
Atherosclerosis in HIV Patients: What Do We Know so Far?

 ,
,  ,
, 
Abstract
:1. Atherosclerosis
2. Cardiovascular Disease in HIV Patients
3. Underlying Cellular Mechanisms
3.1. Oxidative Stress
3.2. ER Stress
3.3. NLRP3 Inflammasome Activation
3.4. Autophagy Inhibition
4. Managing Atherosclerosis in HIV
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Rafieian-Kopaei, M.; Setorki, M.; Doudi, M.; Baradaran, A.; Nasri, H. Atherosclerosis: Process, indicators, risk factors and new hopes. Int. J. Prev. Med. 2014, 5, 927–946. [Google Scholar] [PubMed]
- Linton, M.R.F.; Yancey, P.G.; Davies, S.S.; Jerome, W.G.; Linton, E.F.; Song, W.L.; Doran, A.C.; Vickers, K.C. The Role of Lipids and Lipoproteins in Atherosclerosis. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. Available online: https://www.ncbi.nlm.nih.gov/books/NBK343489/ (accessed on 2 November 2021).
- Nelson, R.H. Hyperlipidemia as a Risk Factor for Cardiovascular Disease. Prim. Care 2013, 40, 195–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, J.-J.; Chien, S. Effects of Disturbed Flow on Vascular Endothelium: Pathophysiological Basis and Clinical Perspectives. Physiol. Rev. 2011, 91, 327–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, H.; Cassorla, G.; Pertsoulis, N.; Patel, V.; Vicaretti, M.; Marmash, N.; Hitos, K.; Fletcher, J.P.; Medbury, H.J. Human classical monocytes display unbalanced M1/M2 phenotype with increased atherosclerotic risk and presence of disease. Int. Angiol. 2017, 36, 145–155. [Google Scholar] [CrossRef]
- Italiani, P.; Boraschi, D.; Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques, A.R.A.; Ramos, C.; Machado-Oliveira, G.; Vieira, O.V. Lysosome (Dys)function in Atherosclerosis—A Big Weight on the Shoulders of a Small Organelle. Front. Cell Dev. Biol. 2021, 9, 658995. [Google Scholar] [CrossRef]
- Prame Kumar, K.; Nicholls, A.J.; Wong, C.H.Y. Partners in crime: Neutrophils and monocytes/macrophages in inflammation and disease. Cell Tissue Res. 2018, 371, 551–565. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, N.; Kobayashi, K. Macrophages in Inflammation. Curr. Drug Target Inflamm. Allergy 2005, 4, 281–286. [Google Scholar] [CrossRef]
- Olejarz, W.; Łacheta, D.; Kubiak-Tomaszewska, G. Matrix Metalloproteinases as Biomarkers of Atherosclerotic Plaque Instability. Int. J. Mol. Sci. 2020, 21, 3946. [Google Scholar] [CrossRef]
- Newby, A.C. Metalloproteinases and Vulnerable Atherosclerotic Plaques. Trends Cardiovasc. Med. 2007, 17, 253–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quillard, T.; Franck, G.; Mawson, T.; Folco, E.; Libby, P. Mechanisms of erosion of atherosclerotic plaques. Curr. Opin. Lipidol. 2017, 28, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Libby, P.; Tabas, I. Inflammation and plaque vulnerability. J. Intern. Med. 2015, 278, 483–493. [Google Scholar] [CrossRef]
