
Inside out Approach to Rotator State in Hydrogen-Bonded System—Experimental and Theoretical Cross-Examination in n-Octanol

 , , , and
, , , and
Abstract
1. Introduction
2. Experimental and Theoretical Methods
2.1. Experimental Procedures: Infrared Spectroscopy (IR) and the Nonlinear Dielectric Effect (NDE)
2.2. Computational Methodology
2.2.1. Classical Molecular Dynamics (MD)
2.2.2. Car–Parrinello Molecular Dynamics (CPMD) in the Crystalline Phase
2.2.3. Path Integral Molecular Dynamics in the Crystalline Phase
2.2.4. Post-Processing of the Car–Parrinello and Path Integral MD
3. Results and Discussion
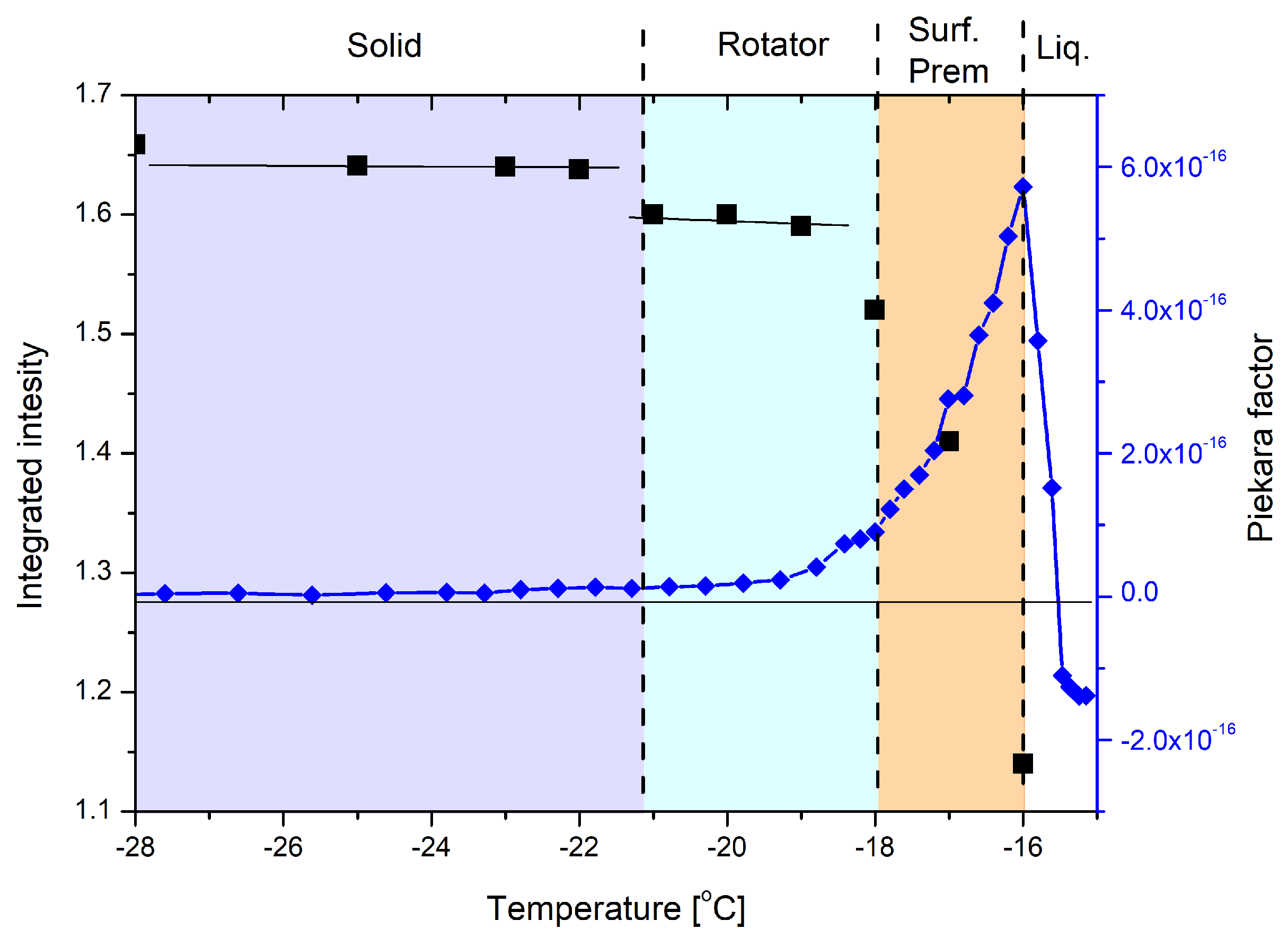
3.1. Experimental Data Analysis
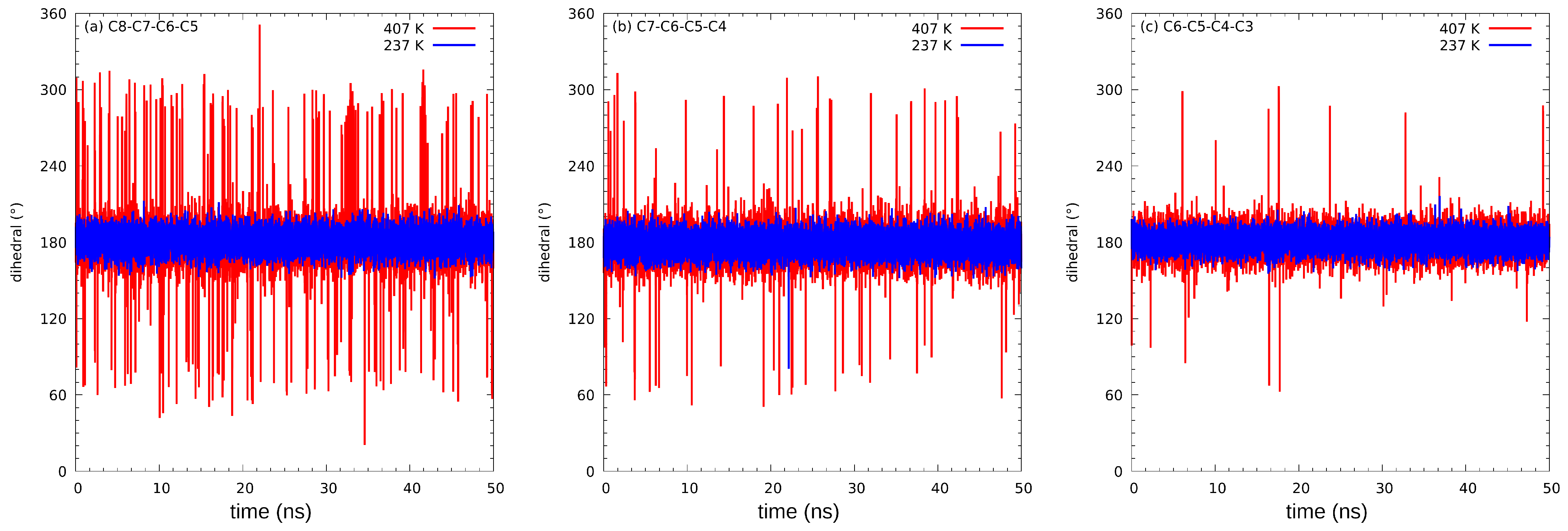
3.2. Classical Molecular Dynamics (MD) Results
3.3. Car–Parrinello Molecular Dynamics (CPMD)
3.4. Path Integral Molecular Dynamics (PIMD)
3.5. Cross-Examination of Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HB | hydrogen bond |
| IR | infrared spectroscopy |
| FTIR | Fourier transform infrared spectroscopy |
| NDE | nonlinear dielectric effect |
| MD | molecular dynamics |
| CPMD | Car–Parrinello molecular dynamics |
| PIMD | path integral molecular dynamics |
| CCDC | Cambridge Crystallographic Data Centre |
| RMSD | root mean square deviation |
| RDF | radial distribution function |
| PBC | periodic boundary conditions |
| DFT | density functional theory |
| pmf | potential of mean force |
References
- Faraday, M. Lecture given at the Royal Institution, London, June 7, 1850. Athenaeum 1850, 1181, 640–641. [Google Scholar]
- Faraday, M., XXIV. On regelation, and on the conservation of force. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1859, 17, 162–169. [Google Scholar] [CrossRef]
- Mukerji, I.; Williams, A.P. UV Resonance Raman and Circular Dichroism Studies of a DNA Duplex Containing an A3T3 Tract: Evidence for a Premelting Transition and Three-Centered H-Bonds. Biochemistry 2001, 41, 69–77. [Google Scholar] [CrossRef][Green Version]
- Li, Y.; Somorjai, G.A. Surface Premelting of Ice. J. Phys. Chem. C 2007, 111, 9631–9637. [Google Scholar] [CrossRef]
- Björneholm, O.; Hansen, M.H.; Hodgson, A.; Liu, L.M.; Limmer, D.T.; Michaelides, A.; Pedevilla, P.; Rossmeisl, J.; Shen, H.; Tocci, G.; et al. Water at Interfaces. Chem. Rev. 2016, 116, 7698–7726. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.N.; Berger, R.; Butt, H.J. Surface Premelting and Interfacial Interactions of Semi-Clathrate Hydrate. J. Phys. Chem. C 2019, 123, 24080–24086. [Google Scholar] [CrossRef]
