2.1. Characterization of Pure Polymer Samples
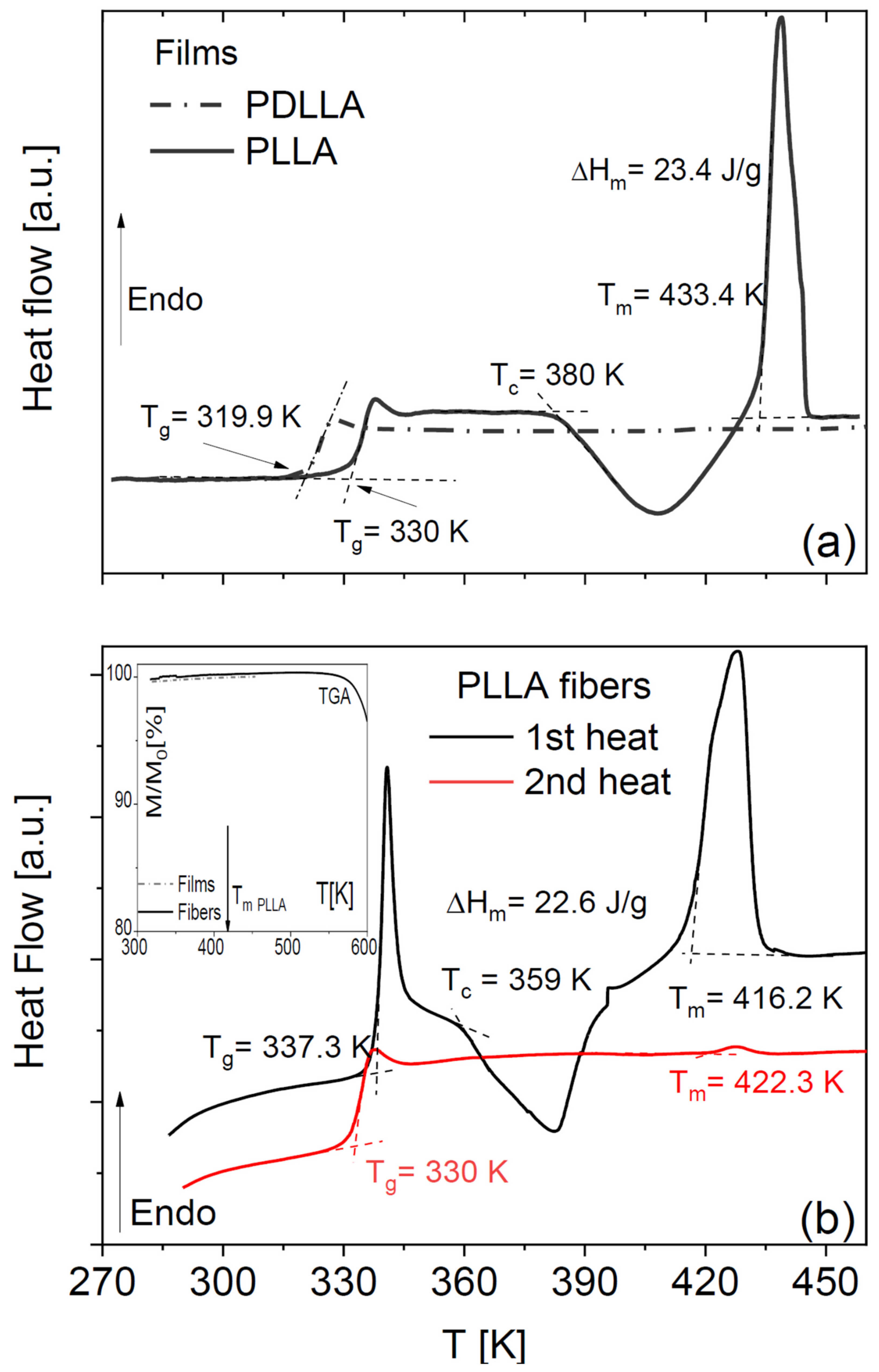
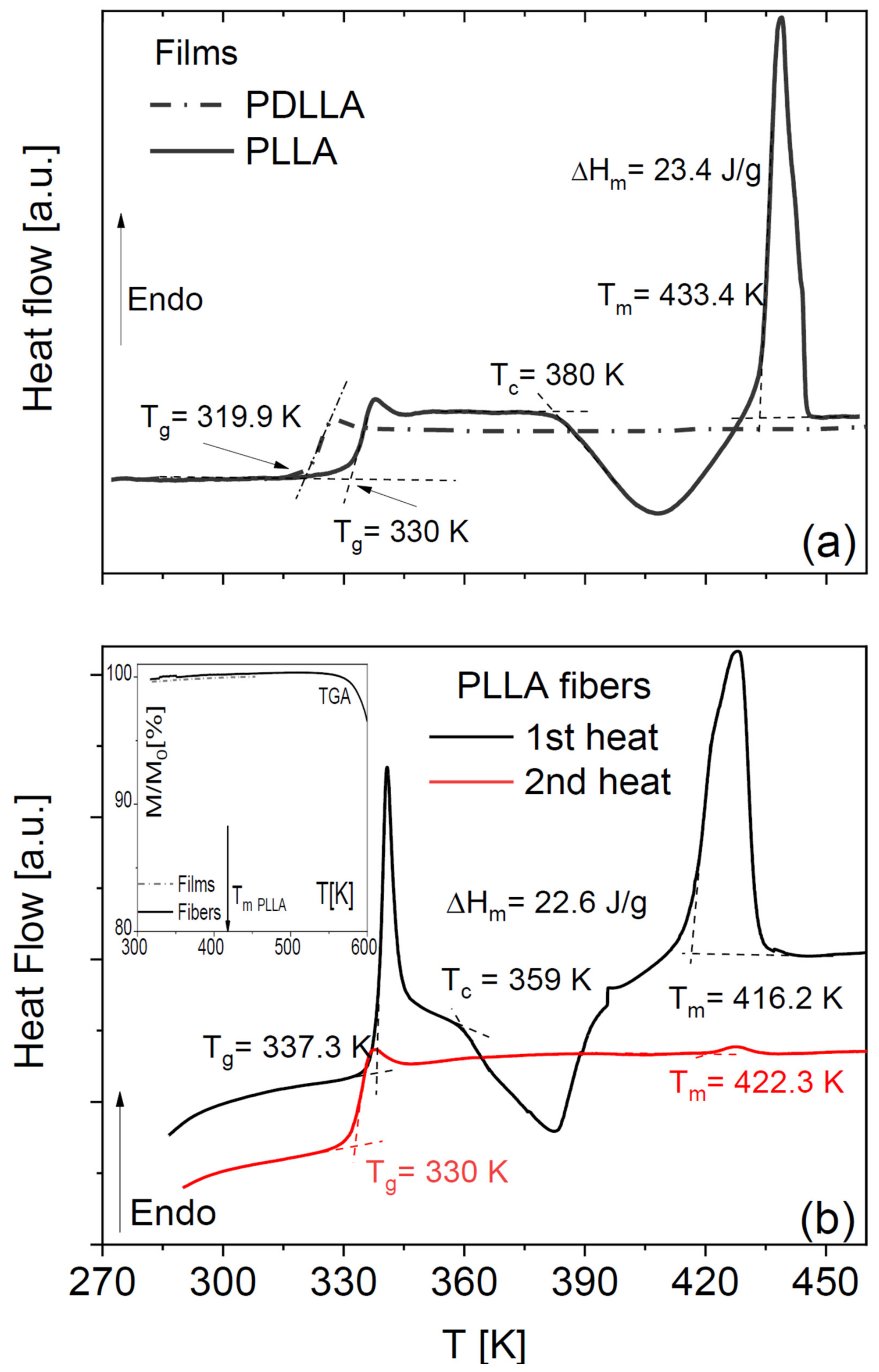
Pure amorphous PLLA films obtained by annealing a solvent-cast sample display the characteristic step-like increase in specific heat across its glass transition and then undergo (partial) cold crystallization, followed by melting at higher temperature (
Figure 1a). Such peaks were absent in our home-synthesized racemic PDLLA polymer (same panel), which shows that it is completely amorphous as expected. It can be observed that the
Tg of the fully amorphous PDLLA films is lower by 10 K with respect to that of the amorphous PLLA films obtained right after cooling from the melt. This difference in glass transition temperature is due to the different molecular weights of the samples (see
Section 4).
The DSC thermograms of as-deposited scaffolds of PLLA microfibres (
Figure 1b) have qualitatively the same line shape as the PLLA films but differ in the onset melting temperature (
Tm = 433 K for films and
Tm = 416 K for the fibre scaffolds) and in the onset temperature of crystallization, which were ca. 380 and 360 K for the films and the microfibres, respectively. Comparison of the enthalpy of cold crystallization and of melting with the melting enthalpy of fully crystalline PLLA (see Equation (3) in
Section 4) allows concluding that the as-deposited fibres have a crystalline fraction of at most 2%, and the annealed ones a crystalline fraction of 21%, which is comparable with that of the cold-crystallized PLLA films.
The absence of any residual volatile solvents in either the membranes or the microfibres was checked by thermogravimetry analysis (see the inset to
Figure 1b for representative traces for both morphologies), and by IR characterization (not shown).
Visual comparison of the DSC thermograms of the fibre scaffolds shows a significant ageing effect in the as-deposited fibres, visible both in the higher glass transition temperature (
Tg) and in the pronounced enthalpy recovery peak accompanying the first-heat scan. The higher
Tg value could also be partially due to the non-zero crystalline fraction of the as-deposited fibres, since an increase in
Tg with increasing crystalline fraction is often observed in semicrystalline polymers [
27]. On the other hand, the
Tg values of both PLLA fibres and films right after cooling them from above the melting point were identical (330 K). This shows that, to compare the
Tg values of different samples, care must be taken to achieve a similar degree of ageing. In the following, to compare different sample morphologies, we will consider dielectric measurements carried out on samples with a similar thermal history.