- Poznyak, A.; Grechko, A.V.; Poggio, P.; Myasoedova, V.A.; Alfieri, V.; Orekhov, A.N. The Diabetes Mellitus–Atherosclerosis Connection: The Role of Lipid and Glucose Metabolism and Chronic Inflammation. Int. J. Mol. Sci. 2020, 21, 1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, D.S.M.; Da Silva, M.J.L.V. Cardiovascular Disease in the Setting of Human Immunodeficiency Virus Infection. Curr. Cardiol. Rev. 2018, 14, 25–41. [Google Scholar] [CrossRef] [PubMed]
- Althoff, K.N.; Smit, M.; Reiss, P.; Justice, A.C. HIV and ageing: Improving quantity and quality of life. Curr. Opin. HIV AIDS 2016, 11, 527–536. [Google Scholar] [CrossRef]
- Cahill, S.; Valadéz, R. Growing Older With HIV/AIDS: New Public Health Challenges. Am. J. Public Health 2013, 103, e7–e15. [Google Scholar] [CrossRef]
- Kaplan, R.C.; Hanna, D.B.; Kizer, J.R. Recent Insights Into Cardiovascular Disease (CVD) Risk Among HIV-Infected Adults. Curr. HIV/AIDS Rep. 2016, 13, 44–52. [Google Scholar] [CrossRef]
- Triant, V.A. Cardiovascular Disease and HIV Infection. Curr. HIV/AIDS Rep. 2013, 10, 199–206. [Google Scholar] [CrossRef]
- Siedner, M.J. START or SMART? Timing of Antiretroviral Therapy Initiation and Cardiovascular Risk for People with Human Immunodeficiency Virus Infection. Open Forum Infect. Dis. 2016, 3, ofw032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacson, J.C.; Barnes, R.P.; Bahrami, H. Coronary Artery Disease in HIV-Infected Patients: Downside of Living Longer. Curr. Atheroscler. Rep. 2017, 19, 18. [Google Scholar] [CrossRef]
- Baker, J.V.; Henry, W.K.; Neaton, J.D. The consequences of HIV infection and antiretroviral therapy use for cardiovascular disease risk: Shifting paradigms. Curr. Opin. HIV AIDS 2009, 4, 176–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freiberg, M.S.; Chang, C.C.H.; Kuller, L.H.; Skanderson, M.; Lowy, E.; Kraemer, K.L.; Butt, A.A.; Goetz, M.B.; Leaf, D.; Oursler, K.A.; et al. HIV Infection and the Risk of Acute Myocardial Infarction. JAMA Intern. Med. 2013, 173, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Triant, V.A. Epidemiology of coronary heart disease in patients with human immunodeficiency virus. Rev. Cardiovasc. Med. 2014, 15, S1–S8. [Google Scholar]
- Krishnan, S.; Wilson, E.M.P.; Sheikh, V.; Rupert, A.; Mendoza, D.; Yang, J.; Lempicki, R.; Migueles, S.A.; Sereti, I. Evidence for innate immune system activation in HIV type 1-infected elite controllers. J. Infect. Dis. 2014, 209, 931–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltran, L.M.; Rubio-Navarro, A.; Amaro-Villalobos, J.M.; Egido, J.; García-Puig, J.; Moreno, J.A. Influence of immune activation and inflammatory response on cardiovascular risk associated with the human immunodeficiency virus. Vasc. Health Risk Manag. 2015, 11, 35–48. [Google Scholar] [CrossRef] [Green Version]