- Slater, B.; Michaelides, A. Surface premelting of water ice. Nat. Rev. Chem. 2019, 3, 172–188. [Google Scholar] [CrossRef]
- Niblett, S.P.; Limmer, D.T. Ion Dissociation Dynamics in an Aqueous Premelting Layer. J. Phys. Chem. B 2021, 125, 2174–2181. [Google Scholar] [CrossRef]
- Wilson, P.; Arthur, J.; Haymet, A. Ice Premelting during Differential Scanning Calorimetry. Biophys. J. 1999, 77, 2850–2855. [Google Scholar] [CrossRef]
- Dosch, H.; Lied, A.; Bilgram, J. Glancing-angle X-ray scattering studies of the premelting of ice surfaces. Surf. Sci. 1995, 327, 145–164. [Google Scholar] [CrossRef]
- Bluhm, H.; Ogletree, D.F.; Fadley, C.S.; Hussain, Z.; Salmeron, M. The premelting of ice studied with photoelectron spectroscopy. J. Phys. Condens. Matter. 2002, 14, L227–L233. [Google Scholar] [CrossRef]
- Mizuno, Y.; Hanafusa, N. Studies of surface properties of ice using nuclear magnetic resonance. J. Phys. Colloq. 1987, 48, C1-511–C1-517. [Google Scholar] [CrossRef]
- Zerbi, G.; Conti, G.; Minoni, G.; Pison, S.; Bigotto, A. Premelting phenomena in fatty acids: An infrared and Raman study. J. Phys. Chem. 1987, 91, 2386–2393. [Google Scholar] [CrossRef]
- Hnatiuk, K.I.; Alekseev, A.N.; Alekseev, S.A.; Grabovsky, Y.E.; Yablochkova, K.S.; Lazarenko, M.V.; Lazarenko, M.M. Investigation of the structure and mechanisms of thermal motion in nanostructured undecylenic acid. Mol. Cryst. Liq. Cryst. 2020, 701, 16–27. [Google Scholar] [CrossRef]
- VanHook, A.; Silver, L. Premelting Anomalies of Some Long Chain Normal Paraffin Hydrocarbons. J. Chem. Phys. 1942, 10, 686–690. [Google Scholar] [CrossRef]
- Zerbi, G.; Magni, R.; Gussoni, M.; Moritz, K.H.; Bigotto, A.; Dirlikov, S. Molecular mechanics for phase transition and melting of n-alkanes: A spectroscopic study of molecular mobility of solid n-nonadecane. J. Chem. Phys. 1981, 75, 3175–3194. [Google Scholar] [CrossRef]
- Pocheć, M.; Orzechowski, K.; Rutkowski, K. Indicators of premelting in 1-decanol and 1-nonanol studied by FTIR spectroscopy. Surf. Interfaces 2022, 28, 101676. [Google Scholar] [CrossRef]
- Ozaki, Y.; Liu, Y.; Noda, I. Two-Dimensional Near-Infrared Correlation Spectroscopy Study of Premelting Behavior of Nylon 12. Macromolecules 1997, 30, 2391–2399. [Google Scholar] [CrossRef]
- Wang, J.; El-Sayed, M.A. Temperature Jump-Induced Secondary Structural Change of the Membrane Protein Bacteriorhodopsin in the Premelting Temperature Region: A Nanosecond Time-Resolved Fourier Transform Infrared Study. Biophys. J. 1999, 76, 2777–2783. [Google Scholar] [CrossRef]