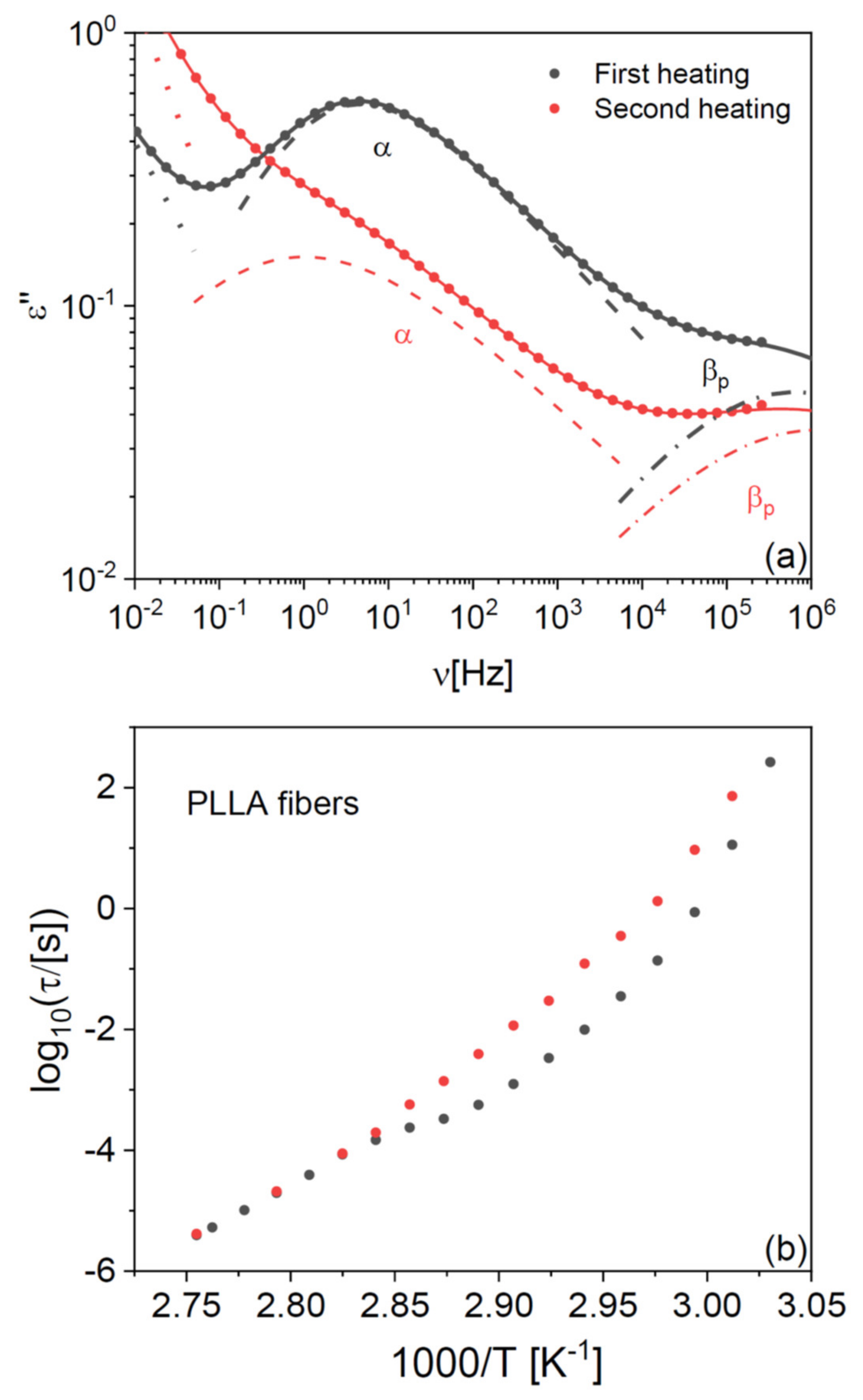
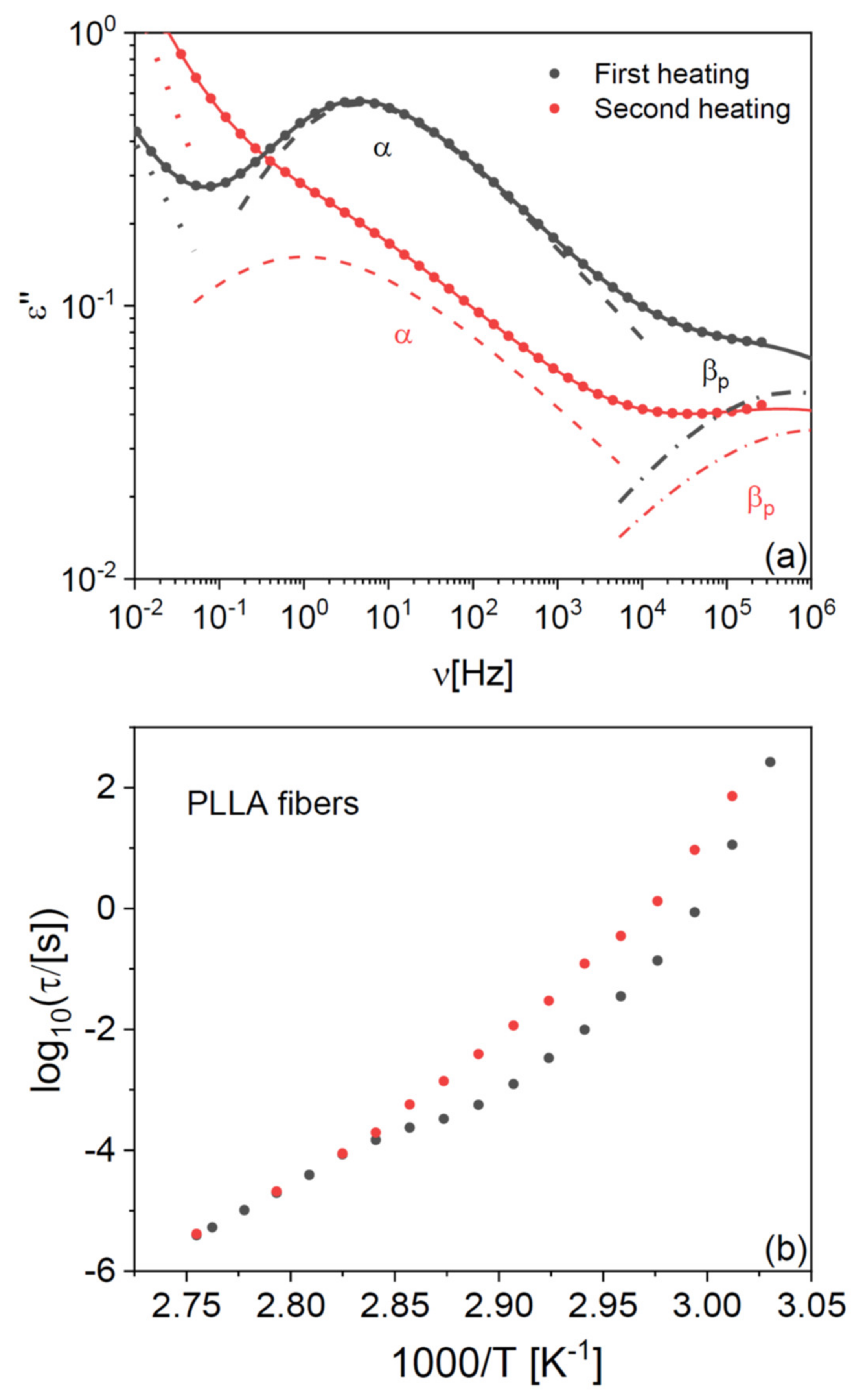
In both PLLA films and fibres, the cold crystallization and melting peaks were almost completely absent in the second-heating thermograms, indicating that PLLA has only weak tendency to crystallize after melting also in microfibre form. To further investigate the impact of the crystalline fraction on the glass transition, we studied the effect of partial (cold) crystallization on the dielectric segmental mobility (structural α relaxation) of the PLLA fibres. For this purpose, two series of isothermal dielectric loss spectra were acquired on the scaffold of pure PLLA fibres while increasing the temperature in a step-like fashion, first on the as-deposited scaffold, and then on the same sample heated up to 373 K (i.e., above the cold crystallization temperature observed in
Figure 1b) and cooled down again to below
Tg. Representative spectra at the temperature of 338 K, just above the
Tg of the pure fibres, are shown in
Figure 2a together with their fit with the imaginary part of Equation (4) (
Section 4). The most intense spectral feature is the α relaxation, which is accompanied by a secondary relaxation at higher frequency (discussed in
Section 2.4). The intensity of the α loss feature is clearly lower in the partially crystallized fibres, as expected from the reduction in the amorphous fraction. At the same time, the relaxation frequency (peak position of the loss feature) is lower, as expected due to the enhanced chain rigidity imposed by the presence of crystallites in semicrystalline polymers [
28].
This effect is more clearly visible in
Figure 2b, which displays the Arrhenius plot (logarithm of the relaxation time vs. inverse absolute temperature) of the structural relaxation time τ
α (see Equation (5) in
Section 4), as determined from the fits of both series of isothermal spectra. The relaxation time was higher by almost a decade after partial crystallization compared to the as-deposited fibre, as visible by the vertical separation of the experimental points in
Figure 2b.
