
Augmentation of Transcriptomic Data for Improved Classification of Patients with Respiratory Diseases of Viral Origin



Abstract
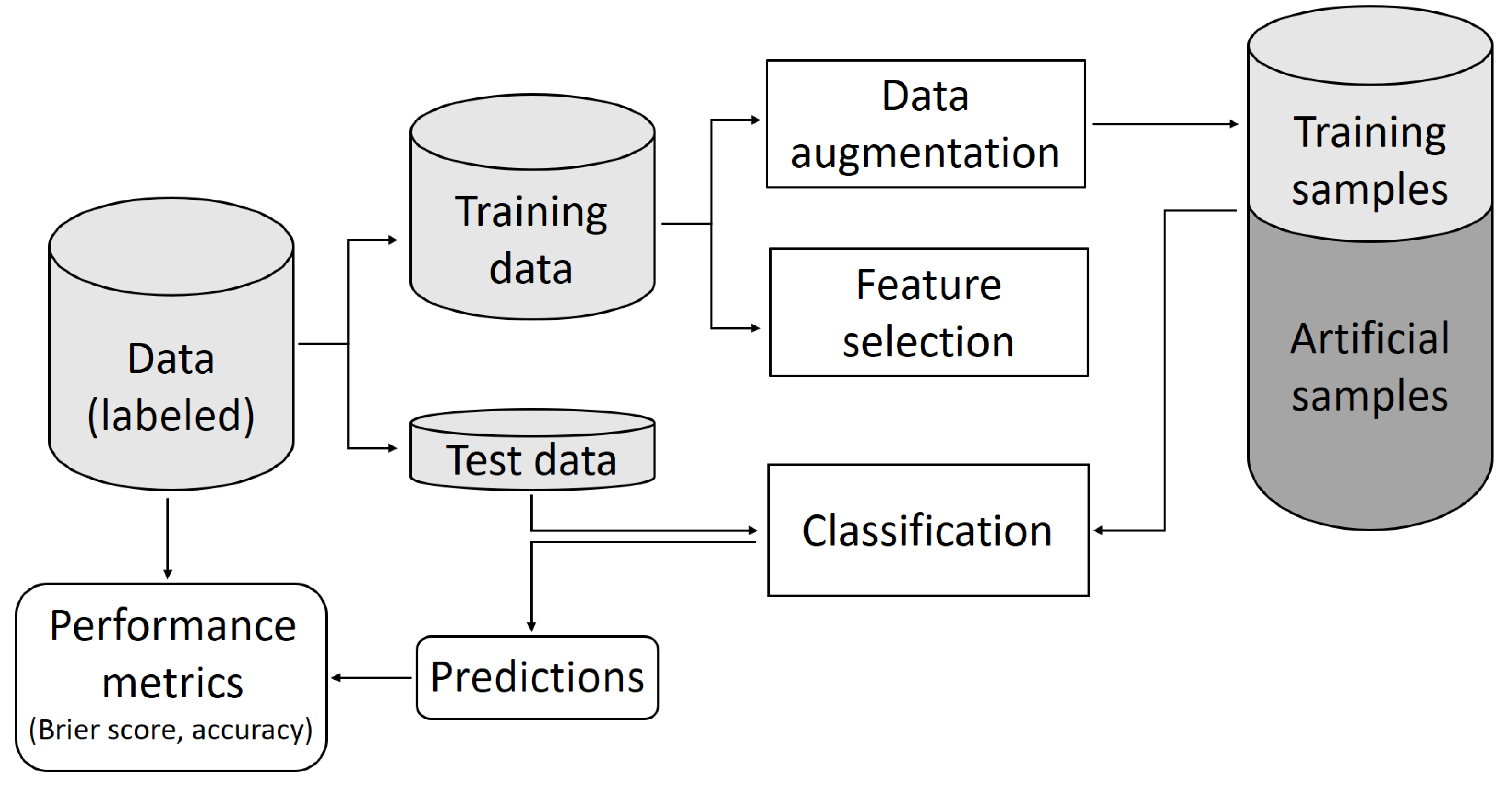
:1. Introduction
2. Results
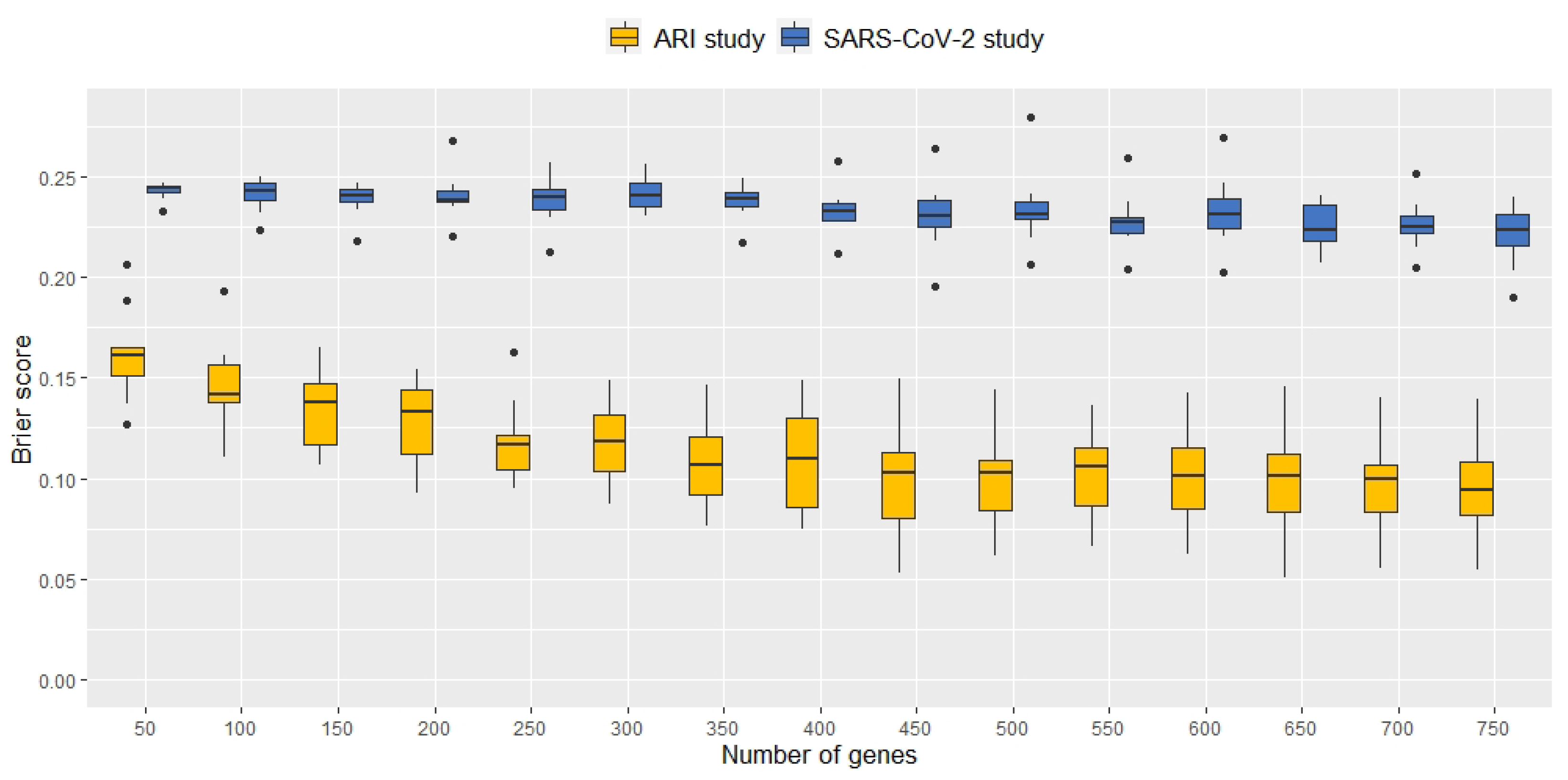
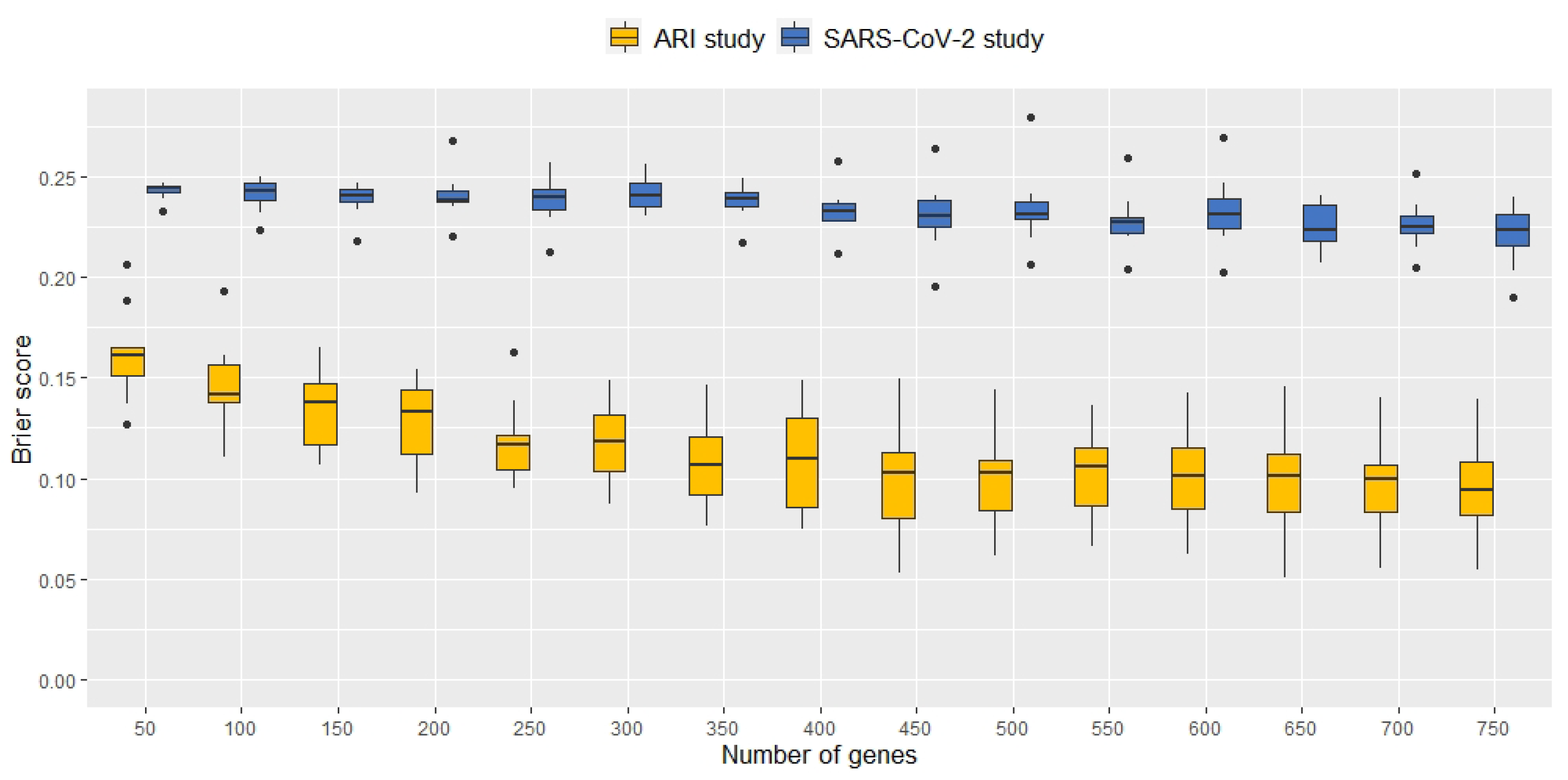
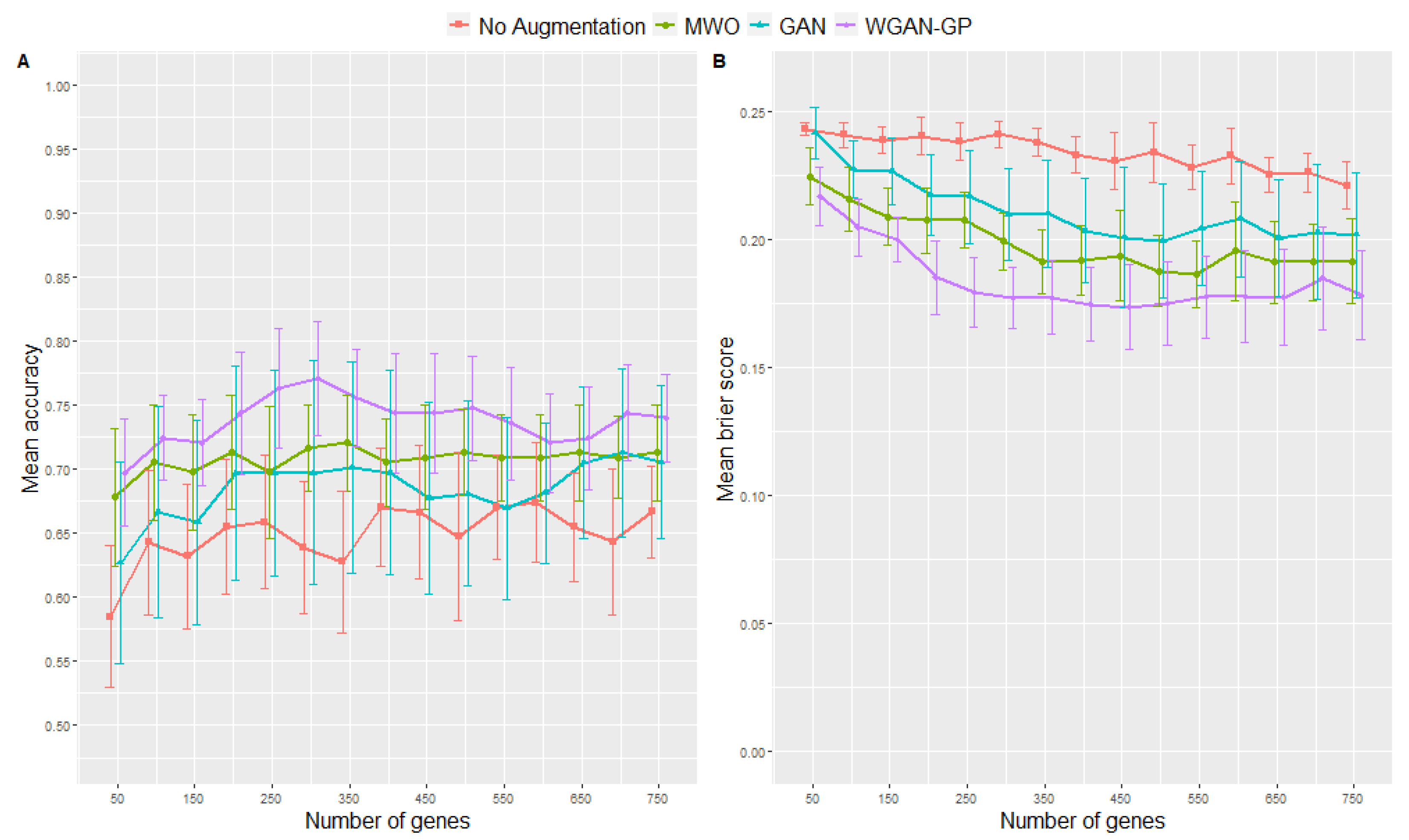
2.1. Signature Selection and Classifier Performance without Data Augmentation
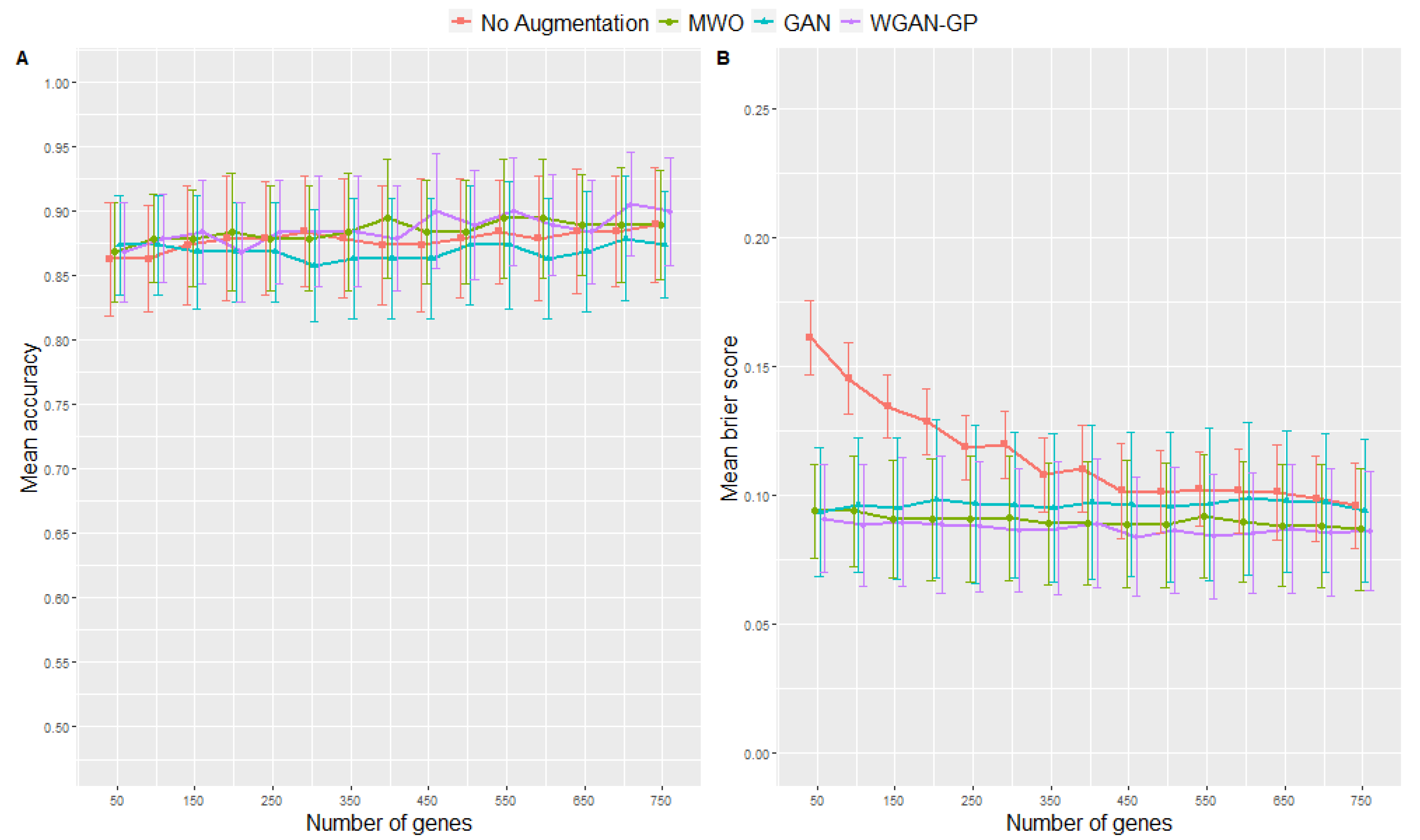
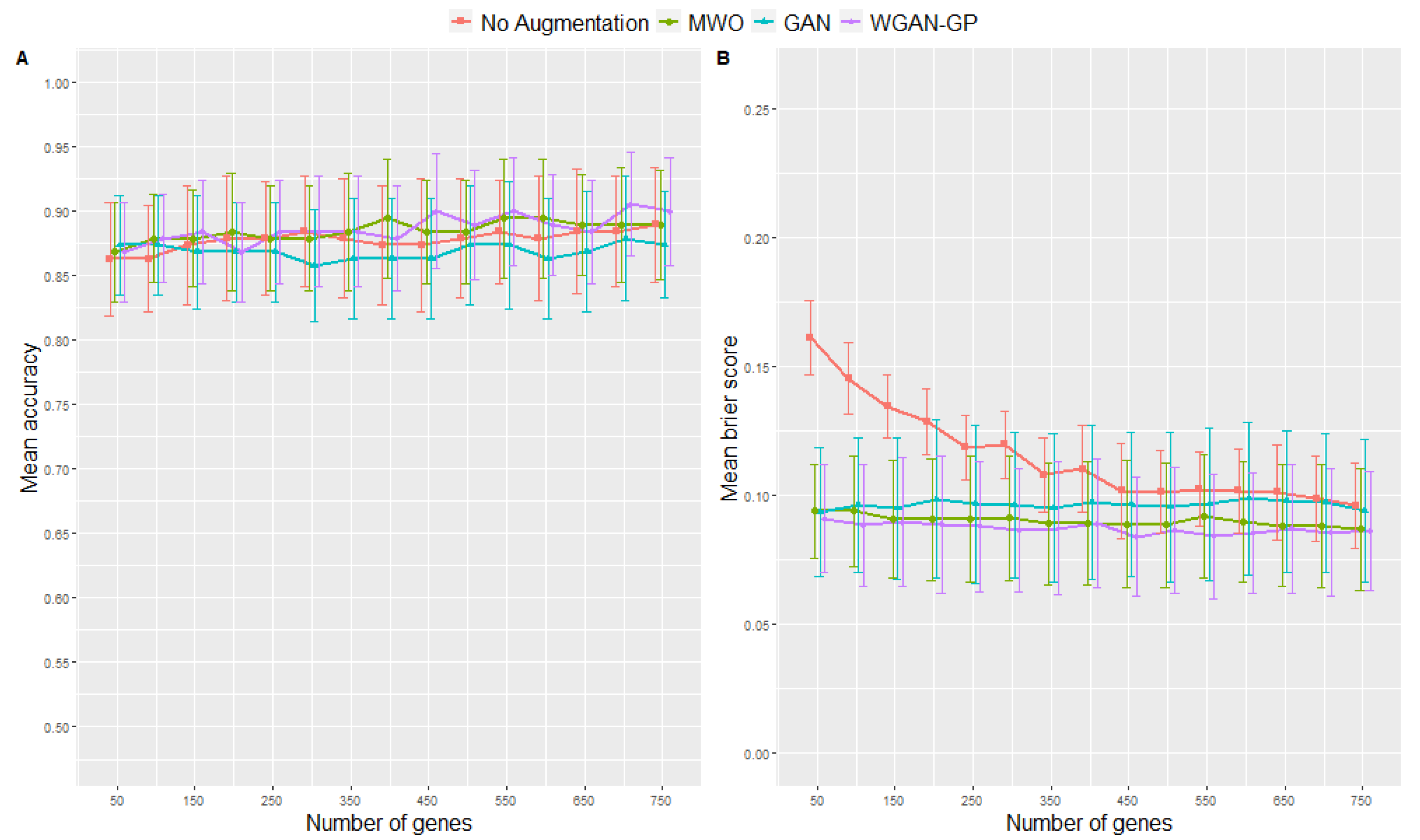
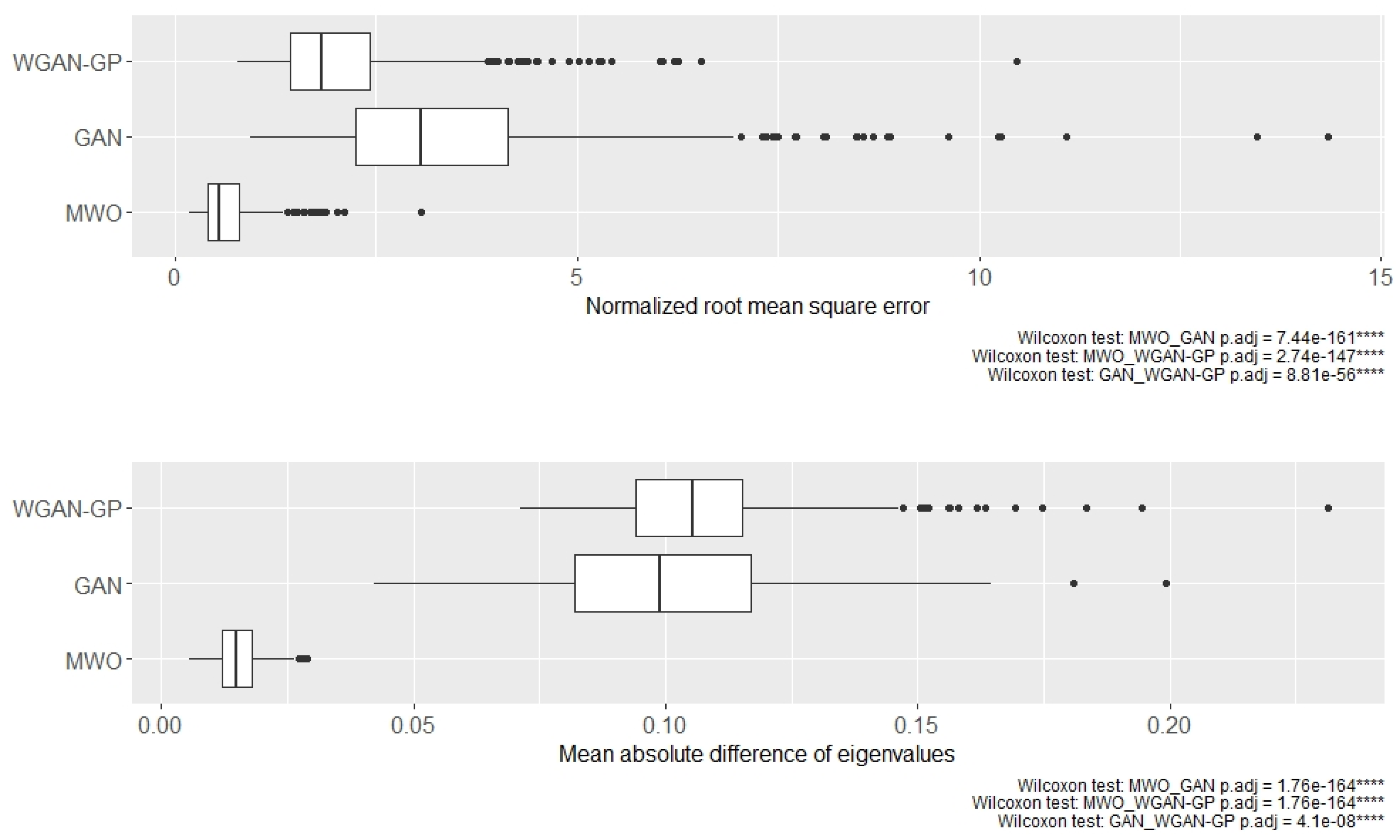
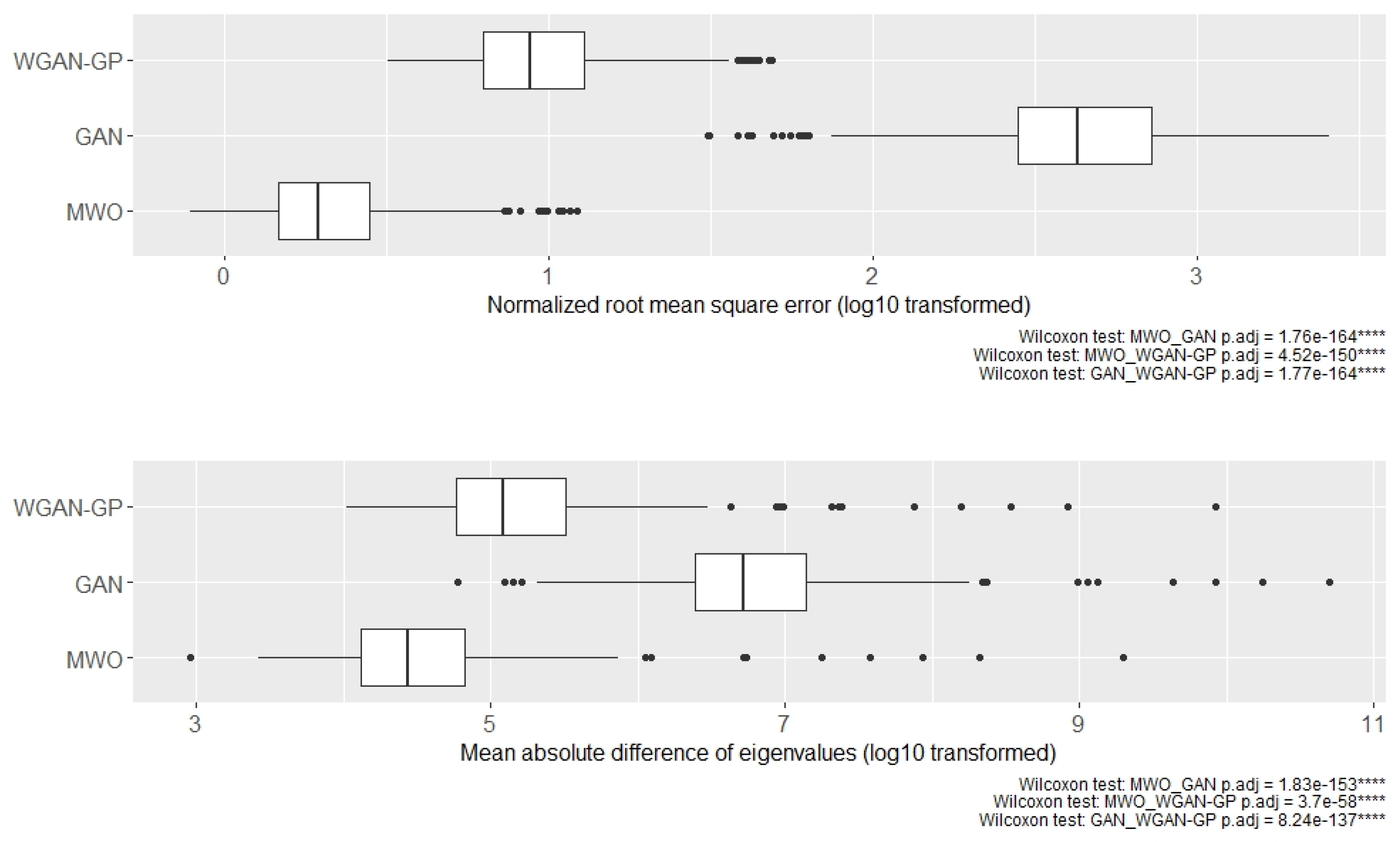
2.2. Classification Performance of Molecular Signatures Selected in the Microarray Ari Study
2.3. Classification Performance of Molecular Signatures Selected in the RNA-Seq SARS-CoV-2 Study
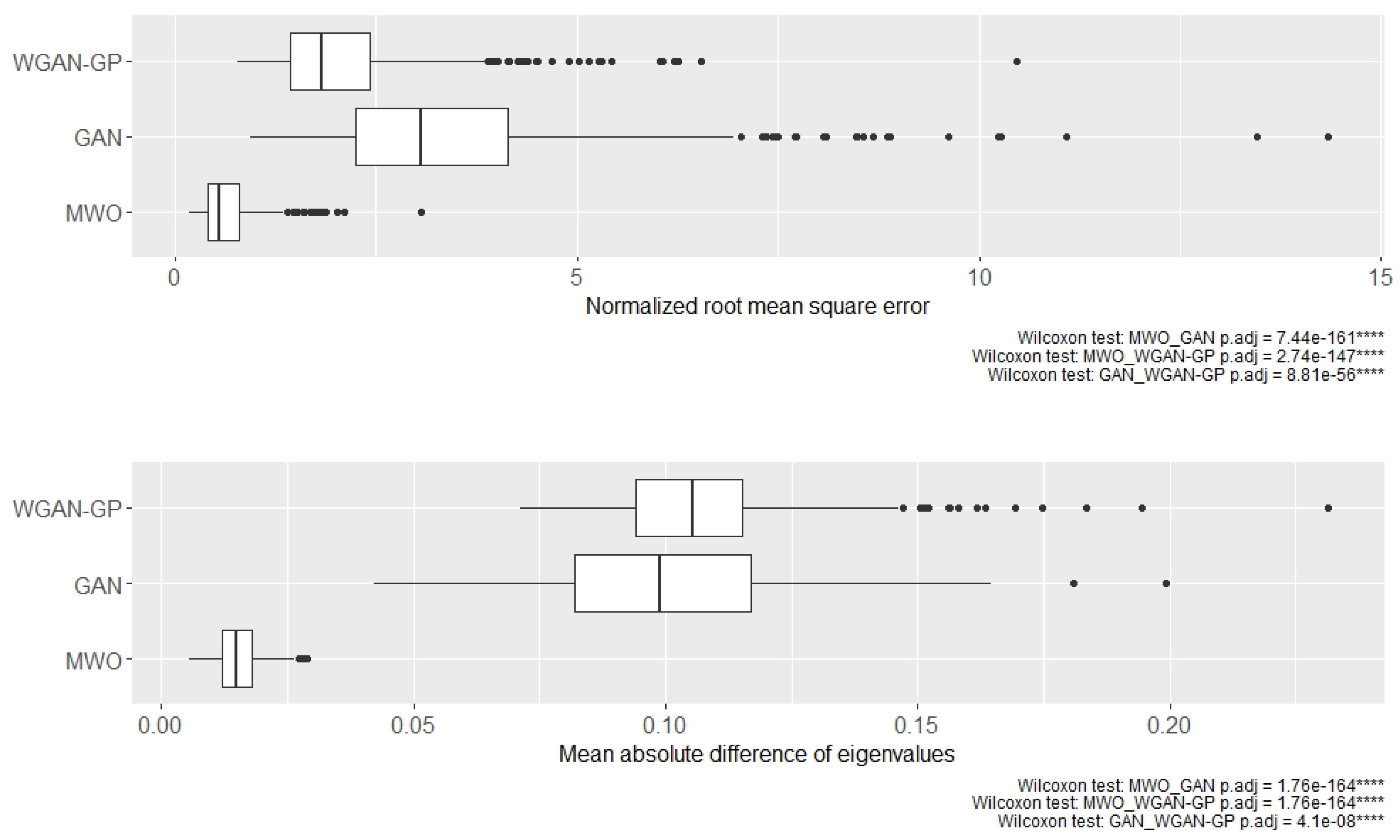
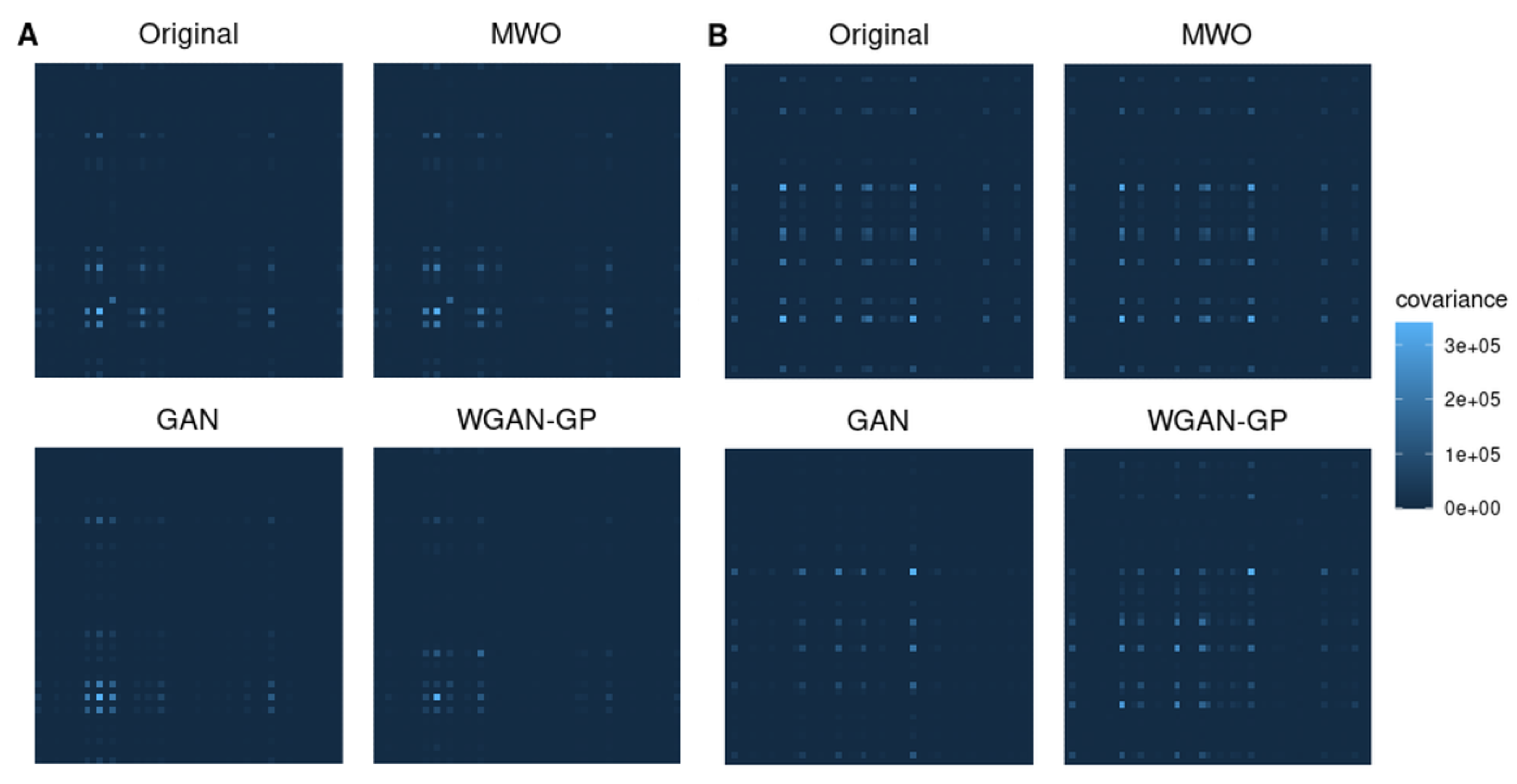
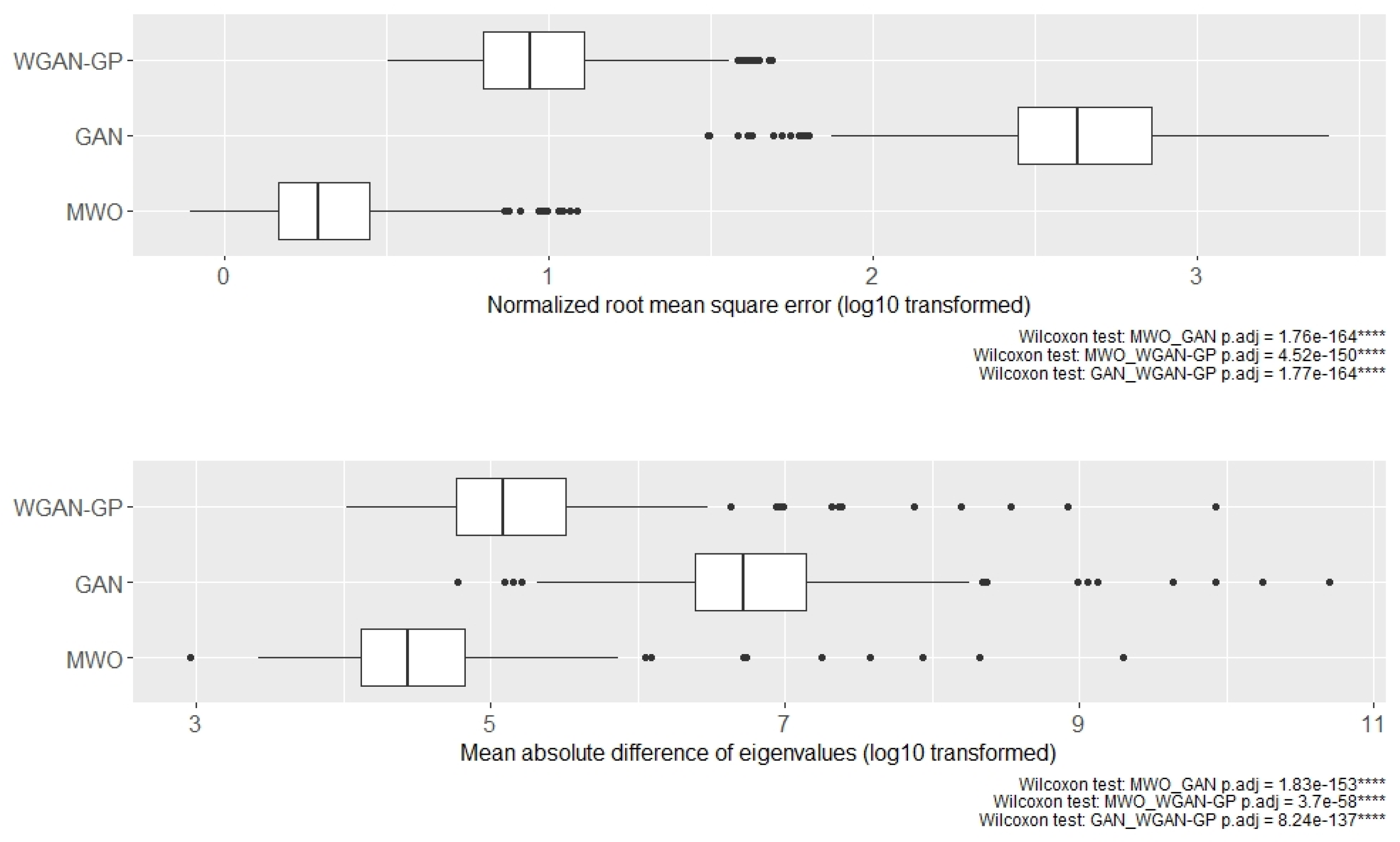
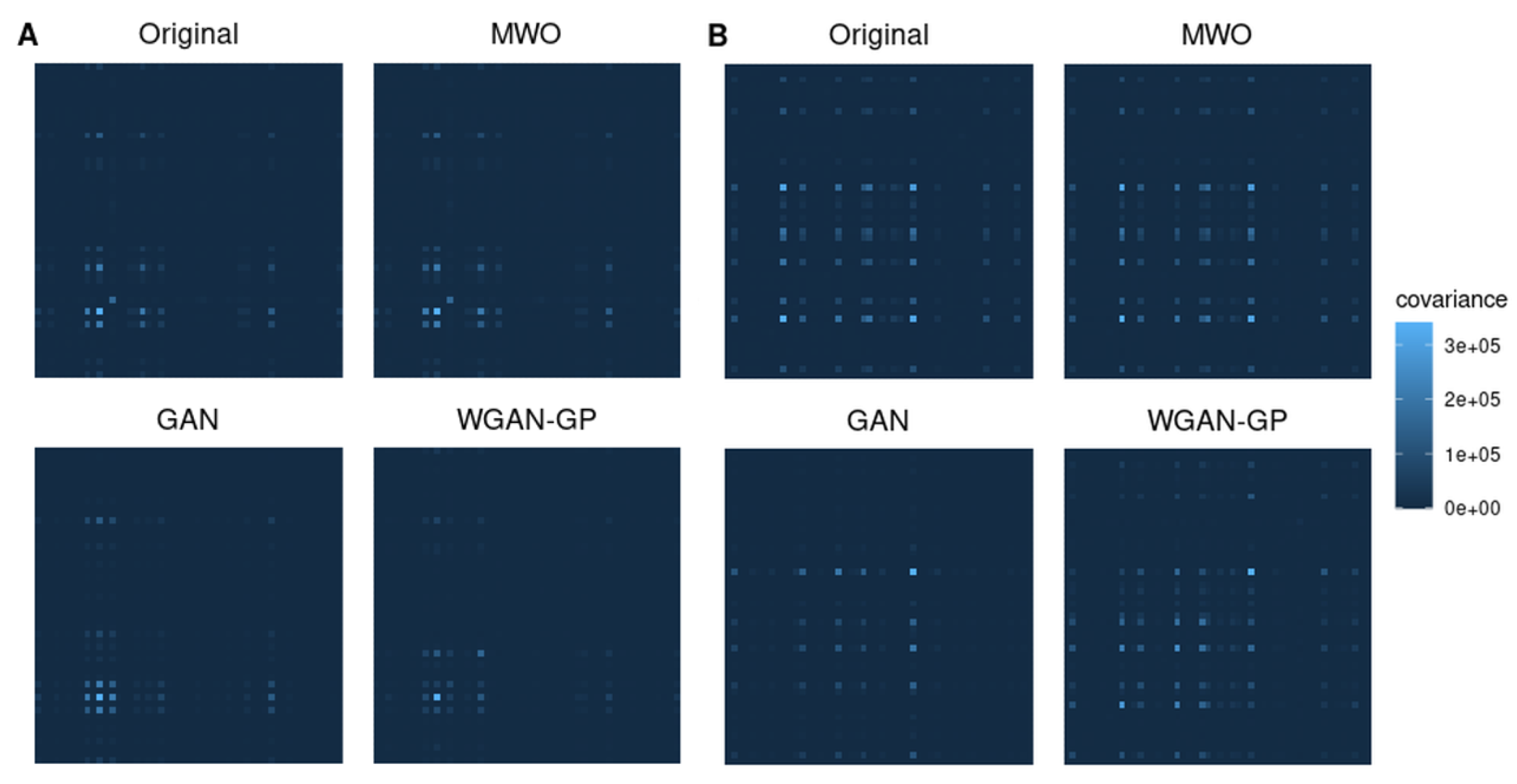
2.4. Molecular Signatures of Microarray and RNA-Seq Study, and Overlap with Originally Published Signatures
- Microarray example, biological processes: antigen processing and presentation of endogenous peptide antigen (GO:0002483); antigen processing and presentation of endogenous peptide antigen via MHC class I (GO:0019885); antigen processing and presentation (GO:0019882); antigen processing and presentation of peptide antigen via MHC class I (GO:0002474); regulation of dendritic cell differentiation (GO:2001198); dendritic cell differentiation (GO:0097028); macrophage cytokine production (GO:0010934); antigen processing and presentation of endogenous antigen (GO:0019883); phagocytosis, engulfment (GO:0006911); antigen processing and presentation of peptide antigen (GO:0048002)
- Microarray example, cellular component: phagocytic vesicle (GO:0045335)
- Microarray example, molecular function: MHC protein binding (GO:0042287); MHC class I protein binding (GO:0042288); MHC protein complex binding (GO:0023023)
- RNA-seq example, biological processes: macrophage activation (GO:0042116); T-cell differentiation involved in immune response (GO:0002292); leukocyte activation involved in immune response (GO:0002366); regulation of macrophage activation (GO:0043030); CD4-positive, alpha-beta T-cell differentiation involved in immune response (GO:0002294); cell activation involved in immune response (GO:0002263); alpha-beta T-cell activation involved in immune response (GO:0002287); alpha-beta T-cell differentiation involved in immune response (GO:0002293); regulation of interleukin-1 production (GO:0032652); regulation of