- Hsue, P.Y.; Hunt, P.W.; Schnell, A.; Kalapus, S.C.; Hoh, R.; Ganz, P.; Martin, J.N.; Deeks, S.G. Role of viral replication, antiretroviral therapy, and immunodeficiency in HIV-associated atherosclerosis. AIDS 2009, 23, 1059–1067. [Google Scholar] [CrossRef]
- Kearns, A.; Gordon, J.; Burdo, T.H.; Qin, X. HIV-1–Associated Atherosclerosis: Unraveling the Missing Link. J. Am. Coll. Cardiol. 2017, 69, 3084–3098. [Google Scholar] [CrossRef]
- Cornwell, A.; Palli, R.; Singh, M.V.; Benoodt, L.; Tyrell, A.; Abe, J.-I.; Schifitto, G.; Maggirwar, S.B.; Thakar, J. Molecular characterization of atherosclerosis in HIV positive persons. Sci. Rep. 2021, 11, 3232. [Google Scholar] [CrossRef]
- Crowe, S.M.; Westhorpe, C.L.V.; Mukhamedova, N.; Jaworowski, A.; Sviridov, D.; Bukrinsky, M. The macrophage: The intersection between HIV infection and atherosclerosis. J. Leukoc. Biol. 2010, 87, 589–598. [Google Scholar] [CrossRef] [Green Version]
- Campbell, J.H.; Hearps, A.C.; Martin, G.E.; Williams, K.C.; Crowe, S.M. The importance of monocytes and macrophages in HIV pathogenesis, treatment, and cure. AIDS 2014, 28, 2175–2187. [Google Scholar] [CrossRef]
- Laurence, J.; Elhadad, S.; Ahamed, J. HIV-associated cardiovascular disease: Importance of platelet activation and cardiac fibrosis in the setting of specific antiretroviral therapies. Open Heart 2018, 5, e000823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsue, P.Y.; Waters, D.D. HIV infection and coronary heart disease: Mechanisms and management. Nat. Rev. Cardiol. 2019, 16, 745–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, J.-X.; Kong, Q.; Ma, X. Current advances in circulating inflammatory biomarkers in atherosclerosis and related cardio-cerebrovascular diseases. Chronic Dis. Transl. Med. 2017, 3, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Goncharov, N.V.; Nadeev, A.D.; Jenkins, R.O.; Avdonin, P.V. Markers and Biomarkers of Endothelium: When Something Is Rotten in the State. Oxidative Med. Cell. Longev. 2017, 2017, 9759735. [Google Scholar] [CrossRef] [PubMed]
- Sáez-Cirión, A.; Jacquelin, B.; Barré-Sinoussi, F.; Müller-Trutwin, M. Immune responses during spontaneous control of HIV and AIDS: What is the hope for a cure? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130436. [Google Scholar] [CrossRef] [Green Version]
- Anand, A.R.; Rachel, G.; Parthasarathy, D. HIV Proteins and Endothelial Dysfunction: Implications in Cardiovascular Disease. Front. Cardiovasc. Med. 2018, 5, 185. [Google Scholar] [CrossRef]
