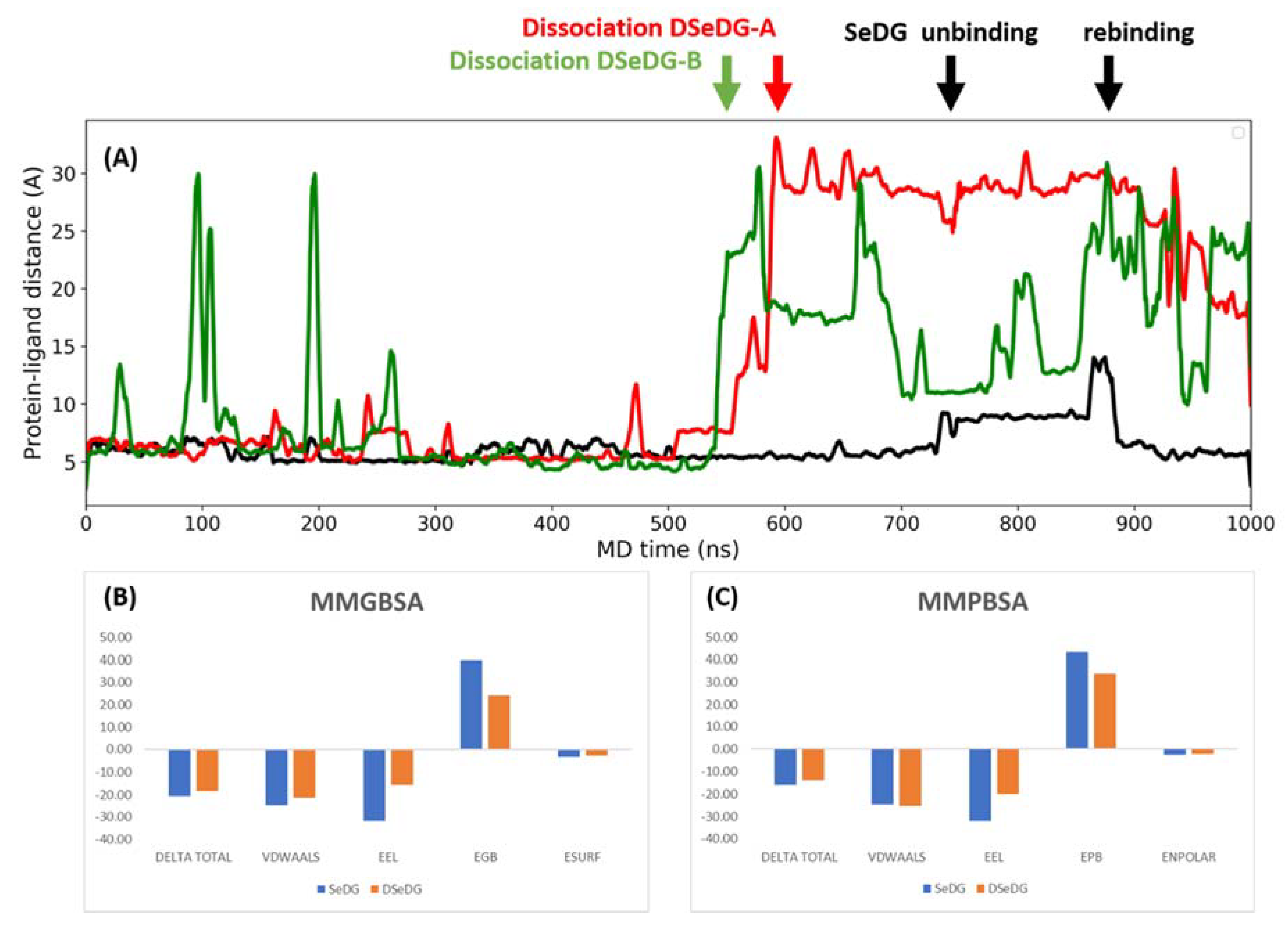

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kd (µM) | SeDG | DSeDG |
|---|---|---|
| ITC [40] | 93.1 ± 2.6 | 2800 ± 180 |
| Fluorescence anisotropy titration | 35.2 ± 7.2 | n.a. |
| 15N-1H HSQC titration | 123 ± 5.0 | 8500 ± 2500 |
| MMGBSA | MMPBSA | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Residue | SeDG | DSeDG A | DSeDG B | Residue | SeDG | DSeDG A | DSeDG B | ||
| ligand | −10.98 | −7.54 | −8.84 | ligand | −14.91 | −11.84 | −12.61 | ||
| TRP | 181 | −2.95 | −1.60 | −2.06 | TRP | 181 | −2.87 | −1.28 | −1.70 |
| ASN | 174 | −2.30 | −0.77 | −1.15 | HIE | 158 | −1.14 | −0.46 | −0.24 |
| HIE | 158 | −1.80 | −0.83 | −0.75 | ASN | 174 | −1.07 | −0.48 | −0.54 |
| ARG | 162 | −1.48 | −0.42 | −0.98 | ASN | 160 | −1.01 | −0.43 | −0.63 |
| ASN | 160 | −1.48 | −0.66 | −0.87 | ARG | 162 | −0.92 | −0.38 | −0.72 |
| VAL | 172 | −1.04 | −0.39 | −0.52 | VAL | 172 | −0.53 | −0.25 | −0.20 |
| ALA | 146 | −0.71 | −0.32 | −0.27 | ALA | 146 | −0.50 | −0.24 | −0.22 |
| CYS | 173 | −0.61 | −0.11 | −0.22 | ILE | 145 | −0.31 | −0.10 | −0.14 |
| ARG | 144 | −0.56 | −0.18 | −0.31 | CYS | 173 | −0.28 | −0.05 | −0.04 |
| GLU | 184 | −0.36 | −0.40 | −0.12 | PHE | 159 | −0.23 | −0.07 | −0.08 |
| PHE | 159 | −0.36 | −0.12 | −0.14 | GLU | 165 | −0.17 | −0.04 | −0.03 |
| VAL | 155 | −0.28 | −0.28 | −0.08 | PRO | 161 | −0.17 | −0.06 | −0.09 |
| ILE | 145 | −0.26 | −0.08 | −0.12 | LEU | 147 | −0.16 | −0.06 | −0.05 |
| ARG | 183 | −0.21 | −0.23 | −0.06 | VAL | 155 | −0.14 | −0.20 | −0.06 |
| SER | 237 | −0.21 | −0.03 | −0.07 | ASP | 239 | −0.13 | −0.02 | 0.05 |