interleukin-1 beta production (GO:0032651); myeloid leukocyte activation (GO:0002274); regulation of cytokine production(GO:0001817); regulation of interleukin-17 production (GO:0032660); immune response (GO:0006955); T-cell activation involved in immune response (GO:0002286); response to molecule of bacterial origin (GO:0002237); cytokine production (GO:0001816); response to lipopolysaccharide (GO:0032496); cellular response to oxidative stress (GO:0034599); NIK/NF-kappaB signaling (GO:0038061); regulation of microglial cell activation (GO:1903978); T-helper cell differentiation (GO:0042093); microglial cell activation (GO:0001774); leukocyte activation involved in inflammatory response (GO:0002269); interleukin-1 production (GO:0032612); reactive oxygen species metabolic process (GO:0072593); leukocyte activation (GO:0045321); interleukin-1 beta production (GO:0032611); plasma membrane bounded cell projection assembly (GO:0120031); positive regulation of cytokine production (GO:0001819); interleukin-17 production (GO:0032620); mitochondrial translation (GO:0032543); cytokine-mediated signaling pathway (GO:0019221); CD4-positive, alpha-beta T-cell differentiation (GO:0043367); lymphocyte activation involved in immune response (GO:0002285); cell projection assembly (GO:0030031)
- RNA-seq example, molecular function: antigen binding (GO:0003823); virus receptor activity (GO:0001618); immune receptor activity (GO:0140375); cytokine receptor binding (GO:0005126); double-stranded RNA binding (GO:0003725); cytokine receptor activity (GO:0004896); chemokine binding (GO:0019956)
3. Discussion
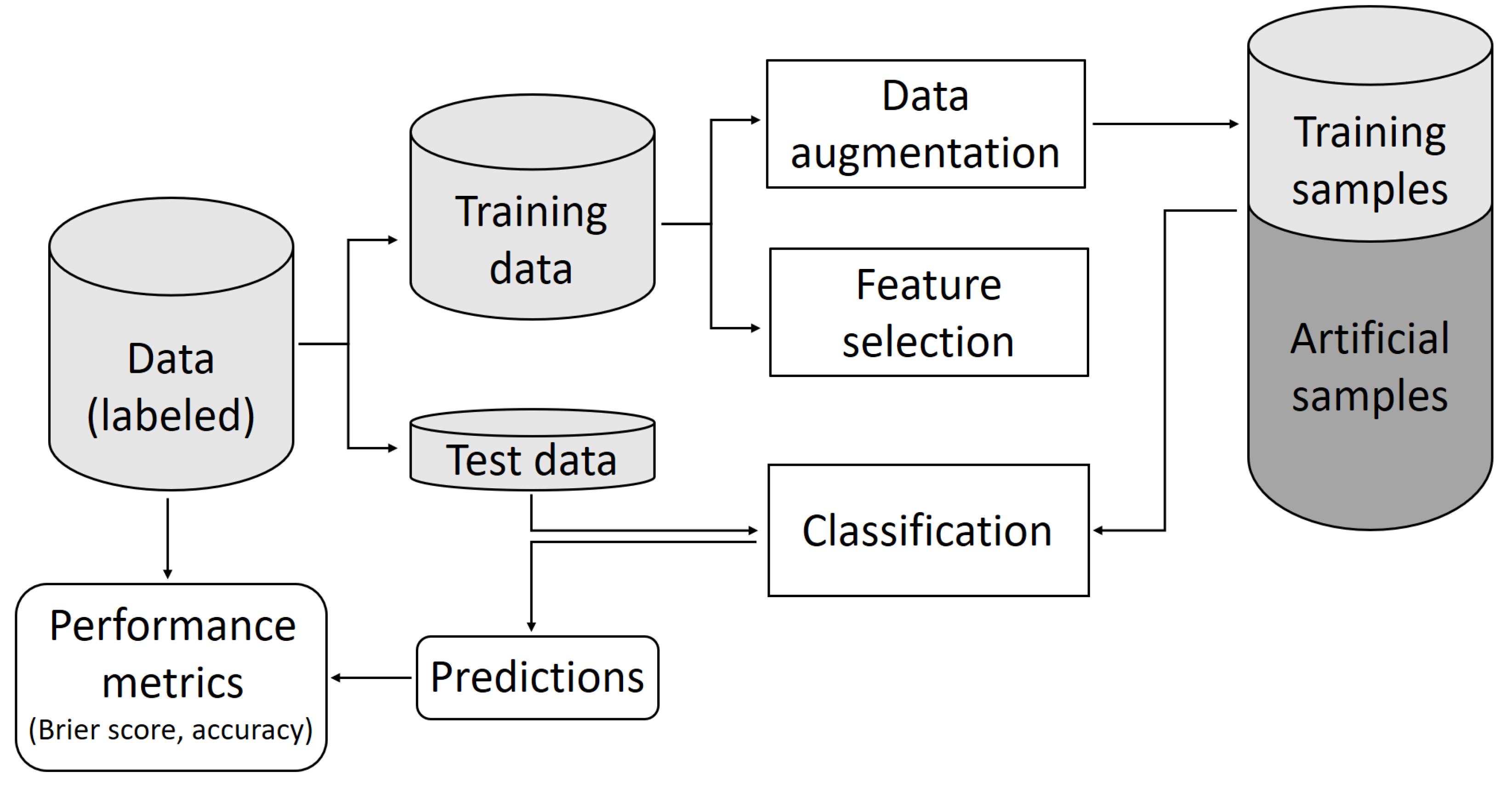
4. Materials and Methods
4.1. Example 1: Microarray Data from Infection Study
4.2. Example 2: RNA-Seq Data from SARS-CoV-2 Study
4.3. K-Folds Cross-Validation
4.4. Classification with Artificial Neural Networks
4.5. Incremental Feature Selection
4.6. Augmentation by Weighted Mixed Observations
4.7. Augmentation by Gan
4.8. Augmentation by Wasserstein Gan with Gradient Penalty
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ANN | Artificial neural network |
| ARI | Acute respiratory illness |
| CI | Confidence interval |
| GAN | Generative adversarial network |
| GEO | Gene-expression omnibus |
| GO | Gene ontology |
| ML | Machine learning |
| MWO | Mixed weighted observations |
| ReLU | Rectified linear unit |
| WGAN | Wasserstein generative adversarial network |
| WGAN-GP | Wasserstein generative adversarial network with gradient penalty |
References