- Juan, C.A.; Pérez de la Lastra, J.M.; Plou, F.J.; Pérez-Lebeña, E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int. J. Mol. Sci. 2021, 22, 4642. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxidative Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.-A.; Runge, M.S.; Madamanchi, N.R. Oxidative stress, NADPH oxidases, and arteries. Hamostaseologie 2016, 36, 77–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrotta, I.; Aquila, S. The Role of Oxidative Stress and Autophagy in Atherosclerosis. Oxidative Med. Cell. Longev. 2015, 2015, 130315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxidative Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Ivanov, A.V.; Valuev-Elliston, V.T.; Ivanova, O.N.; Kochetkov, S.N.; Starodubova, E.S.; Bartosch, B.; Isaguliants, M.G. Oxidative Stress during HIV Infection: Mechanisms and Consequences. Oxidative Med. Cell Longev. 2016, 2016, 8910396. [Google Scholar] [CrossRef] [Green Version]
- Porter, K.M.; Sutliff, R.L. HIV-1, reactive oxygen species, and vascular complications. Free Radic. Biol. Med. 2012, 53, 143–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.S.; Kaufman, R.J. Endoplasmic Reticulum Stress and Oxidative Stress in Cell Fate Decision and Human Disease. Antioxidative Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Walter, P.; Yen, T.B. Endoplasmic Reticulum Stress in Disease Pathogenesis. Annu. Rev. Pathol. Mech. Dis. 2008, 3, 399–425. [Google Scholar] [CrossRef]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Ivanova, E.A.; Orekhov, A.N. The Role of Endoplasmic Reticulum Stress and Unfolded Protein Response in Atherosclerosis. Int. J. Mol. Sci. 2016, 17, 193. [Google Scholar] [CrossRef]
- Zhou, A.X.; Tabas, I. The UPR in atherosclerosis. Semin. Immunopathol. 2013, 35, 321–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabas, I. The Role of Endoplasmic Reticulum Stress in the Progression of Atherosclerosis. Circ. Res. 2010, 107, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Sobenin, I.A.; Orekhov, A.N.; Bobryshev, Y.V. Role of Endoplasmic Reticulum Stress in Atherosclerosis and Diabetic Macrovascular Complications. BioMed Res. Int. 2014, 2014, 610140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, R.; Yang, L.; Niu, F.; Buch, S. HIV Tat-Mediated Induction of Human Brain Microvascular Endothelial Cell Apoptosis Involves Endoplasmic Reticulum Stress and Mitochondrial Dysfunction. Mol. Neurobiol. 2016, 53, 132–142. [Google Scholar] [CrossRef]
- Fribley, A.; Zhang, K.; Kaufman, R.J. Regulation of Apoptosis by the Unfolded Protein Response. Methods Mol. Biol. 2009, 559, 191–204. [Google Scholar] [CrossRef] [Green Version]
- Heron, S.E.; Elahi, S. HIV Infection and Compromised Mucosal Immunity: Oral Manifestations and Systemic Inflammation. Front. Immunol. 2017, 8, 241. [Google Scholar] [CrossRef] [Green Version]
- Mogensen, T.H. Pathogen Recognition and Inflammatory Signaling in Innate Immune Defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [Green Version]
- Suresh, R.; Mosser, D.M. Pattern recognition receptors in innate immunity, host defense, and immunopathology. Adv. Physiol. Educ. 2013, 37, 284–291. [Google Scholar] [CrossRef]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 2018, 9, 2379. [Google Scholar] [CrossRef] [Green Version]