The dielectric characterization allows studying the effect of crystallization on the structural relaxation in real time. In fact, the temperature dependence of τ
α for the as-deposited scaffold exhibited a cross-over in correspondence with the cold crystallization process, which was instead absent in the second temperature ramp on the same sample. Such cross-over in the relaxation time provides direct spectroscopic evidence of the slow-down of the cooperative α relaxation of PLLA upon partial crystallization of the fibre. The same effect has been observed in PLLA films [
29], and as mentioned it is due to the enhanced rigidity of the amorphous fraction in the proximity to crystalline domains, as compared to the fully amorphous polymer [
28].
Partial crystallization of the fibres also affects the dynamic glass transition temperature, defined by convention as the temperature at which the structural relaxation time reaches 100 s (i.e., log
10(τ
α/[s]) = 2). As will be shown in detail below, a fit procedure shows that the
Tg of the partially crystallized microfibres is only 1 K higher than that of as-deposited ones. The small impact of partial crystallization on the
Tg of PLLA fibres is consistent with previous results on PLLA films [
29].
2.2. Morphological and Calorimetric Characterization of Binary Samples
As mentioned in
Section 4, the solvent-cast PLLA films were obtained by annealing at 435 K, i.e., above the melting point of the pure polymer. This was necessary both to remove all the solvent, and to ensure a fully amorphous and homogeneous sample, by cooling the PLLA matrix (or the PLLA-drug mixture) from the molten state [
4]. The lack of any left-over solvents in annealed PLLA films was confirmed by thermogravimetry analysis and IR spectroscopy (not shown). Such annealing step is, however, unsuitable to achieve polymer dispersions of organic molecules that are thermally labile [
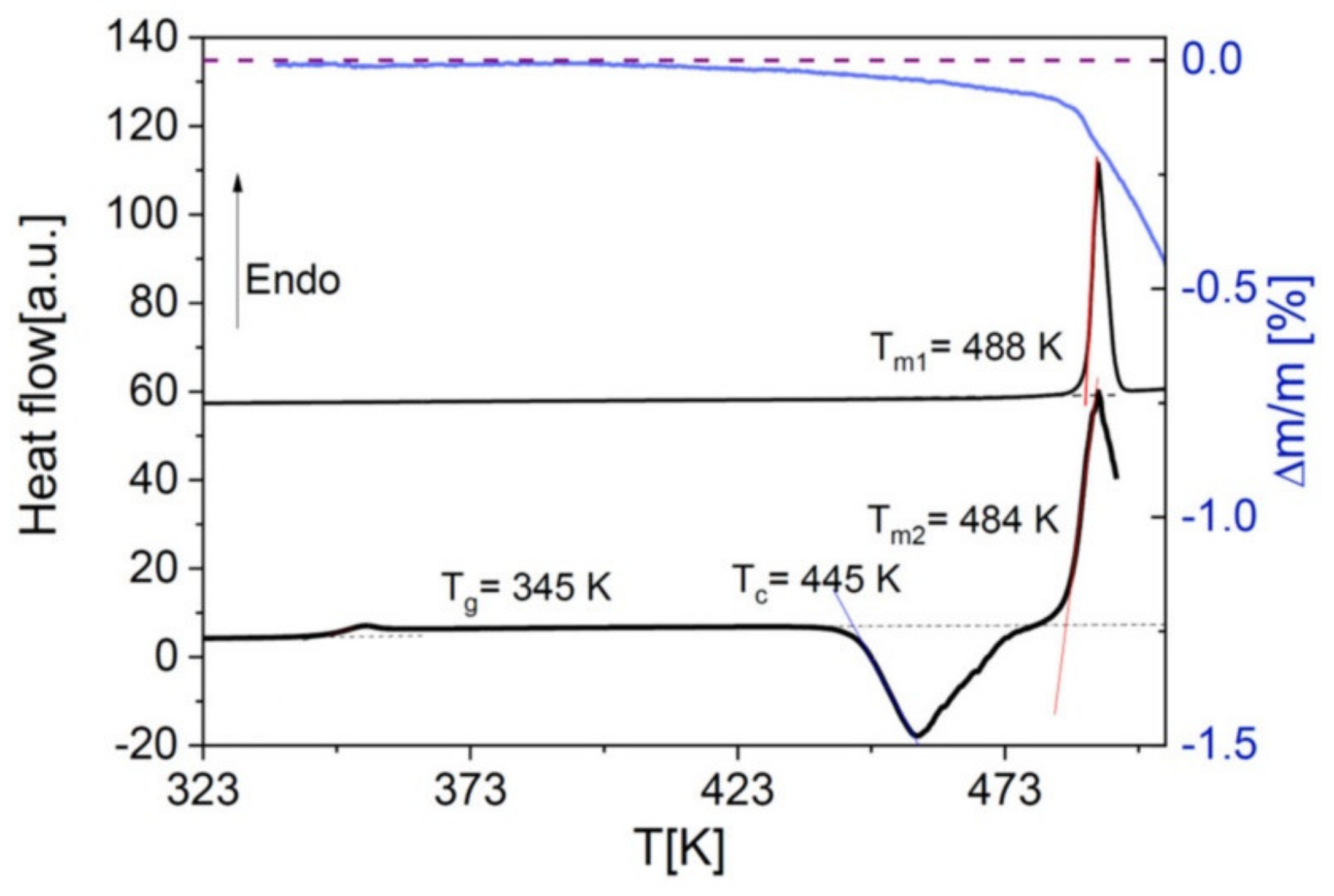
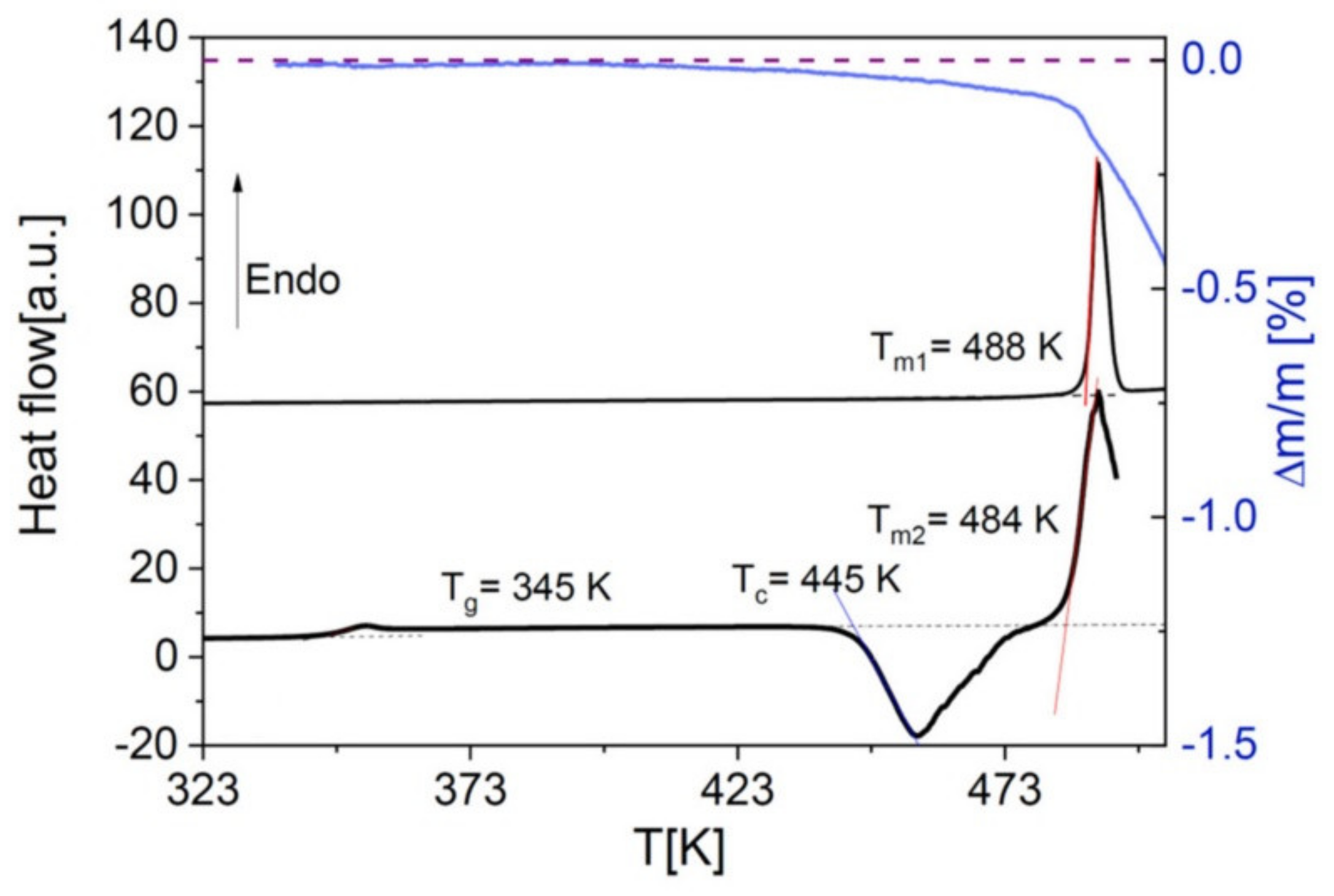
30] such as Nordazepam, which decomposes prior to melting as visible in the thermogravimetry curve of
Figure 3.
For this reason, we employed two different strategies (see
Section 5 for details on sample preparation), namely: (
i) casting fully amorphous films of the racemic PDLLA polymer, which only required heating to 393 K for the full evaporation of the solvent (instead of heating above the melting point of semicrystalline PLLA) [
4]; (
ii) electrospinning, directly from a co-dissolution, NOR-doped PLLA microfibres [
31] with a small-enough diameter to ensure solvent evaporation during the formation of the fibres themselves.