- Bhattacharya, S.; Rosenberg, A.F.; Peterson, D.R.; Grzesik, K.; Baran, A.M.; Ashton, J.M.; Gill, S.R.; Corbett, A.M.; Holden-Wiltse, J.; Topham, D.J.; et al. Transcriptomic biomarkers to discriminate bacterial from nonbacterial infection in adults hospitalized with respiratory illness. Sci. Rep. 2017, 7, 6548. [Google Scholar] [CrossRef] [PubMed]
- de Lamballerie, C.N.; Pizzorno, A.; Dubois, J.; Julien, T.; Padey, B.; Bouveret, M.; Traversier, A.; Legras-Lachuer, C.; Lina, B.; Boivin, G.; et al. Characterization of cellular transcriptomic signatures induced by different respiratory viruses in human reconstituted airway epithelia. Sci. Rep. 2019, 9, 11493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forno, E.; Celedón, J.C. Epigenomics and transcriptomics in the prediction and diagnosis of childhood asthma: Are we there yet? Front. Pediatr. 2019, 7, 115. [Google Scholar] [CrossRef] [PubMed]
- Mejias, A.; Dimo, B.; Suarez, N.M.; Garcia, C.; Suarez-Arrabal, M.C.; Jartti, T.; Blankenship, D.; Jordan-Villegas, A.; Ardura, M.I.; Xu, Z.; et al. Whole blood gene expression profiles to assess pathogenesis and disease severity in infants with respiratory syncytial virus infection. PLoS Med. 2013, 10, e1001549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furey, T.S.; Cristianini, N.; Duffy, N.; Bednarski, D.W.; Schummer, M.; Haussler, D. Support vector machine classification and validation of cancer tissue samples using microarray expression data. Bioinformatics 2000, 16, 906–914. [Google Scholar] [CrossRef]
- Guo, Y.; Hastie, T.; Tibshirani, R. Regularized linear discriminant analysis and its application in microarrays. Biostatistics 2007, 8, 86–100. [Google Scholar] [CrossRef] [Green Version]
- Díaz-Uriarte, R.; De Andres, S.A. Gene selection and classification of microarray data using random forest. BMC Bioinform. 2006, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Ng, D.L.; Granados, A.C.; Santos, Y.A.; Servellita, V.; Goldgof, G.M.; Meydan, C.; Sotomayor-Gonzalez, A.; Levine, A.G.; Balcerek, J.; Han, L.M.; et al. A diagnostic host response biosignature for COVID-19 from RNA profiling of nasal swabs and blood. Sci. Adv. 2021, 7, eabe5984. [Google Scholar] [CrossRef]
- Tibshirani, R.; Hastie, T.; Narasimhan, B.; Chu, G. Class prediction by nearest shrunken centroids, with applications to DNA microarrays. Stat. Sci. 2003, 104–117. [Google Scholar] [CrossRef]
- Nilsson, R.; Björkegren, J.; Tegnér, J. On reliable discovery of molecular signatures. BMC Bioinform. 2009, 10, 38. [Google Scholar] [CrossRef] [Green Version]