- Grebe, A.; Hoss, F.; Latz, E. NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ. Res. 2018, 122, 1722–1740. [Google Scholar] [CrossRef]
- Hoseini, Z.; Sepahvand, F.; Rashidi, B.; Sahebkar, A.; Masoudifar, A.; Mirzaei, H. NLRP3 inflammasome: Its regulation and involvement in atherosclerosis. J. Cell. Physiol. 2018, 233, 2116–2132. [Google Scholar] [CrossRef] [PubMed]
- Cai, R.; Liu, L.; Luo, B.; Wang, J.; Shen, J.; Shen, Y.; Zhang, R.; Chen, J.; Lu, H. Caspase-1 Activity in CD4 T Cells Is Downregulated Following Antiretroviral Therapy for HIV-1 Infection. AIDS Res. Hum. Retrovir. 2017, 33, 164–171. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Gao, J.; Taxman, D.J.; Ting, J.P.Y.; Su, L. HIV-1 Infection Induces Interleukin-1β Production via TLR8 Protein-dependent and NLRP3 Inflammasome Mechanisms in Human Monocytes. J. Biol. Chem. 2014, 289, 21716–21726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Zhang, S.; Xiao, Y.; Zhang, W.; Wu, S.; Qin, T.; Yue, Y.; Qian, W.; Li, L. NLRP3 Inflammasome and Inflammatory Diseases. Oxidative Med. Cell. Longev. 2020, 2020, 4063562. [Google Scholar] [CrossRef]
- Valdor, R.; Macian, F. Autophagy and the regulation of the immune response. Pharmacol. Res. 2012, 66, 475–483. [Google Scholar] [CrossRef] [Green Version]
- Parzych, K.R.; Klionsky, D.J. An Overview of Autophagy: Morphology, Mechanism, and Regulation. Antioxidants Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [Green Version]
- Poznyak, A.V.; Nikiforov, N.G.; Wu, W.-K.; Kirichenko, T.V.; Orekhov, A.N. Autophagy and Mitophagy as Essential Components of Atherosclerosis. Cells 2021, 10, 443. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, Y.; Li, Y.; Ren, X.; Zhang, X.; Hu, D.; Gao, Y.; Xing, Y.; Shang, H. Oxidative Stress-Mediated Atherosclerosis: Mechanisms and Therapies. Front. Physiol. 2017, 8, 600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prestes, E.B.; Bruno, J.C.P.; Travassos, L.H.; Carneiro, L.A.M. The Unfolded Protein Response and Autophagy on the Crossroads of Coronaviruses Infections. Front. Cell. Infect. Microbiol. 2021, 11, 668034. [Google Scholar] [CrossRef]
- Cabrera-Rodríguez, R.; Pérez-Yanes, S.; Estévez-Herrera, J.; Márquez-Arce, D.; Cabrera, C.; Espert, L.; Blanco, J.; Valenzuela-Fernández, A. The Interplay of HIV and Autophagy in Early Infection. Front. Microbiol. 2021, 12, 661446. [Google Scholar] [CrossRef]
- Jaworowski, A.; Hearps, A.C.; Angelovich, T.; Hoy, J.F. How Monocytes Contribute to Increased Risk of Atherosclerosis in Virologically-Suppressed HIV-Positive Individuals Receiving Combination Antiretroviral Therapy. Front. Immunol. 2019, 10, 1378. [Google Scholar] [CrossRef]