Concerning strategy (
i), because the racemic PDLLA polymer does not exhibit a crystalline form, heating was employed solely to extract the solvent from the bulk matrix [
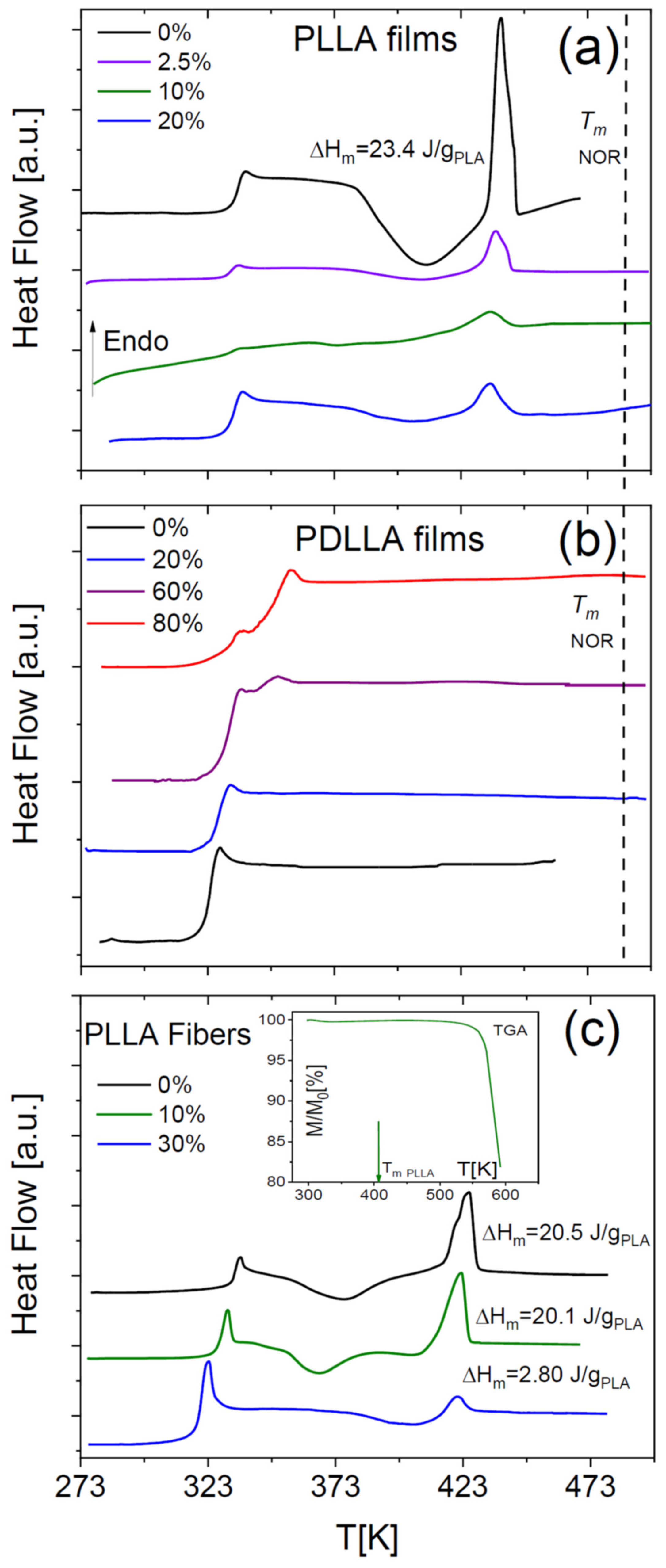
4]. The SEM micrograph of
Figure 4a shows that the film of PDLLA with 20 wt-% NOR is homogeneous, while phase separation is visible in a PDLLA dispersion consisting of 80% NOR in weight (
Figure 4b).
On the other hand, in strategy (
ii) the relatively small diameter of the microfibres (of the order of 600–700 nm, see the bottom panels of
Figure 4) allowed full evaporation of the solvent during the electrospinning process and the subsequent storage under low vacuum conditions at room temperature (see
Section 4), without any need for a subsequent annealing step. The lack of solvents is confirmed by the TGA trace of the scaffold of as-deposited microfibres, where no significant mass loss was detected below the onset of decomposition of the fibres (see the inset to
Figure 1b for the pure PLLA fibres, and inset to
Figure 5c for the fibres loaded with 10 wt-% NOR). Moreover, as it can be observed in the SEM micrographs of the as-spun microfibres (bottom panels of
Figure 4), the fibres appear rather homogeneous, so that macroscopic phase separation can be excluded.
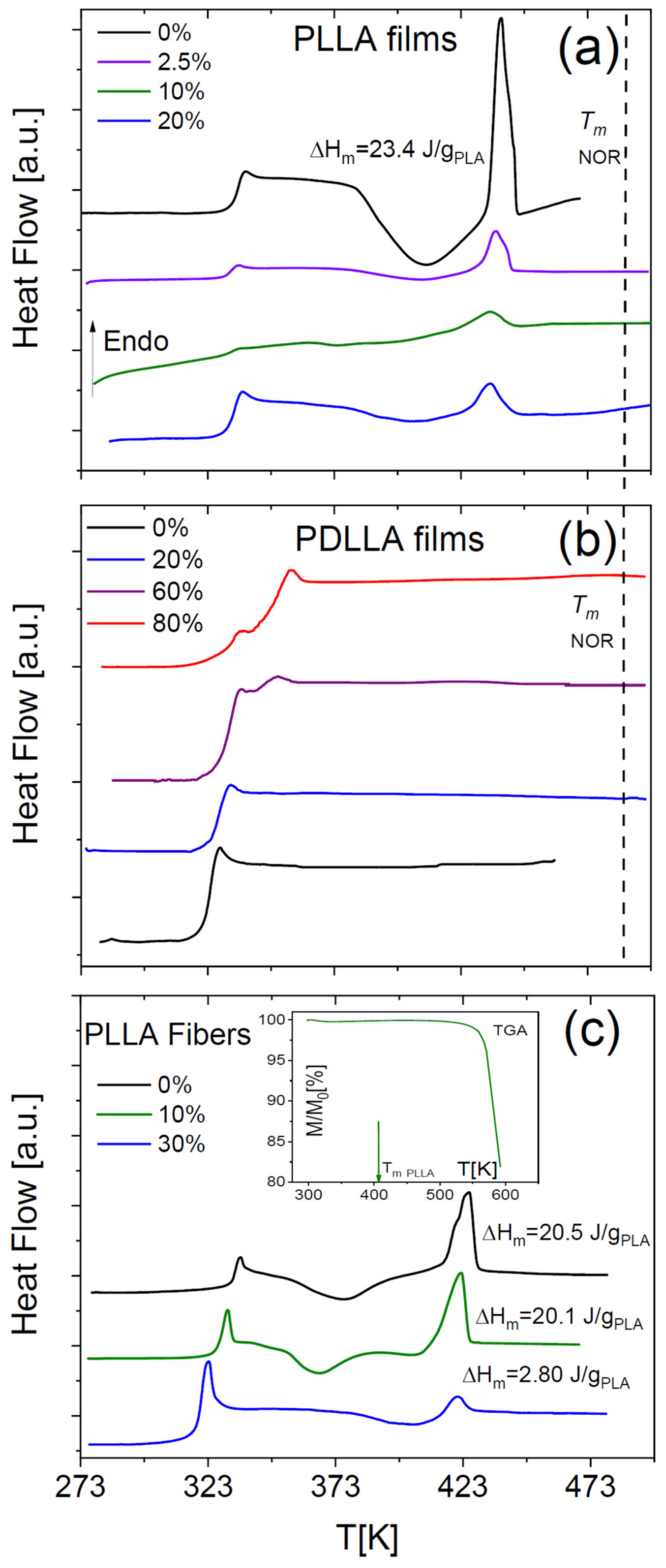
Figure 5a shows the DSC thermograms of NOR-doped PLLA films, acquired after supercooling the samples from 453 K down to their glass state. This temperature was chosen to be slightly higher than the melting onset of pure PLLA in order to ensure that the sample did not contain any PLLA spherulites or lamellae prior to supercooling. As visible in
Figure 5a, the films exhibited a melting endotherm at a temperature similar to that of pure PLLA, preceded by an exothermic process signalling phase separation of the mixtures by crystallization of the polymer component. On the other hand, at higher temperature no melting peak ascribable to the pure drug could be observed (even at the relatively high drug concentration of 40%), which indicates that NOR is fully dissolved in the molten polymer matrix at these temperatures. In other words, even samples that are phase-separated at room temperature actually are homogeneous (viscoelastic) liquids at higher
T. This validates our procedure for obtaining homogeneous dispersions in polylactide.
The DSC traces of binary films of NOR in racemic PDLLA are displayed in
Figure 5b. These films exhibited a single
Tg value at a low enough drug concentration (at least up to 20%), indicative of the formation of homogeneous amorphous solid dispersions (see also
Figure 4a). On the contrary, films at higher drug loading were characterized by amorphous phase separation, with the occurrence of two distinct glass transitions (compare with the SEM micrograph in
Figure 4b).
Our DSC and SEM characterization shows that both enantiomeric forms of the polymer lead to amorphous, homogeneous binary dispersions at low enough drug concentration. This result is analogous to that obtained with another small-molecule drug (the chloramphenicol antibiotic) dispersed in the same two polylactide polymers [
4].