- di Iulio, J.; Bartha, I.; Spreafico, R.; Virgin, H.W.; Telenti, A. Transfer transcriptomic signatures for infectious diseases. Proc. Natl. Acad. Sci. USA 2021, 118, e2022486118. [Google Scholar] [CrossRef] [PubMed]
- Oshansky, C.M.; Zhang, W.; Moore, E.; Tripp, R.A. The host response and molecular pathogenesis associated with respiratory syncytial virus infection. Future Microbiol. 2009, 4, 279–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, R.; Liu, L.; Wang, Y. Viral proteins recognized by different TLRs. J. Med Virol. 2021, 93, 6116–6123. [Google Scholar] [CrossRef] [PubMed]
- Gralinski, L.E.; Baric, R.S. Molecular pathology of emerging coronavirus infections. J. Pathol. 2015, 235, 185–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, T.; Troup, D.B.; Wilhite, S.E.; Ledoux, P.; Rudnev, D.; Evangelista, C.; Kim, I.F.; Soboleva, A.; Tomashevsky, M.; Edgar, R. NCBI GEO: Mining tens of millions of expression profiles—Database and tools update. Nucleic Acids Res. 2007, 35, D760–D765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsalik, E.L.; Henao, R.; Nichols, M.; Burke, T.; Ko, E.R.; McClain, M.T.; Hudson, L.L.; Mazur, A.; Freeman, D.H.; Veldman, T.; et al. Host gene expression classifiers diagnose acute respiratory illness etiology. Sci. Transl. Med. 2016, 8, 322ra11. [Google Scholar] [CrossRef] [Green Version]
- Goodfellow, I.; Bengio, Y.; Courville, A. Deep Learning; MIT Press: Cambridge, MA, USA, 2016. [Google Scholar]
- Min, S.; Lee, B.; Yoon, S. Deep learning in bioinformatics. Briefings Bioinform. 2017, 18, 851–869. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhou, Y.; Liu, X.; Dong, F.; Wang, C.; Wang, Z. Wasserstein GAN-based small-sample augmentation for new-generation artificial intelligence: A case study of cancer-staging data in biology. Engineering 2019, 5, 156–163. [Google Scholar] [CrossRef]
- Taylor, L.; Nitschke, G. Improving deep learning using generic data augmentation. arXiv 2017, arXiv:1708.06020. [Google Scholar]
- Shorten, C.; Khoshgoftaar, T.M. A survey on image data augmentation for deep learning. J. Big Data 2019, 6, 60. [Google Scholar] [CrossRef]
- Goodfellow, I.J.; Pouget-Abadie, J.; Mirza, M.; Xu, B.; Warde-Farley, D.; Ozair, S.; Courville, A.; Bengio, Y. Generative adversarial networks. arXiv 2014, arXiv:1406.2661. [Google Scholar] [CrossRef]
- Chaudhari, P.; Agrawal, H.; Kotecha, K. Data augmentation using MG-GAN for improved cancer classification on gene expression data. Soft Comput. 2019, 24, 11381–11391. [Google Scholar] [CrossRef]