- Sadtchenko, V.; Ewing, G.E. A new approach to the study of interfacial melting of ice: Infrared spectroscopy. Can. J. Phys. 2003, 81, 333–341. [Google Scholar] [CrossRef]
- Shallard-Brown, H.A.; Watkin, D.J.; Cowley, A.R. n-Octanol. Acta Crystallogr. E 2005, 61, o213–o214. [Google Scholar] [CrossRef]
- Jeffrey, G.A. An Introduction to Hydrogen Bonding; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Müller-Dethlefs, K.; Hobza, P. Noncovalent Interactions: A Challenge for Experiment and Theory. Chem. Rev. 2000, 100, 143–167. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R. Hydrogen Bridges in Crystal Engineering: Interactions without Borders. Acc. Chem. Res. 2002, 35, 565–573. [Google Scholar] [CrossRef]
- Pocheć, M.; Małycha, K.; Orzechowski, K. Nonlinear Dielectric Effect in the Vicinity of Solid–Liquid Phase Transition of 1-Decanol. J. Phys. Chem. C 2019, 123, 16929–16933. [Google Scholar] [CrossRef]
- Pocheć, M.; Niu, H.; Ren, L.; Bai, S.; Orzechowski, K. Premelting Phenomena in n-Alcohols from Nonanol to Dodecanol. J. Phys. Chem. C 2020, 124, 21013–21017. [Google Scholar] [CrossRef]
- Herweg, J. Die elektrischen Dipole in flüssigen Dielektricis. Z. Phys. 1920, 3, 36–47. [Google Scholar] [CrossRef]
- Herweg, J.; Pötzsch, W. Die elektrischen Dipole in flüssigen Dielektricis. Z. Phys. 1922, 8, 1–12. [Google Scholar] [CrossRef]
- Piekara, A.; Piekara, B. Saturation électrique dans les liquides purs et leurs mélanges. Compt. Rend. Acad. Sci. 1936, 203, 852. [Google Scholar]
- Wagner, K.W. Erklärung der dielektrischen Nachwirkungsvorgänge auf Grund Maxwellscher Vorstellungen. Arch. Elektrotech. 1914, 2, 371–387. [Google Scholar] [CrossRef]
- Piekara, A. A theory of electric polarization, electro-optical Kerr effect and electric saturation in liquids and solutions. Proc. Math. Phys. Eng. Sci. 1939, 172, 360–383. [Google Scholar]
- Orzechowski, K. Electrical properties of an ethanol–dodecane mixture near the upper critical solution point. J. Chem. Soc. Faraday Trans. 1994, 90, 2757–2763. [Google Scholar] [CrossRef]
- Rzoska, S.J. Kerr effect and the nonlinear dielectric effect on approaching the critical consolute point. Phys. Rev. E 1993, 48, 1136–1141. [Google Scholar] [CrossRef] [PubMed]
- Car, R.; Parrinello, M. Unified Approach for Molecular Dynamics and Density-Functional Theory. Phys. Rev. Lett. 1985, 55, 2471–2474. [Google Scholar] [CrossRef] [PubMed]