The DSC traces of the electrospun NOR-PLLA microfibres are shown in
Figure 5c. A single
Tg feature was observed at least up to a drug weight fraction of 30%, followed by a cold crystallization exotherm and corresponding melting endotherm, indicating partial crystallization of the PLLA fraction in the microfibres upon heating. In the case of 30% drug weight fraction, the melting enthalpy was considerably smaller than that of films or that of the microfibre with 10% drug weight fraction. This indicates that the crystalline fraction of the 30% fibre, even after cold crystallization, was significantly lower (<2%) than that of pure semicrystalline PLLA (21%) and shows that the crystallization of PLA was clearly hindered by the presence of drug molecules. No melting transition of the drug component was observed in any NOR-loaded PDLLA sample, indicating again that at high temperature NOR is dissolved in the molten polymer matrix. These results indicate that the drug and polylactide are highly miscible, at least at high temperatures.
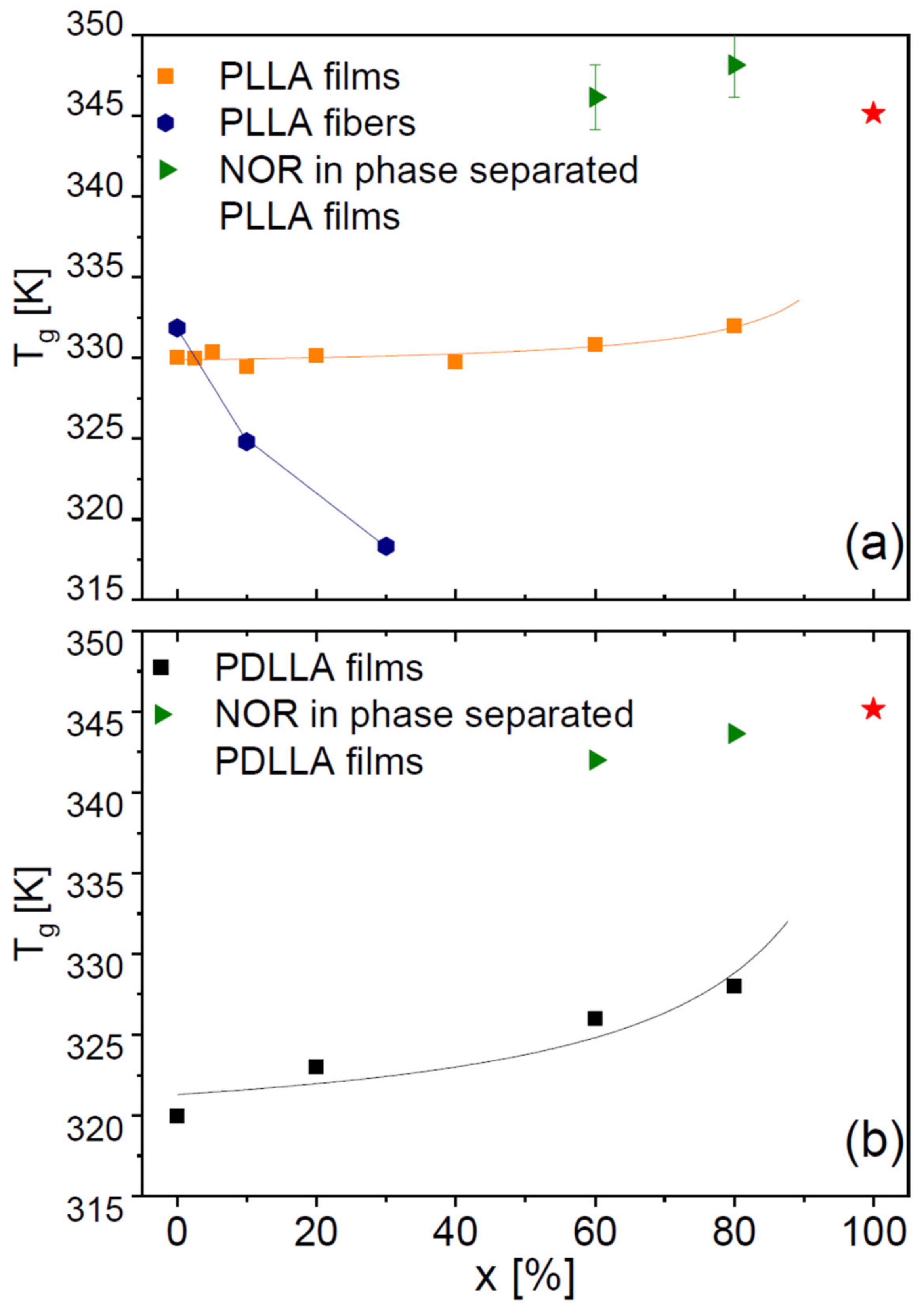
Figure 6 displays the onset glass transition temperature of all dispersions obtained with PLLA (both films and microfibres, panel a) and with PDLLA (panel b). In the case of the PLLA films, NOR has a weak antiplasticizing effect, the
Tg of the binary films being slightly higher than that of the pure polymer (
Figure 6a). The antiplasticizing effect of the drug is more pronounced in the PDLLA films (b). This is qualitatively in agreement with the expected behaviour of a binary mixture based, e.g., on the Gordon–Taylor equation [
2], because the
Tg of our home-synthesized PDLLA polymer is lower and thus further from that of NOR compared with PLLA: the greater the difference with the
Tg of the organic molecule, the more visible the antiplasticizing effect for a given loading. In the PDLLA films with high NOR loading, which display amorphous phase-separation characterized by two distinct
Tg values, the
Tg of the component richer in Nordazepam is lower than that of the pure drug, again expected if the polymer acts, due to its lower
Tg, as an antiplasticizer for the drug.
On the contrary, in the PLLA microfibres (
Figure 6a, blue circles)
Tg is observed to
decrease with increasing drug content, as can be gathered directly also from visual inspection of
Figure 5c. This cannot be an effect of the solvent of the electrospun dissolution, not only due to the systematic dependence on drug concentration, but also because no mass loss is observed in TGA experiments (see the insets to
Figure 1b and
Figure 5c), indicating that no solvent is present (as also confirmed by IR, not shown). Similarly, it cannot be due to the crystalline fraction of the as-deposited microfibres, because the presence of a crystalline fraction increases the
Tg compared to the fully amorphous polymer (by 1 K in the case of PLLA fibres, see
Section 2.3) [
28], while the observed
Tg of the microfibre dispersions is actually lower than that of fully amorphous PLLA samples. In fact, as visible in
Figure 6a, the
Tg of PLLA microfibres with 30% drug loading is lower by 14 K with respect to the pure fibres, and lower by more than 10 K with respect to that of the fully amorphous PLLA films (both with and without drug) obtained by cooling from above the melting point of the polymer.
Thus, we find that Nordazepam actually has a genuine plasticizing effect on PLLA microfibres, while it has an antiplasticizing effect on the polylactide films. Such dependence upon the morphology of the sample is striking, and might reflect the peculiar chain morphology in the microfibres, where the PLLA chains are at least partially oriented in a parallel fashion (in the direction of the fibre axis) rather than randomly coiled as in the films [
31]. We have acquired IR spectra of all samples (not shown) and did not find any significant difference in vibrational frequency of the IR bands between films and fibres, which indicates that drug-polymer interactions are similar in both morphologies. We suggest therefore that the antiplasticizing effect of the drug on the microfibres (and thus the breakdown of the Gordon–Taylor prediction) could be due to the structural changes induced by the drug molecules, which disrupt the quasi-parallel ordering of the macromolecular chains in the fibre morphology.
Concerning the antiplasticizing effect of the drug on the films, we point out that increasing the polymer’s
Tg by adding a genuine antiplasticizing agent with higher
Tg, may be an interesting strategy to metastabilize amorphous or semicrystalline polymer dispersions in which demixing occurs by cold crystallization of the polymer component. In fact, the antiplasticizer effect of the drug entails that the segmental mobility of the polymer (see
Section 2.3) is slower in the presence of the drug, and therefore that the nucleation and growth rates [
32] of lamellae/spherulites are similarly slower. If phase-separation of the dispersions occurs by cold crystallization of the polymer component, as for example is the case of the chloramphenicol antibiotic dispersed in polylactide [
4] but also of our dispersions (
Figure 5a), an antiplasticizing effect of the drug would thus increase the kinetic stability of the amorphous mixture not only thanks to the entropy of mixing, but also by slowing down both the fluctuations leading to nucleation and the growth rate (which for temperatures just above the glass transition temperature is correlated with the structural (α) relaxation frequency [
32]). It should also be noted, moreover, that a polymer in the (supercooled) viscoelastic liquid state might offer the advantage of a higher solubility of the molecular drug, as seems to be the case for the binary Nordazepam-PLA system.
2.3. Dielectric Spectroscopy of Binary Samples: Structural Relaxation
To fully explore the impact of the molecular filler, we carried out temperature-dependent dielectric spectroscopy experiments on binary samples. At low drug content, all samples displayed a single structural (α) relaxation, as expected for a homogenous amorphous phase.