- Home—Gene—NCBI. National Center for Biotechnology Information. Available online: https://www.ncbi.nlm.nih.gov/gene (accessed on 15 April 2020).
- Pfaender, S.; Mar, K.B.; Michailidis, E.; Kratzel, A.; Boys, I.N.; V’kovski, P.; Fan, W.; Kelly, J.N.; Hirt, D.; Ebert, N.; et al. LY6E impairs coronavirus fusion and confers immune control of viral disease. Nat. Microbiol. 2020, 5, 1330–1339. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Ghosh, A.; Sarkar, S.N. OASL—A new player in controlling antiviral innate immunity. Curr. Opin. Virol. 2015, 12, 15–19. [Google Scholar] [CrossRef] [Green Version]
- Murphy, T.L.; Tussiwand, R.; Murphy, K.M. Specificity through cooperation: BATF–IRF interactions control immune-regulatory networks. Nat. Rev. Immunol. 2013, 13, 499–509. [Google Scholar] [CrossRef]
- Rose, C.E., Jr.; Sung, S.S.J.; Fu, S.M. Significant involvement of CCL2 (MCP-1) in inflammatory disorders of the lung. Microcirculation 2003, 10, 273–288. [Google Scholar] [CrossRef]
- Alexa, A.; Rahnenführer, J.; Lengauer, T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 2006, 22, 1600–1607. [Google Scholar] [CrossRef] [Green Version]
- Giles, P.J.; Kipling, D. Normality of oligonucleotide microarray data and implications for parametric statistical analyses. Bioinformatics 2003, 19, 2254–2262. [Google Scholar] [CrossRef] [Green Version]
- Kruppa, J.; Kramer, F.; Beißbarth, T.; Jung, K. A simulation framework for correlated count data of features subsets in high-throughput sequencing or proteomics experiments. Stat. Appl. Genet. Mol. Biol. 2016, 15, 401–414. [Google Scholar] [CrossRef]
- Goutte, C.; Gaussier, E. A probabilistic interpretation of precision, recall and F-score, with implication for evaluation. In Proceedings of the European Conference on Information Retrieval, Santiago de Compostela, Spain, 21–23 March 2005; Springer: Berlin/Heidelberg, Germany, 2005; pp. 345–359. [Google Scholar]
- Roberson, E.; Mesa, R.A.; Morgan, G.A.; Cao, L.; Marin, W.; Pachman, L.M. Transcriptomes of peripheral blood mononuclear cells from juvenile dermatomyositis patients show elevated inflammation even when clinically 2 inactive. Sci. Rep. 2022, 12, 275. [Google Scholar] [CrossRef] [PubMed]
- Mahmud, S.H.; Al-Mustanjid, M.; Akter, F.; Rahman, M.S.; Ahmed, K.; Rahman, M.H.; Chen, W.; Moni, M.A. Bioinformatics and system biology approach to identify the influences of SARS-CoV-2 infections to idiopathic pulmonary fibrosis and chronic obstructive pulmonary disease patients. Briefings Bioinform. 2021, 22, bbab115. [Google Scholar] [CrossRef] [PubMed]