- Seedat, F.; Raal, F.; Martinson, N.; Variava, E. Lipid and lipoprotein levels in HIV-infected adults with sepsis compared to healthy HIV-infected controls. Afr. J. Infect. Dis. 2020, 14, 1–9. [Google Scholar] [CrossRef]
- Da Cunha, J.; Maselli, L.M.; Stern, A.C.; Spada, C.; Bydlowski, S.P. Impact of antiretroviral therapy on lipid metabolism of human immunodeficiency virus-infected patients: Old and new drugs. World J. Virol. 2015, 4, 56–77. [Google Scholar] [CrossRef] [PubMed]
- Shafran, S.D.; Mashinter, L.D.; Roberts, S.E. The effect of low-dose ritonavir monotherapy on fasting serum lipid concentrations. HIV Med. 2005, 6, 421–425. [Google Scholar] [CrossRef]
- Rubins, H.B.; Robins, S.J. Effect of reduction of plasma triglycerides with gemfibrozil on high-density-lipoprotein-cholesterol concentrations. J. Intern. Med. 1992, 231, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Attie, A.D.; Krauss, R.M.; Gray-Keller, M.P.; Brownlie, A.; Miyazaki, M.; Kastelein, J.J.; Lusis, A.J.; Stalenhoef, A.F.H.; Stoehr, J.P.; Hayden, M.R.; et al. Relationship between stearoyl-CoA desaturase activity and plasma triglycerides in human and mouse hypertriglyceridemia. J. Lipid Res. 2002, 43, 1899–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feeney, E.R.; Mallon, P.W. HIV and HAART-Associated Dyslipidemia. Open Cardiovasc. Med. J. 2011, 5, 49–63. [Google Scholar] [CrossRef]
- Malvestutto, C.D.; Aberg, J.A. Management of dyslipidemia in HIV-infected patients. Clin. Lipidol. 2011, 6, 447–462. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Brown, T.T. Lipid Disorders in People with HIV. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Hershman, J.M., Hofland, J., Kalra, S., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. Available online: https://www.ncbi.nlm.nih.gov/books/NBK567198/ (accessed on 2 November 2021).
- Maggi, P.; Di Biagio, A.; Rusconi, S.; Cicalini, S.; D’Abbraccio, M.; D’Ettorre, G.; Martinelli, C.; Nunnari, G.; Sighinolfi, L.; Spagnuolo, V.; et al. Cardiovascular risk and dyslipidemia among persons living with HIV: A review. BMC Infect. Dis. 2017, 17, 551. [Google Scholar] [CrossRef] [Green Version]
- Quinn, T.C. HIV epidemiology and the effects of antiviral therapy on long-term consequences. AIDS 2008, 22 (Suppl. 3), S7–S12. [Google Scholar] [CrossRef] [Green Version]
- Rosen, S.; Fox, M.; Gill, C. Patient Retention in Antiretroviral Therapy Programs in Sub-Saharan Africa: A Systematic Review. PLoS Med. 2007, 4, e298. [Google Scholar] [CrossRef] [Green Version]