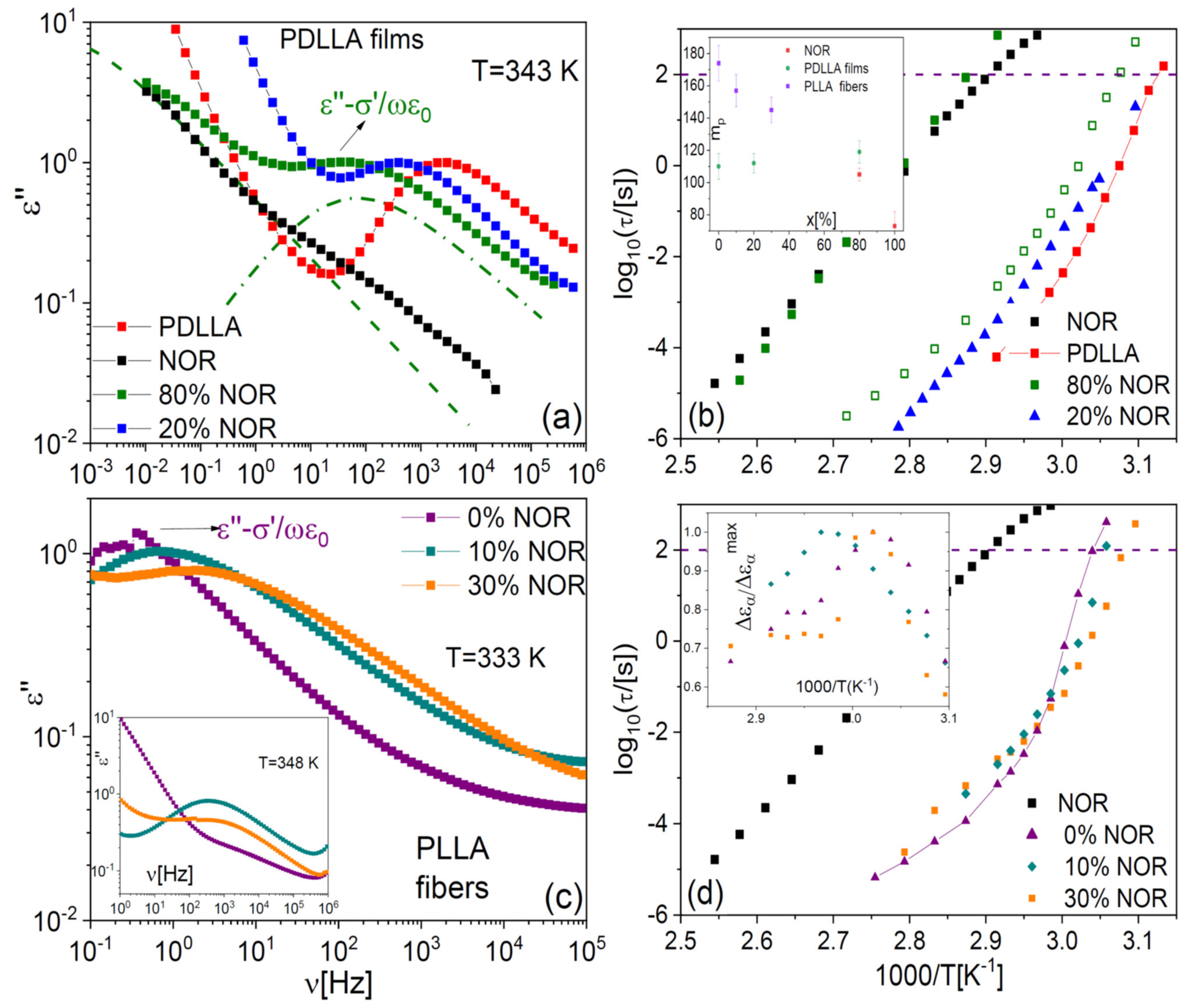
Figure 7a displays the loss spectra of PDLLA films at different drug loading, at the same fixed temperature of 343 K. The vertical scale of each loss spectrum was normalized to the height of the dielectric loss feature that corresponds to the α relaxation, peaked in the kHz frequency range at this temperature. The structural relaxation is observed to shift to lower frequency with increasing drug content. Such a shift implies that the α relaxation of the PDLLA chains in the fibres slows down as a result of polymer-molecule interactions. The effect is consistent with the observed increase in the
Tg of the films with increasing NOR content and confirms that the small-molecule drug acts as a genuine antiplasticizer for the polylactide films.
All dielectric loss spectra were fitted with model functions (see
Section 4) to extract quantitative information on the characteristic time of relaxation processes. The Arrhenius plot of the structural relaxation time of polylactide films is displayed in
Figure 7b. The genuine antiplasticizer effect of the drug on the polylactide films is again evident in the observed shift of the dynamic
Tg. It is worthwhile to note, however, that the antiplasticizing effect of the drug can be visualized more clearly by comparing the dielectric relaxation times in the Arrhenius plot, and that it is a consistent effect at all temperatures above
Tg. This is a clear advantage of dielectric spectroscopy compared to the DSC study of
Tg values at a single fixed temperature-scanning rate. This advantage, together with the direct visualization of the impact of the drug on the structural dynamics offered by the loss spectra, shows the importance of performing dynamic studies to characterise the (anti)plasticizer effect of a polymer filler.
As visible in
Figure 7a, the spectrum of the sample containing 80% in weight of NOR exhibited two main dielectric losses, of which the one at lower frequency matches the spectral position of the structural relaxation of pure NOR, also shown for comparison purposes. The latter spectrum was normalized so that the loss maximum of the α relaxation of pure NOR had the same intensity as the low-frequency feature of the 80% sample. The presence of two primary relaxations agrees with the observation of two distinct glass transitions in this phase-separated sample. The observed relaxation times indicate that one of the phases consisted of drug-loaded polymer domains, and the other of almost pure amorphous NOR. The loss feature of the sample with 20% drug had a dielectric strength (intensity) comparable to that of pure PDLLA, while the intensity of the lower-frequency feature in the sample with 80% drug was directly comparable to that of pure amorphous NOR, as it may be expected (not shown).
The spectra of the PLLA microfibres at the temperature of 333 K (
Figure 7c) exhibited, compared with the films, an opposite shift of the segmental relaxation with increasing NOR content. The observed shift of the α relaxation toward higher frequency in the microfibre with NOR is the expected effect of a polymer plasticizer, in agreement with the DSC results presented in
Section 2.2. It should be noted, however, that at higher temperature the α relaxation shifted to lower frequency with increasing NOR content, as visible in the isothermal spectra at 348 K displayed in the inset to
Figure 7c. In other words, in the microfibres at high
T, the drug displays again its “normal” antiplasticizer effect. Both effects are also visible in the Arrhenius plot of the structural relaxation time of the as-deposited fibres, shown in
Figure 7d, where it may be observed that the sign of the shift changes from that of a plasticizer to that of a (genuine) antiplasticizer at the temperature of ca. 335 K.
If the plasticizing effect of the drug on PLLA microfibres is the result of the local structural modifications (disorder) induced in roughly parallel macromolecular chains, as we suggest, then the cross-over to the expected antiplasticizer effect may be rationalized considering that the activation of the segmental motion entails a dynamic disorder that makes irrelevant the (static) disorder induced by the drug molecules in the glassy state of the polymer.
As shown in the inset to