- Gollapalli, P.; B.S, S.; Rimac, H.; Patil, P.; Nalilu, S.K.; Kandagalla, S.; Shetty, P. Pathway enrichment analysis of virus-host interactome and prioritization of novel compounds targeting the spike glycoprotein receptor binding domain–human angiotensin-converting enzyme 2 interface to combat SARS-CoV-2. J. Biomol. Struct. Dyn. 2020; ePub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Fan, Y.; Mu, D.; Song, F.; Tian, F.; Yin, Q. Transcriptomic Analysis Exploring the Molecular Mechanisms of Hanchuan Zupa Granules in Alleviating Asthma in Rat. Evid.-Based Complement. Altern. Med. 2021, 2021, 5584099. [Google Scholar] [CrossRef]
- Liu, J.; Yang, X.; Zhang, L.; Yang, B.; Rao, W.; Li, M.; Dai, N.; Yang, Y.; Qian, C.; Zhang, L.; et al. Microarray analysis of the expression profile of immune-related gene in rapid recurrence early-stage lung adenocarcinoma. J. Cancer Res. Clin. Oncol. 2020, 146, 2299–2310. [Google Scholar] [CrossRef]
- Liu, H.; Setiono, R. Incremental feature selection. Appl. Intell. 1998, 9, 217–230. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef] [Green Version]
- Love, M.; Anders, S.; Huber, W. Differential analysis of count data—The DESeq2 package. Genome Biol. 2014, 15, 10–1186. [Google Scholar]
- Zeng, X.; Martinez, T.R. Distribution-balanced stratified cross-validation for accuracy estimation. J. Exp. Theor. Artif. Intell. 2000, 12, 1–12. [Google Scholar] [CrossRef]
- Kohavi, R. A study of cross-validation and bootstrap for accuracy estimation and model selection. In Proceedings of the 14th International Joint Conference on Artificial intelligence, Montreal, QC, Canada, 20–25 August 1995; Volume 14, pp. 1137–1145. [Google Scholar]
- Brier, G.W. Verification of forecasts expressed in terms of probability. Mon. Weather. Rev. 1950, 78, 1–3. [Google Scholar] [CrossRef]
- Agarap, A.F. Deep learning using rectified linear units (relu). arXiv 2018, arXiv:1803.08375. [Google Scholar]
- Srivastava, N.; Hinton, G.; Krizhevsky, A.; Sutskever, I.; Salakhutdinov, R. Dropout: A simple way to prevent neural networks from overfitting. J. Mach. Learn. Res. 2014, 15, 1929–1958. [Google Scholar]
- Paszke, A.; Gross, S.; Massa, F.; Lerer, A.; Bradbury, J.; Chanan, G.; Killeen, T.; Lin, Z.; Gimelshein, N.; Antiga, L.; et al. Pytorch: An imperative style, high-performance deep learning library. Adv. Neural Inf. Process. Syst. 2019, 32, 8026–8037. [Google Scholar]
- Falcon, W.A. PyTorch Lightning. GitHub. 2019, Volume 3. Available online: https://github.com/PyTorchLightning/pytorch-lightning (accessed on 15 February 2021).