- Dimala, C.A.; Blencowe, H.; Choukem, S.P. The association between antiretroviral therapy and selected cardiovascular disease risk factors in sub-Saharan Africa: A systematic review and meta-analysis. PLoS ONE 2018, 13, e0201404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joy, T.; Keogh, H.M.; Hadigan, C.; Lee, H.; Dolan, S.E.; Fitch, K.; Liebau, J.; Lo, J.; Johnsen, S.; Hubbard, J.; et al. Dietary fat intake and relationship to serum lipid levels in HIV-infected patients with metabolic abnormalities in the HAART era. AIDS 2007, 21, 1591–1600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobson, T.A.; Ito, M.K.; Maki, K.C.; Orringer, C.E.; Bays, H.E.; Jones, P.H.; McKenney, J.M.; Grundy, S.M.; Gill, E.A.; Wild, R.A.; et al. National Lipid Association Recommendations for Patient-Centered Management of Dyslipidemia: Part 1—Full Report. J. Clin. Lipidol. 2015, 9, 129–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, N.D.; Young, D.; Zhao, Y.; Nguyen, H.; Caballes, J.; Khan, I.; Sanchez, R.J. Prevalence of the American College of Cardiology/American Heart Association statin eligibility groups, statin use, and low-density lipoprotein cholesterol control in US adults using the National Health and Nutrition Examination Survey 2011. J. Clin. Lipidol. 2016, 10, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Cholesterol Treatment Trialists’ (CTT) Collaboration; Mihaylova, B.; Emberson, J.; Blackwell, L.; Keech, A.; Simes, J.; Barnes, E.H.; Voysey, M.; Gray, A.; Collins, R.; et al. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: Meta-analysis of individual data from 27 randomised trials. Lancet 2012, 380, 581–590. [Google Scholar] [CrossRef]
- Eckard, A.R.; Mccomsey, G.A. The Role of Statins in the Setting of HIV Infection. Curr. HIV/AIDS Rep. 2015, 12, 305–312. [Google Scholar] [CrossRef] [Green Version]
- Chastain, D.B.; Stover, K.R.; Riche, D.M. Evidence-based review of statin use in patients with HIV on antiretroviral therapy. J. Clin. Transl. Endocrinol. 2017, 8, 6–14. [Google Scholar] [CrossRef]
- Calza, L.; Manfredi, R.; Colangeli, V.; Pocaterra, D.; Pavoni, M.; Chiodo, F. Rosuvastatin, pravastatin, and atorvastatin for the treatment of hypercholesterolaemia in HIV-infected patients receiving protease inhibitors. Curr. HIV Res. 2008, 6, 572–578. [Google Scholar] [CrossRef]
- Ward, N.C.; Watts, G.F.; Eckel, R.H. Statin Toxicity. Circ. Res. 2019, 124, 328–350. [Google Scholar] [CrossRef]
- Mosepele, M.; Molefe-Baikai, O.J.; Grinspoon, S.K.; Triant, V.A. Benefits and Risks of Statin Therapy in the HIV-Infected Population. Curr. Infect. Dis. Rep. 2018, 20, 20. [Google Scholar] [CrossRef] [PubMed]
- Aberg, J.A.; A Sponseller, C.A.; Ward, D.J.; Kryzhanovski, V.A.; Campbell, S.E.; Thompson, M.A. Pitavastatin versus pravastatin in adults with HIV-1 infection and dyslipidaemia (INTREPID): 12 week and 52 week results of a phase 4, multicentre, randomised, double-blind, superiority trial. Lancet HIV 2017, 4, e284–e294, Erratum in Lancet HIV 2017, 4, e283. [Google Scholar] [CrossRef]