Figure 7d, the dielectric strength of the samples displayed a maximum in correspondence with the cross-over temperature of 335 K. Different aspects concur to determine the dielectric strength of the cooperative relaxation, especially in polymers [
5]; it is significant that the change in the temperature dependence of the dielectric intensity occurs precisely at the same temperature as the crossover between the opposite plasticizing behaviours.
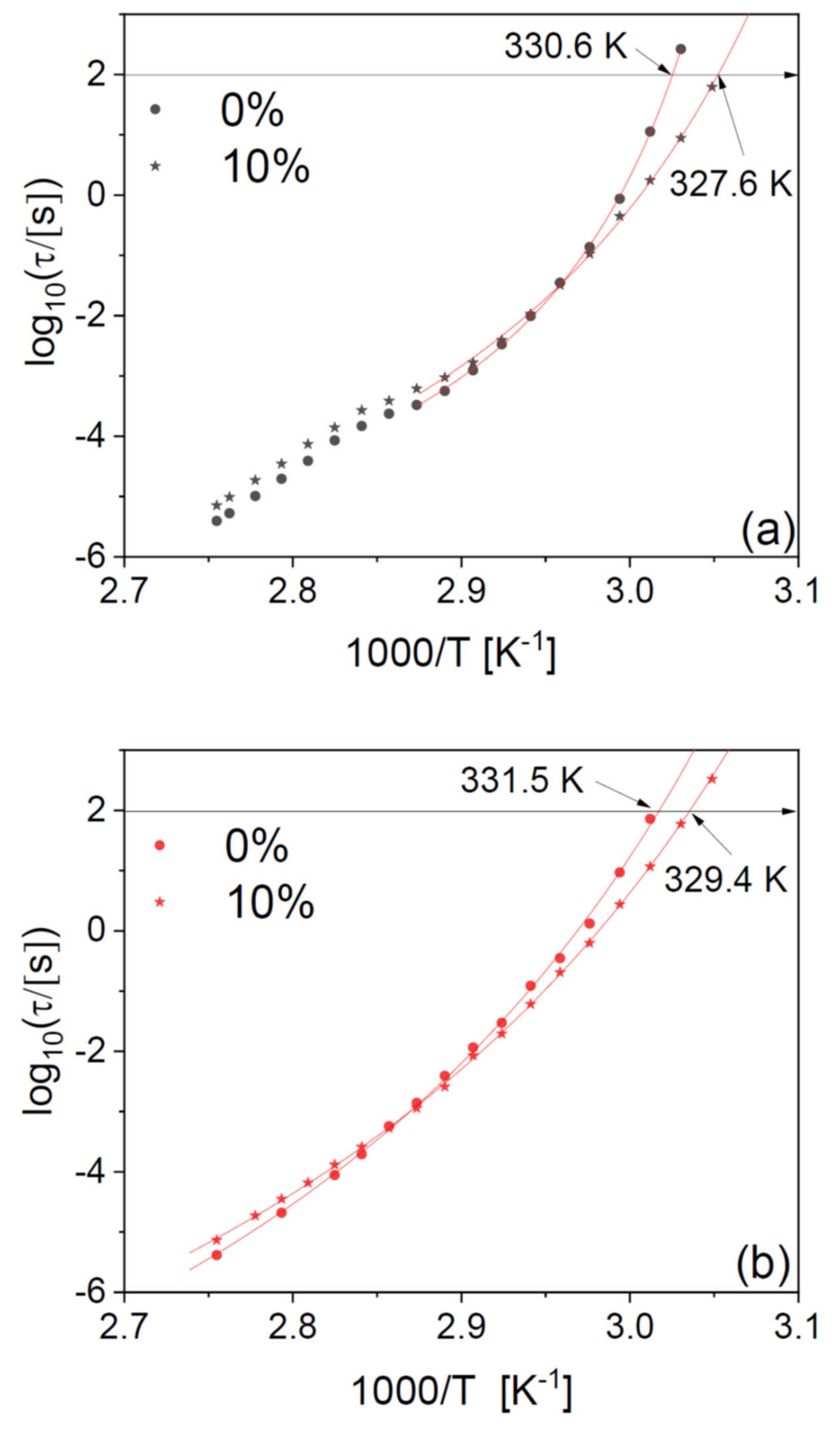
It should be remembered that the PLLA microfibres undergo at least partial crystallization upon heating (see
Figure 5c), and this is expected to have an impact on the relaxation time. Therefore, to investigate further the effect of the drug filler, we compare in panels (a) and (b) of
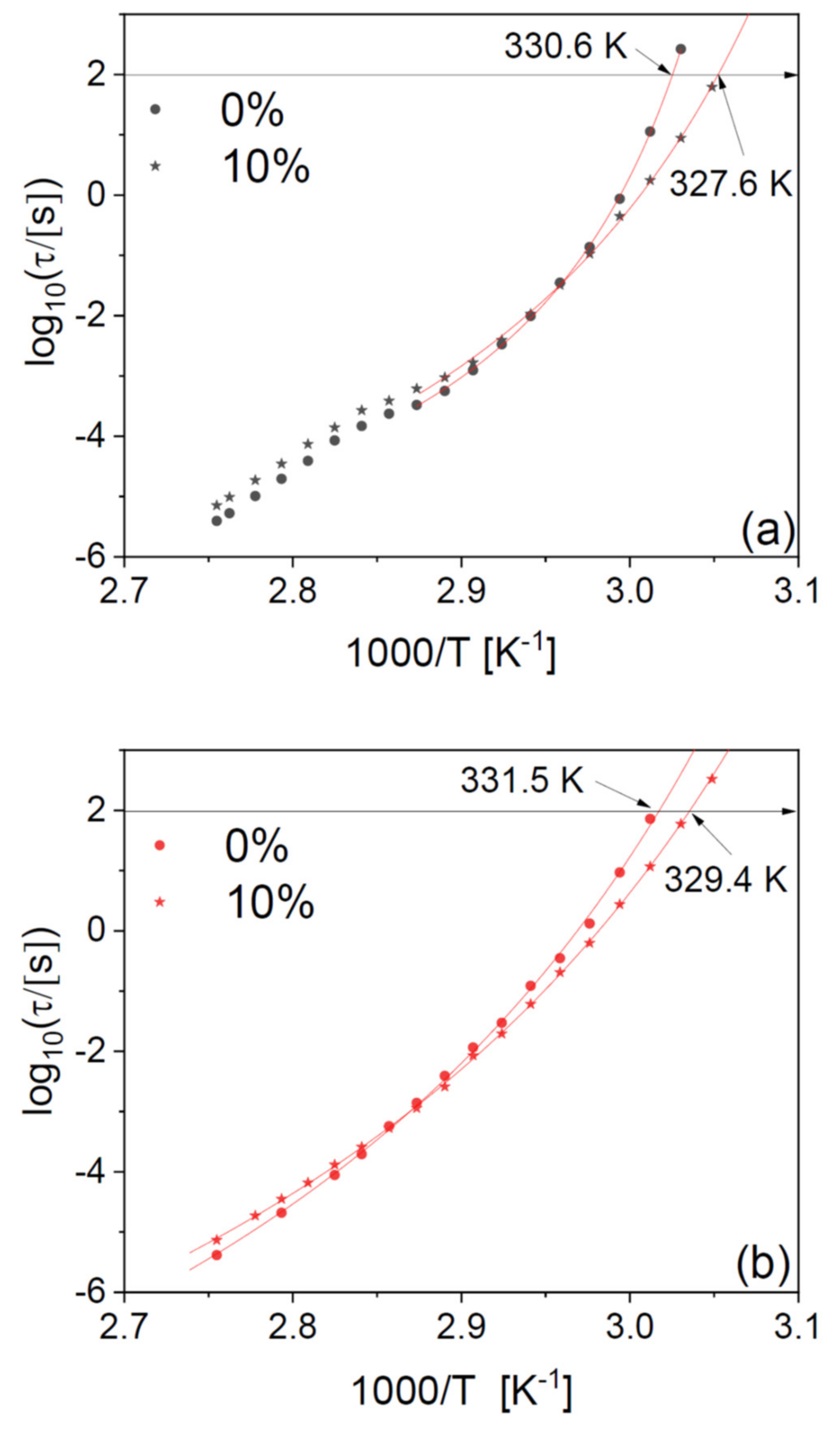
Figure 8 the structural relaxation times of a PLLA microfibre scaffold at 10-wt% drug loading and those of a pure PLLA fibre, for two series of isothermal spectra acquired while increasing the temperature to 363 K, first on the as-deposited scaffolds (a), and then right afterwards, after cooling again to below
Tg (b). The second series of spectra displays slightly higher dynamic
Tg (by 1–2 K) with respect to the as-deposited scaffold, due to the effect of partial crystallization already discussed for the pure polymer fibres.
It can be observed that the plasticizing effect near Tg is present in both series of spectra, as is the cross-over between plasticizing and antiplasticizing behaviour at higher temperature. In particular, the plasticizer effect of the drug on the dynamic Tg is consistent (3 K) between the first and the second series of spectra, that is, it is maintained also after partial crystallization of the fibres.
In agreement with our findings on the pure fibres in
Figure 2, the relaxation time of the drug-loaded fibres has a smooth temperature dependence in the second heat-up series (
Figure 8b), while it clearly displays a discontinuity near the cold-crystallization temperature in the first one (
Figure 8a). To discuss this discontinuity in more detail, we have fitted the Arrhenius plot of the observed relaxation times with continuous functions. The temperature dependence of the structural relaxation time in molecular and macromolecular glass formers is typically described by the Vogel–Fulcher–Tammann (VFT) function, which is given by [
33,
34,
35]:
Here, τ
∞ is the segmental time extrapolated at a very high temperature,
D is the so-called fragility strength coefficient, related with the kinetic fragility defined later on, and
TVF is the Vogel–Fulcher temperature. The VFT Equation (1) is mathematically equivalent to the Williams–Landell–Ferry equation. The temperature dependence of the relaxation time of the second series of spectra acquired on NOR-loaded microfibres (
Figure 8b) could be well modelled with a single VFT function. In contrast, the as-deposited scaffold (
Figure 8a) displayed two different temperature dependences above and below the cold crystallization temperature, due to the dynamic cross-over caused by the presence of crystalline domains (as already discussed in reference to
Figure 2) [
29].
It is interesting to analyse the so-called kinetic fragility index of the samples, which as mentioned in the introduction is a measure of how fast the relaxation time varies with temperature in the proximity of the glass transition. The kinetic fragility is defined as [
23]:
A high value of kinetic fragility implies that the molecular mobility changes fast when the temperature varies approaching
Tg; in general, this entails that a polymer dispersion with higher fragility is more sensitive to temperature fluctuations and therefore potentially more prone to crystallization. The fragility index, displayed in the inset to
Figure 7b, is found to increase with the drug loading in the films (namely, from 110 to 119), despite the fact that the fragility of the Nordazepam glass former is actually lower than that of the amorphous polymer. On the contrary, the fragility decreases (from 174 to 145) with drug loading in the electrospun microfibres. In other words, the kinetic fragility exhibits the same behaviour with drug concentration as the glass transition temperature, and in particular, it has opposite dependence in the two studied sample morphologies. It is also interesting to note that the kinetic fragility of the pure PLLA microfibres is larger than that of the pure PLLA films, which may be due to the different chain morphology.