- Smyth, G.K. Limma: Linear models for microarray data. In Bioinformatics and Computational Biology Solutions using R and Bioconductor; Springer: Cham, Switzerland, 2005; pp. 397–420. [Google Scholar]
- Creswell, A.; White, T.; Dumoulin, V.; Arulkumaran, K.; Sengupta, B.; Bharath, A.A. Generative adversarial networks: An overview. IEEE Signal Process. Mag. 2018, 35, 53–65. [Google Scholar] [CrossRef] [Green Version]
- Sathya, R.; Abraham, A. Comparison of supervised and unsupervised learning algorithms for pattern classification. Int. J. Adv. Res. Artif. Intell. 2013, 2, 34–38. [Google Scholar] [CrossRef] [Green Version]
- Viñas, R.; Andrés-Terré, H.; Liò, P.; Bryson, K. Adversarial generation of gene expression data. bioRxiv 2019, 38, 836254. [Google Scholar] [CrossRef]
- Marouf, M.; Machart, P.; Bansal, V.; Kilian, C.; Magruder, D.S.; Krebs, C.F.; Bonn, S. Realistic in silico generation and augmentation of single-cell RNA-seq data using generative adversarial networks. Nat. Commun. 2020, 11, 166. [Google Scholar] [CrossRef] [Green Version]
- Kingma, D.P.; Ba, J. Adam: A method for stochastic optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Arjovsky, M.; Chintala, S.; Bottou, L. Wasserstein generative adversarial networks. In Proceedings of the International Conference on Machine Learning, PMLR, Sydney, Australia, 6–11 August 2017; pp. 214–223. [Google Scholar]
- Weng, L. From gan to wgan. arXiv 2019, arXiv:1904.08994. [Google Scholar]
- Gulrajani, I.; Ahmed, F.; Arjovsky, M.; Dumoulin, V.; Courville, A. Improved training of wasserstein gans. arXiv 2017, arXiv:1704.00028. [Google Scholar]
- Persson, A. WGAN-GP. GitHub. 2021. Available online: https://github.com/aladdinpersson/Machine-Learning-Collection/tree/master/ML/Pytorch\/GANs/4.%20WGAN-GP (accessed on 5 April 2021).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Number of Top Genes | Accuracy | Brier Score | Sensitivity | Specificity |
|---|---|---|---|---|---|
| No | 50 | 0.86 ± 0.0440 | 0.16 ± 0.0143 | 0.92 ± 0.0515 | 0.76 ± 0.0998 |
| Augmentation | 100 | 0.86 ± 0.0413 | 0.15 ± 0.0140 | 0.92 ± 0.0487 | 0.78 ± 0.1090 |
| 150 | 0.87 ± 0.0466 | 0.13 ± 0.0121 | 0.92 ± 0.0514 | 0.79 ± 0.1090 | |
| 200 | 0.88 ± 0.0488 | 0.13 ± 0.0128 | 0.93 ± 0.0535 | 0.79 ± 0.1090 | |
| 250 | 0.88 ± 0.0436 | 0.12 ± 0.0124 | 0.93 ± 0.0411 | 0.79 ± 0.1037 | |
| 300 | 0.88 ± 0.0429 | 0.12 ± 0.0129 | 0.92 ± 0.0514 | 0.82 ± 0.0887 | |
| 350 | 0.88 ± 0.0463 | 0.11 ± 0.0144 | 0.92 ± 0.0515 | 0.81 ± 0.0939 | |
| 400 | 0.87 ± 0.0466 | 0.11 ± 0.0168 | 0.92 ± 0.0487 | 0.81 ± 0.1028 | |
| 450 | 0.87 ± 0.0515 | 0.10 ± 0.0184 | 0.92 ± 0.0515 | 0.79 ± 0.1118 | |
| 500 | 0.88 ± 0.0463 | 0.10 ± 0.0160 | 0.92 ± 0.0514 | 0.81 ± 0.0998 | |
| 550 | 0.88 ± 0.0401 | 0.10 ± 0.0145 | 0.92 ± 0.0487 | 0.84 ± 0.0748 | |
| 600 | 0.88 ± 0.0488 | 0.10 ± 0.0164 | 0.92 ± 0.0515 | 0.81 ± 0.0939 | |
| 650 | 0.88 ± 0.0481 | 0.10 ± 0.0184 | 0.92 ± 0.0515 | 0.82 ± 0.0919 | |
| 700 | 0.88 ± 0.0429 | 0.10 ± 0.0168 | 0.92 ± 0.0515 | 0.82 ± 0.0783 | |
| 750 | 0.89 ± 0.0447 | 0.10 ± 0.0166 | 0.93 ± 0.0411 | 0.82 ± 0.0919 |
| Method | Number of Top Genes | Accuracy | Brier Score | Sensitivity | Specificity |
|---|---|---|---|---|---|
| MWO | 50 | 0.87 ± 0.0384 | 0.09 ± 0.0181 | 0.91 ± 0.0451 | 0.81 ± 0.0805 |
| 100 | 0.88 ± 0.0346 | 0.09 ± 0.0216 | 0.91 ± 0.0451 | 0.83 ± 0.0798 | |
| 150 | 0.88 ± 0.0378 | 0.09 ± 0.0228 | 0.92 ± 0.0487 | 0.82 ± 0.0783 | |
| 200 | 0.88 ± 0.0456 | 0.09 ± 0.0235 | 0.92 ± 0.0487 | 0.84 ± 0.0953 | |
| 250 | 0.88 ± 0.0408 | 0.09 ± 0.0245 | 0.91 ± 0.0451 | 0.84 ± 0.0953 | |
| 300 | 0.88 ± 0.0408 | 0.09 ± 0.0243 | 0.91 ± 0.0451 | 0.84 ± 0.0953 | |
| 350 | 0.88 ± 0.0456 | 0.09 ± 0.0236 | 0.92 ± 0.0487 | 0.84 ± 0.0953 | |
| 400 | 0.89 ± 0.0461 | 0.09 ± 0.0239 | 0.92 ± 0.0487 | 0.86 ± 0.0760 | |
| 450 | 0.88 ± 0.0401 | 0.09 ± 0.0247 | 0.92 ± 0.0487 | 0.84 ± 0.0748 | |
| 500 | 0.88 ± 0.0401 | 0.09 ± 0.0242 | 0.91 ± 0.0451 | 0.85 ± 0.0815 | |
| 550 | 0.89 ± 0.0461 | 0.09 ± 0.0240 | 0.92 ± 0.0487 | 0.86 ± 0.0760 | |
| 600 | 0.89 ± 0.0461 | 0.09 ± 0.0235 | 0.92 ± 0.0487 | 0.86 ± 0.0760 | |
| 650 | 0.89 ± 0.0391 | 0.09 ± 0.0238 | 0.91 ± 0.0451 | 0.86 ± 0.0760 | |
| 700 | 0.89 ± 0.0447 | 0.09 ± 0.0240 | 0.92 ± 0.0487 | 0.85 ± 0.0815 | |
| 750 | 0.89 ± 0.0420 | 0.09 ± 0.0237 | 0.91 ± 0.0451 | 0.86 ± 0.0760 | |
| GAN | 50 | 0.87 ± 0.0383 | 0.09 ± 0.0248 | 0.9 ± 0.0484 | 0.84 ± 0.0748 |
| 100 | 0.87 ± 0.0383 | 0.10 ± 0.0261 | 0.90 ± 0.0484 | 0.84 ± 0.0748 | |
| 150 | 0.87 ± 0.0442 | 0.09 ± 0.0275 | 0.90 ± 0.0484 | 0.82 ± 0.0919 | |
| 200 | 0.87 ± 0.0384 | 0.10 ± 0.0307 | 0.89 ± 0.0497 | 0.84 ± 0.0748 | |
| 250 | 0.87 ± 0.0384 | 0.10 ± 0.0308 | 0.89 ± 0.0497 | 0.84 ± 0.0748 | |
| 300 | 0.86 ± 0.0436 | 0.10 ± 0.0283 | 0.88 ± 0.0504 | 0.82 ± 0.0919 | |
| 350 | 0.86 ± 0.0466 | 0.10 ± 0.0286 | 0.88 ± 0.0504 | 0.84 ± 0.0889 | |
| 400 | 0.86 ± 0.0466 | 0.10 ± 0.0297 | 0.88 ± 0.0504 | 0.84 ± 0.0889 | |
| 450 | 0.86 ± 0.0466 | 0.10 ± 0.0280 | 0.87 ± 0.0561 | 0.85 ± 0.0848 | |
| 500 | 0.87 ± 0.0466 | 0.10 ± 0.0291 | 0.88 ± 0.0504 | 0.87 ± 0.0897 | |
| 550 | 0.87 ± 0.0491 | 0.10 ± 0.0298 | 0.89 ± 0.0497 | 0.85 ± 0.0945 | |
| 600 | 0.86 ± 0.0466 | 0.10 ± 0.0297 | 0.88 ± 0.0504 | 0.84 ± 0.0889 | |
| 650 | 0.87 ± 0.0468 | 0.10 ± 0.0275 | 0.88 ± 0.0504 | 0.85 ± 0.0848 | |
| 700 | 0.88 ± 0.0488 | 0.10 ± 0.0270 | 0.89 ± 0.0497 | 0.87 ± 0.0897 | |
| 750 | 0.87 ± 0.0413 | 0.09 ± 0.0277 | 0.88 ± 0.0560 | 0.86 ± 0.0635 | |
| WGAN-GP | 50 | 0.87 ± 0.0384 | 0.09 ± 0.021 | 0.92 ± 0.0487 | 0.79 ± 0.0916 |
| 100 | 0.88 ± 0.0346 | 0.09 ± 0.0235 | 0.91 ± 0.0451 | 0.83 ± 0.0798 | |
| 150 | 0.88 ± 0.0401 | 0.09 ± 0.0249 | 0.92 ± 0.0487 | 0.84 ± 0.0748 | |
| 200 | 0.87 ± 0.0384 | 0.09 ± 0.0264 | 0.91 ± 0.0451 | 0.81 ± 0.0906 | |
| 250 | 0.88 ± 0.0401 | 0.09 ± 0.0254 | 0.91 ± 0.0451 | 0.85 ± 0.0815 | |
| 300 | 0.88 ± 0.0429 | 0.09 ± 0.0240 | 0.92 ± 0.0487 | 0.84 ± 0.0953 | |
| 350 | 0.88 ± 0.0429 | 0.09 ± 0.0260 | 0.92 ± 0.0487 | 0.84 ± 0.0953 | |
| 400 | 0.88 ± 0.0408 | 0.09 ± 0.0250 | 0.91 ± 0.0451 | 0.84 ± 0.0953 | |
| 450 | 0.90 ± 0.0447 | 0.08 ± 0.0231 | 0.93 ± 0.0535 | 0.85 ± 0.0862 | |
| 500 | 0.89 ± 0.0420 | 0.09 ± 0.0245 | 0.92 ± 0.0487 | 0.85 ± 0.0815 | |
| 550 | 0.90 ± 0.0420 | 0.08 ± 0.0241 | 0.92 ± 0.0487 | 0.88 ± 0.0688 | |