- Pawlos, A.; Broncel, M.; Wlazłowska, E.; Jabłonowska, E.; Gorzelak-Pabiś, P. Cardiovascular risk and response to lipid lowering therapy in patients with HIV infection according to different recommendations. PLoS ONE 2020, 15, e0244675, Erratum in PLoS ONE 2021, 22, e0246176. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.T.; Johns, K.W.; Bondy, G.P. Ezetimibe is effective when added to maximally tolerated lipid lowering therapy in patients with HIV. Lipids Health Dis. 2007, 6, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorizate, M.; Kräusslich, H.-G. Role of Lipids in Virus Replication. Cold Spring Harb. Perspect. Biol. 2011, 3, a004820. [Google Scholar] [CrossRef] [Green Version]
- Van Welzen, B.J.; Oomen, P.G.A.; Hoepelman, A.I.M. Dual Antiretroviral Therapy—All Quiet Beneath the Surface? Front. Immunol. 2021, 12, 637910. [Google Scholar] [CrossRef]
- Yuan, G.; Al-Shali, K.Z.; Hegele, R.A. Hypertriglyceridemia: Its etiology, effects and treatment. Can. Med Assoc. J. 2007, 176, 1113–1120. [Google Scholar] [CrossRef] [Green Version]
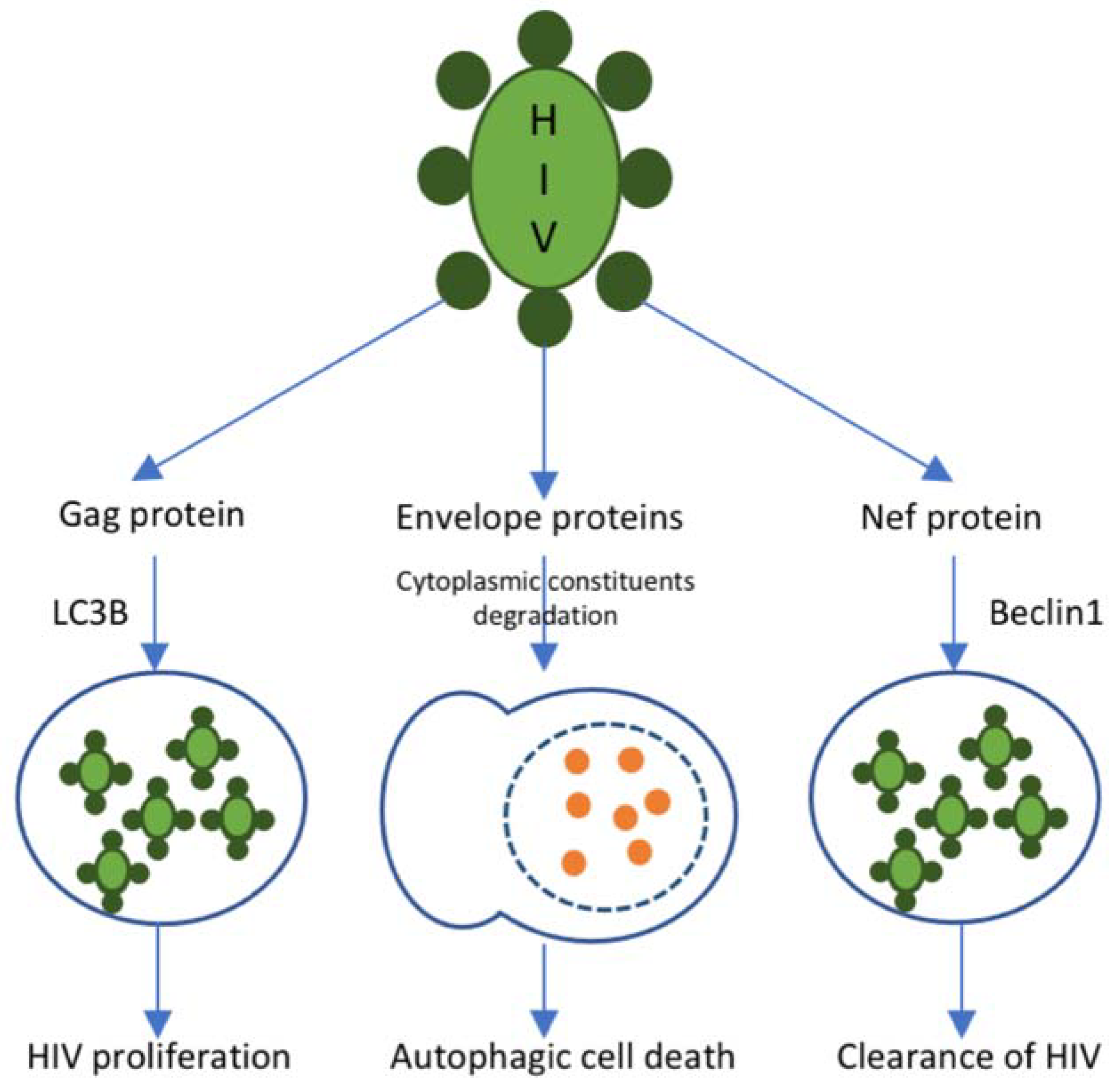

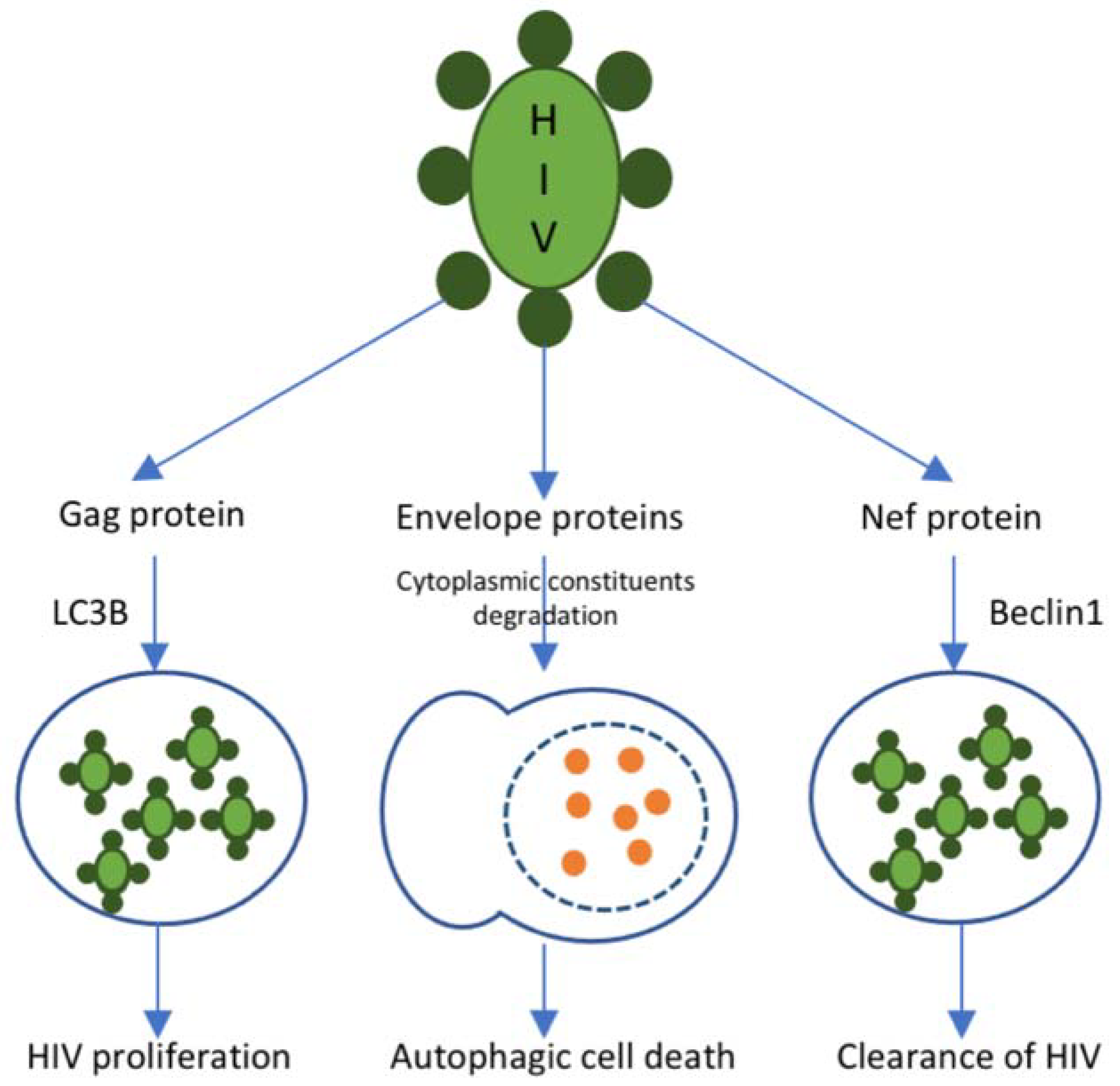{kind=link}
| Drug | Effect on Lipid Profile | Reference |
|---|---|---|
| Ritonavir (protease inhibitor) | HDL-C raised to 2.0 mmol/L; increased level of triglycerides in blood plasma; increased cholesterol level | [75,76,77] |
| Saquinavir (protease inhibitor) | Increased HDL cholesterol levels | [75] |
| Nelfinavir (protease inhibitor) | HDL-C raised to 1.2 mmol/L | [75,76] |
| Indinavir (protease inhibitor) | HDL-C raised to 0.8 mmol/L | [76] |
| Efavirenz (NNRTI) | Increased levels of total cholesterol, HDL cholesterol, LDL cholesterol, and triglycerides | [78,79] |
| Tenofovir alafenamide (NNRTI) | Increased LDL-C levels and HDL-C levels | [74,80,81] |
| Tenofovir disoproxil fumarate (NNRTI) | Hypolipidemic effect | [74,80,81] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poznyak, A.V.; Bezsonov, E.E.; Borisov, E.E.; Grechko, A.V.; Kartuesov, A.G.; Orekhov, A.N. Atherosclerosis in HIV Patients: What Do We Know so Far? Int. J. Mol. Sci. 2022, 23, 2504. https://doi.org/10.3390/ijms23052504
Poznyak AV, Bezsonov EE, Borisov EE, Grechko AV, Kartuesov AG, Orekhov AN. Atherosclerosis in HIV Patients: What Do We Know so Far? International Journal of Molecular Sciences. 2022; 23(5):2504. https://doi.org/10.3390/ijms23052504
Chicago/Turabian StylePoznyak, Anastasia V., Evgeny E. Bezsonov, Evgeny E. Borisov, Andrey V. Grechko, Andrey G. Kartuesov, and Alexander N. Orekhov. 2022. "Atherosclerosis in HIV Patients: What Do We Know so Far?" International Journal of Molecular Sciences 23, no. 5: 2504. https://doi.org/10.3390/ijms23052504
APA StylePoznyak, A. V., Bezsonov, E. E., Borisov, E. E., Grechko, A. V., Kartuesov, A. G., & Orekhov, A. N. (2022). Atherosclerosis in HIV Patients: What Do We Know so Far? International Journal of Molecular Sciences, 23(5), 2504. https://doi.org/10.3390/ijms23052504