| LYS | 176 | 0.14 | 0.00 | 0.07 | LYS | 176 | 0.16 | 0.06 | −0.01 |
| ARG | 224 | 0.15 | −0.19 | 0.10 | ASP | 148 | 0.87 | 0.58 | 0.51 |
| ASP | 148 | 0.16 | −0.09 | 0.09 | GLU | 184 | 1.18 | 0.47 | 1.04 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raics, M.; Balogh, Á.K.; Kishor, C.; Timári, I.; Medrano, F.J.; Romero, A.; Go, R.M.; Blanchard, H.; Szilágyi, L.; E. Kövér, K.; et al. Investigation of the Molecular Details of the Interactions of Selenoglycosides and Human Galectin-3. Int. J. Mol. Sci. 2022, 23, 2494. https://doi.org/10.3390/ijms23052494
Raics M, Balogh ÁK, Kishor C, Timári I, Medrano FJ, Romero A, Go RM, Blanchard H, Szilágyi L, E. Kövér K, et al. Investigation of the Molecular Details of the Interactions of Selenoglycosides and Human Galectin-3. International Journal of Molecular Sciences. 2022; 23(5):2494. https://doi.org/10.3390/ijms23052494
Chicago/Turabian StyleRaics, Mária, Álex Kálmán Balogh, Chandan Kishor, István Timári, Francisco J. Medrano, Antonio Romero, Rob Marc Go, Helen Blanchard, László Szilágyi, Katalin E. Kövér, and et al. 2022. "Investigation of the Molecular Details of the Interactions of Selenoglycosides and Human Galectin-3" International Journal of Molecular Sciences 23, no. 5: 2494. https://doi.org/10.3390/ijms23052494
APA StyleRaics, M., Balogh, Á. K., Kishor, C., Timári, I., Medrano, F. J., Romero, A., Go, R. M., Blanchard, H., Szilágyi, L., E. Kövér, K., & Fehér, K. (2022). Investigation of the Molecular Details of the Interactions of Selenoglycosides and Human Galectin-3. International Journal of Molecular Sciences, 23(5), 2494. https://doi.org/10.3390/ijms23052494