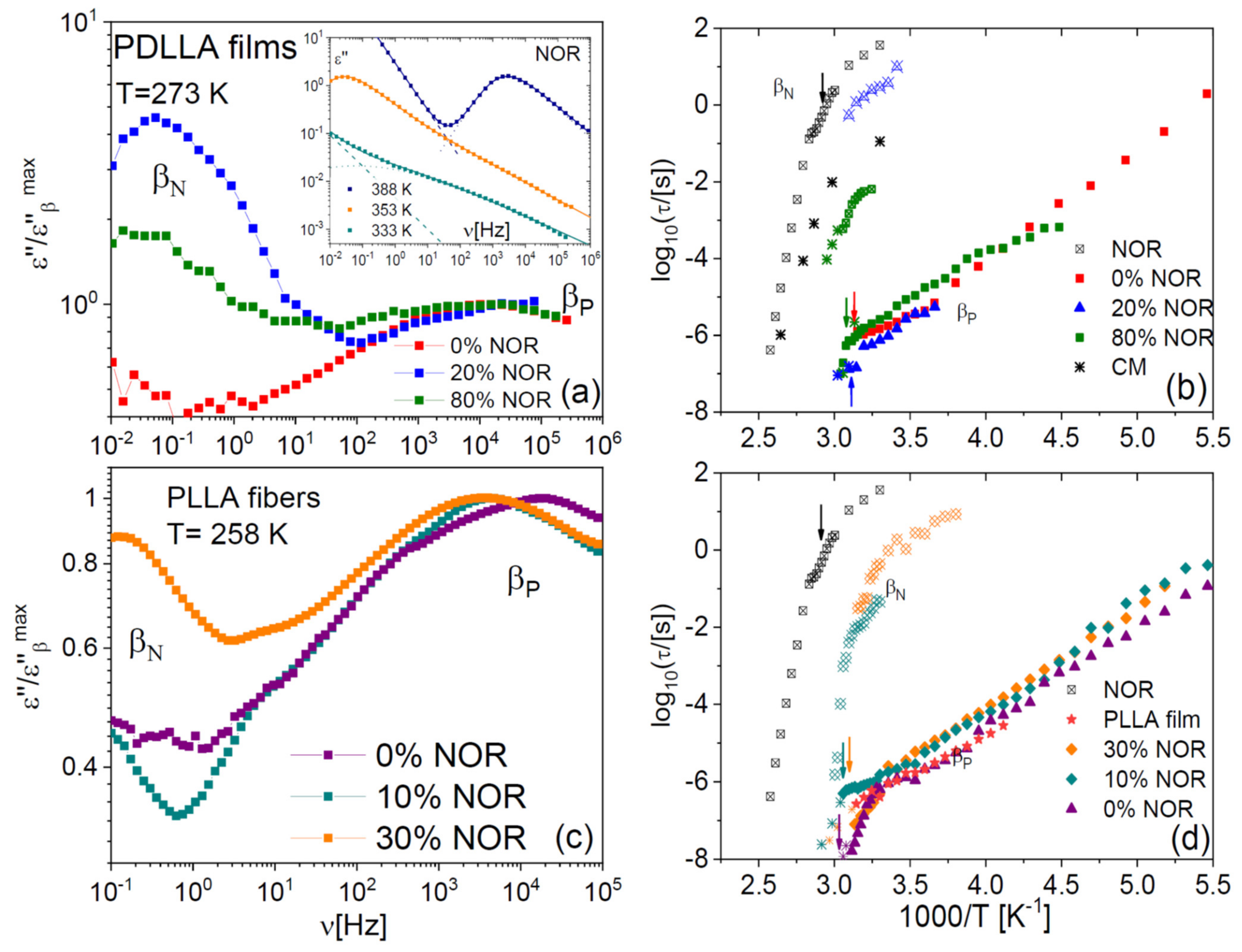
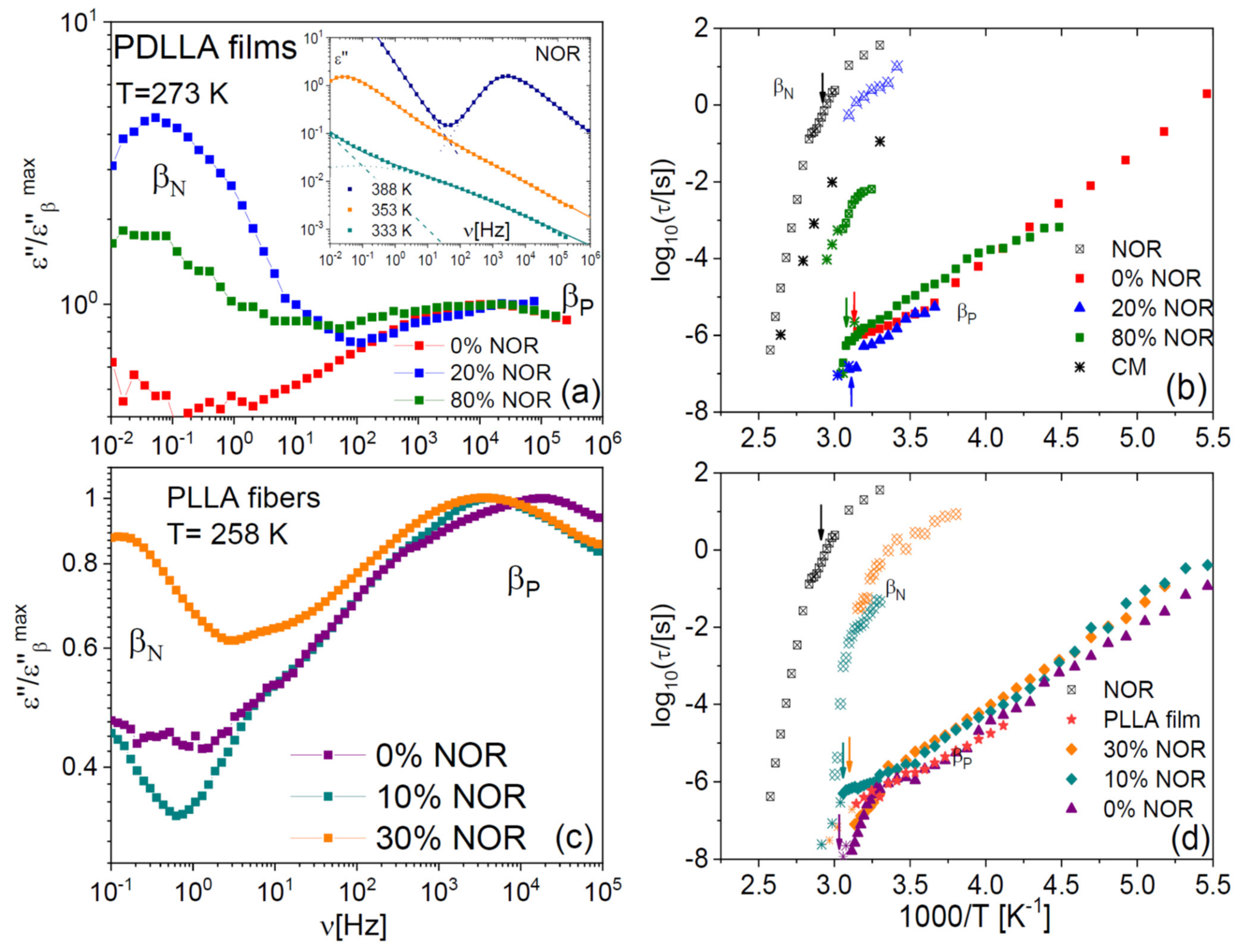
2.4. Johari–Goldstein Relaxations of Binary Samples
The studied binary samples also displayed secondary relaxations at lower temperature/higher frequency. The inset to
Figure 9a displays representative low-temperature spectra of pure NOR. As we reported already in an earlier study [
17], a secondary relaxation, labelled here as β
N, is observed on the high-frequency flank of the α relaxation. Like the α process, the β
N relaxation shifted to higher frequency with increasing temperature. As visible in
Figure 8a,c, a similar relaxation is observed also in the dispersions in polylactide, both in film (a) and microfibre (b) form. The Arrhenius plot of the β
N relaxation is displayed in panels (b) and (d) for both types of samples. The β
N process is significantly affected by the glass transition and by the composition of the dispersion. In particular, the β
N relaxation becomes faster with decreasing drug content, and its relaxation time displays a dynamic cross-over at the glass transition temperatures of the pure drug or of the mixture, respectively, which are indicated by vertical arrows in the right-hand side panels of
Figure 8. In the case of the pure drug, the activation energy of the β
N relaxation changes from 3.5 eV above
Tg to 0.8 eV below. Similar values are found in the binary samples.
As we discussed in a recent publication [
17], in the pure drug, this relaxation corresponds to a non-diffusive relaxation of the whole molecule known as Johari–Goldstein (JG) β relaxation [
36,
37]. These secondary relaxations are observed in fully rigid molecules [
38,
39,
40] and in polymers with monoatomic or methyl side groups such as polybutadiene and 1,4-polyisoprene [
41,
42] and correspond to rigid roto-translations of whole molecules or chain segments. The similar dependence of the β
N and α relaxation times with the chemical composition of the dispersions, and the crossover detected in the β
N relaxation times at
Tg, confirm the JG nature of this relaxation also in the dispersions. By contrast, local inter-conformer conversions are generally observed at a fixed frequency, regardless of the local environment [
5]. In particular, the NOR molecule has two internal degrees of freedom, that correspond respectively to the ring-inversion dynamics of the diazepine moiety and the rotation of the benzyl ring around the σ-bond connecting it with the diazepine ring [
17]. Both motions have a very weak dielectric intensity already in the pure compound, and they moreover occur at a much higher frequency than the β
N process [
17]. These processes could not be detected in our samples due to the low drug content.
It is interesting to note that, in both the films and microfibres, the βN relaxation times change in the same direction with increasing drug loading, namely, the βN process becomes faster with decreasing drug content. This entails that the dispersion of the drug in the polymer matrix (and thus the dilution of inter-drug interactions) always lead to the same (plasticizing) effect on the non-diffusive JG relaxation of the drug, regardless of the sample morphology and thus of the impact of the drug on the segmental mobility.
The pure polylactide polymer also displays a secondary relaxation, which we label as β
P in
Figure 9, and which a previous study has ascribed to local chain-twisting motions of angular amplitude of the order of 11º [
43]. The β
P process is visible in roughly the same spectral range in all samples (both films and microfibres). It can be observed in
Figure 9b,d that also this relaxation exhibits a dynamic cross-over near
Tg, as it is especially visible in the case of the microfibres. By comparing the relaxation times predicted by the Coupling Model [
44,
45] with the β
P relaxation times, we find that they are very similar, except in the pure polymer case. Both these features were encountered also in dispersions in PLLA and PDLLA of another drug (chloramphenicol) [
4].
We thus confirm the conclusion of our previous studies [
4,
30], namely, that the β
P twisting mode of the polymer is the local, non-cooperative version of the segmental α relaxation of the polymer. This assignment is consistent with the idea that the JG relaxations correspond to small-angle single-particle rigid rotations taking place under strong spatial restrictions [
46,
47,
48,
49]. It thus appears that homogeneous “asymmetric” dispersions composed of relatively rigid macromolecular chains and small organic molecules and characterized by a single glass transition and primary relaxation, display not only one but actually
two separate JG-like relaxations, one for each component.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}