| 600 | 0.89 ± 0.0391 | 0.09 ± 0.0235 | 0.92 ± 0.0487 | 0.85 ± 0.0862 | |
| 650 | 0.88 ± 0.0401 | 0.09 ± 0.0248 | 0.91 ± 0.0451 | 0.85 ± 0.0916 | |
| 700 | 0.91 ± 0.0401 | 0.09 ± 0.0247 | 0.92 ± 0.0514 | 0.88 ± 0.0618 | |
| 750 | 0.90 ± 0.0420 | 0.09 ± 0.0231 | 0.92 ± 0.0514 | 0.86 ± 0.0696 |
| Method | Number of Top Genes | Accuracy | Brier Score | Sensitivity | Specificity |
|---|---|---|---|---|---|
| No | 50 | 0.59 ±0.0557 | 0.24 ±0.0027 | 0.24 ±0.1100 | 0.98 ±0.0218 |
| Augmentation | 100 | 0.64 ± 0.0564 | 0.24 ± 0.0050 | 0.52 ± 0.1806 | 0.79 ± 0.2591 |
| 150 | 0.63 ± 0.0563 | 0.24 ± 0.0051 | 0.43 ± 0.1501 | 0.86 ± 0.1902 | |
| 200 | 0.66 ± 0.0527 | 0.24 ± 0.0073 | 0.43 ± 0.0976 | 0.92 ± 0.0544 | |
| 250 | 0.66 ± 0.0524 | 0.24 ± 0.0074 | 0.54 ± 0.1211 | 0.79 ± 0.1842 | |
| 300 | 0.64 ± 0.0512 | 0.24 ± 0.0051 | 0.54 ± 0.1332 | 0.76 ± 0.1813 | |
| 350 | 0.63 ± 0.0550 | 0.24 ± 0.0056 | 0.50 ± 0.1405 | 0.78 ± 0.1825 | |
| 400 | 0.67 ± 0.0464 | 0.23 ± 0.0072 | 0.51 ± 0.0720 | 0.86 ± 0.049 | |
| 450 | 0.67 ± 0.0518 | 0.23 ± 0.0109 | 0.57 ± 0.1160 | 0.78 ± 0.1652 | |
| 500 | 0.65 ± 0.0657 | 0.23 ± 0.0117 | 0.46 ± 0.1185 | 0.86 ± 0.0845 | |
| 550 | 0.67 ± 0.0411 | 0.23 ± 0.0086 | 0.54 ± 0.0630 | 0.82 ± 0.0722 | |
| 600 | 0.67 ± 0.0469 | 0.23 ± 0.0110 | 0.54 ± 0.0663 | 0.82 ± 0.0514 | |
| 650 | 0.66 ± 0.0425 | 0.23 ± 0.0069 | 0.53 ± 0.0516 | 0.80 ± 0.0697 | |
| 700 | 0.64 ± 0.0570 | 0.23 ± 0.0078 | 0.52 ± 0.0482 | 0.78 ± 0.1222 | |
| 750 | 0.67 ± 0.0355 | 0.22 ± 0.0094 | 0.54 ± 0.0547 | 0.81 ± 0.0490 |
| Method | Number of Top Genes | Accuracy | Brier Score | Sensitivity | Specificity |
|---|---|---|---|---|---|
| MWO | 50 | 0.68 ± 0.0533 | 0.22 ± 0.0111 | 0.43 ± 0.0933 | 0.96 ± 0.0365 |
| 100 | 0.71 ± 0.0450 | 0.22 ± 0.0125 | 0.52 ± 0.0654 | 0.92 ± 0.0544 | |
| 150 | 0.70 ± 0.0448 | 0.21 ± 0.0109 | 0.53 ± 0.0729 | 0.89 ± 0.0691 | |
| 200 | 0.71 ± 0.0447 | 0.21 ± 0.0126 | 0.56 ± 0.0651 | 0.89 ± 0.0547 | |
| 250 | 0.70 ± 0.0511 | 0.21 ± 0.0111 | 0.56 ± 0.0595 | 0.86 ± 0.0772 | |
| 300 | 0.72 ± 0.0338 | 0.20 ± 0.0112 | 0.58 ± 0.0481 | 0.88 ± 0.0609 | |
| 350 | 0.72 ± 0.0374 | 0.19 ± 0.0126 | 0.59 ± 0.0505 | 0.87 ± 0.0606 | |
| 400 | 0.71 ± 0.0341 | 0.19 ± 0.0136 | 0.60 ± 0.0466 | 0.82 ± 0.0618 | |
| 450 | 0.71 ± 0.0405 | 0.19 ± 0.0175 | 0.62 ± 0.0480 | 0.81 ± 0.0732 | |
| 500 | 0.71 ± 0.0338 | 0.19 ± 0.0139 | 0.62 ± 0.0537 | 0.82 ± 0.0618 | |
| 550 | 0.71 ± 0.0338 | 0.19 ± 0.0131 | 0.62 ± 0.0487 | 0.81 ± 0.0425 | |
| 600 | 0.71 ± 0.0338 | 0.20 ± 0.0194 | 0.64 ± 0.0406 | 0.79 ± 0.0609 | |
| 650 | 0.71 ± 0.0373 | 0.19 ± 0.0160 | 0.63 ± 0.0475 | 0.81 ± 0.0547 | |
| 700 | 0.71 ± 0.0318 | 0.19 ± 0.0151 | 0.63 ± 0.0518 | 0.80 ± 0.0499 | |
| 750 | 0.71 ± 0.0373 | 0.19 ± 0.0165 | 0.63 ± 0.0475 | 0.81 ± 0.0547 | |
| GAN | 50 | 0.63 ± 0.0792 | 0.24 ± 0.0102 | 0.76 ± 0.1749 | 0.48 ± 0.3068 |
| 100 | 0.67 ± 0.0828 | 0.23 ± 0.0113 | 0.78 ± 0.1292 | 0.53 ± 0.2768 | |
| 150 | 0.66 ± 0.0794 | 0.23 ± 0.0131 | 0.80 ± 0.1247 | 0.50 ± 0.2700 | |
| 200 | 0.70 ± 0.0836 | 0.22 ± 0.0157 | 0.86 ± 0.0934 | 0.52 ± 0.2481 | |
| 250 | 0.70 ± 0.0805 | 0.22 ± 0.0183 | 0.87 ± 0.0776 | 0.50 ± 0.2435 | |
| 300 | 0.70 ± 0.0873 | 0.21 ± 0.0178 | 0.87 ± 0.0974 | 0.50 ± 0.2447 | |
| 350 | 0.70 ± 0.0823 | 0.21 ± 0.0207 | 0.89 ± 0.0840 | 0.49 ± 0.2354 | |
| 400 | 0.70 ± 0.0798 | 0.20 ± 0.0202 | 0.89 ± 0.0840 | 0.48 ± 0.2320 | |
| 450 | 0.68 ± 0.0747 | 0.20 ± 0.0273 | 0.89 ± 0.0760 | 0.43 ± 0.2294 | |
| 500 | 0.68 ± 0.0724 | 0.20 ± 0.0222 | 0.89 ± 0.0760 | 0.44 ± 0.2259 | |
| 550 | 0.67 ± 0.0712 | 0.20 ± 0.0225 | 0.88 ± 0.0862 | 0.43 ± 0.2242 | |
| 600 | 0.68 ± 0.0550 | 0.21 ± 0.0225 | 0.89 ± 0.0814 | 0.45 ± 0.1936 | |
| 650 | 0.71 ± 0.0591 | 0.20 ± 0.0228 | 0.89 ± 0.0760 | 0.49 ± 0.1909 | |
| 700 | 0.71 ± 0.0653 | 0.20 ± 0.0262 | 0.91 ± 0.0628 | 0.49 ± 0.2029 | |
| 750 | 0.71 ± 0.0597 | 0.20 ± 0.0244 | 0.90 ± 0.0633 | 0.48 ± 0.1802 | |
| WGAN-GP | 50 | 0.70 ± 0.0418 | 0.22 ± 0.0114 | 0.48 ± 0.0801 | 0.95 ± 0.0436 |
| 100 | 0.72 ± 0.0333 | 0.20 ± 0.0111 | 0.56 ± 0.0367 | 0.92 ± 0.0544 | |
| 150 | 0.72 ± 0.0334 | 0.20 ± 0.0088 | 0.59 ± 0.0398 | 0.88 ± 0.0609 | |
| 200 | 0.74 ± 0.0474 | 0.18 ± 0.0143 | 0.65 ± 0.0580 | 0.85 ± 0.0722 | |
| 250 | 0.76 ± 0.0462 | 0.18 ± 0.0137 | 0.68 ± 0.0605 | 0.86 ± 0.0646 | |
| 300 | 0.77 ± 0.0448 | 0.18 ± 0.0120 | 0.70 ± 0.0572 | 0.86 ± 0.0691 | |
| 350 | 0.76 ± 0.0379 | 0.18 ± 0.0143 | 0.70 ± 0.0533 | 0.83 ± 0.0568 | |
| 400 | 0.74 ± 0.0469 | 0.17 ± 0.0146 | 0.70 ± 0.0644 | 0.80 ± 0.0606 | |
| 450 | 0.74 ± 0.0469 | 0.17 ± 0.0165 | 0.71 ± 0.0584 | 0.78 ± 0.0697 | |
| 500 | 0.75 ± 0.0404 | 0.17 ± 0.0165 | 0.72 ± 0.0482 | 0.78 ± 0.0653 | |
| 550 | 0.74 ± 0.0440 | 0.18 ± 0.0160 | 0.72 ± 0.0500 | 0.75 ± 0.0689 | |
| 600 | 0.72 ± 0.0385 | 0.18 ± 0.0177 | 0.70 ± 0.0490 | 0.75 ± 0.0596 | |
| 650 | 0.72 ± 0.0402 | 0.18 ± 0.0187 | 0.70 ± 0.0561 | 0.75 ± 0.0596 | |
| 700 | 0.74 ± 0.0373 | 0.18 ± 0.0200 | 0.75 ± 0.0668 | 0.74 ± 0.0665 | |
| 750 | 0.74 ± 0.0344 | 0.18 ± 0.0173 | 0.74 ± 0.0472 | 0.74 ± 0.0618 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kircher, M.; Chludzinski, E.; Krepel, J.; Saremi, B.; Beineke, A.; Jung, K. Augmentation of Transcriptomic Data for Improved Classification of Patients with Respiratory Diseases of Viral Origin. Int. J. Mol. Sci. 2022, 23, 2481. https://doi.org/10.3390/ijms23052481
Kircher M, Chludzinski E, Krepel J, Saremi B, Beineke A, Jung K. Augmentation of Transcriptomic Data for Improved Classification of Patients with Respiratory Diseases of Viral Origin. International Journal of Molecular Sciences. 2022; 23(5):2481. https://doi.org/10.3390/ijms23052481
Chicago/Turabian StyleKircher, Magdalena, Elisa Chludzinski, Jessica Krepel, Babak Saremi, Andreas Beineke, and Klaus Jung. 2022. "Augmentation of Transcriptomic Data for Improved Classification of Patients with Respiratory Diseases of Viral Origin" International Journal of Molecular Sciences 23, no. 5: 2481. https://doi.org/10.3390/ijms23052481
APA StyleKircher, M., Chludzinski, E., Krepel, J., Saremi, B., Beineke, A., & Jung, K. (2022). Augmentation of Transcriptomic Data for Improved Classification of Patients with Respiratory Diseases of Viral Origin. International Journal of Molecular Sciences, 23(5), 2481. https://doi.org/10.3390/ijms23052481