- Marx, D.; Parrinello, M. The Effect of Quantum and Thermal Fluctuations on the Structure of the Floppy Molecule C2H3+. Science 1996, 271, 179–181. [Google Scholar] [CrossRef]
- Tuckerman, M.E.; Marx, D.; Klein, M.L.; Parrinello, M. Efficient and general algorithms for path integral Car-Parrinello molecular dynamics. J. Chem. Phys. 1996, 104, 5579–5588. [Google Scholar] [CrossRef]
- Linde, D.R. CRC Handbook of Chemistry and Physics; Taylor & Francis Ltd.: Abingdon, UK, 2004. [Google Scholar]
- Orzechowski, K.; Burakowski, A. Experimental set-up for measuring the non-linear dielectric effect. Measurement 2019, 131, 219–222. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general Amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: AnN·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Andrea, T.A.; Swope, W.C.; Andersen, H.C. The role of long ranged forces in determining the structure and properties of liquid water. J. Chem. Phys. 1983, 79, 4576–4584. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Case, D.; Aktulga, H.; Belfon, K.; Ben-Shalom, I.; Brozell, S.; Cerutti, D.; Cheatham, T.E., III; Cisneros, G.; Cruzeiro, V.; Darden, T.; et al. Amber 2021; University of California: San Francisco, CA, USA, 2021. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Kittel, C. Introduction to Solid State Physics, 8th ed.; Wiley: Hoboken, NJ, USA, 2019. [Google Scholar]
- Schlegel, H.B. Estimating the hessian for gradient-type geometry optimizations. Theor. Chem. Acc. 1984, 66, 333–340. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Troullier, N.; Martins, J.L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 1991, 43, 1993–2006. [Google Scholar] [CrossRef]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- CPMD Version 4.3-4610, Copyright IBM Corp. (1990–2004) Copyright MPI für Festkoerperforschung Stuttgart (1997–2001). Available online: http://www.cpmd.org/ (accessed on 15 December 2021).
- Forbert, H. A Fortran90 Code to Calculate Power Spectrum of Dipole Moment Trajectory. 2008. Available online: www.cpmd.org/wordpress/index.php/download (accessed on 15 December 2021).
- Williams, T.; Kelley, C. Gnuplot 4.4: An Interactive Plotting Program. 2010. Available online: http://gnuplot.sourceforge.net/ (accessed on 15 December 2021).
- CCDC Structural Database. 2021. Available online: https://www.ccdc.cam.ac.uk/ (accessed on 7 May 2021).
- MacCallum, J.L.; Tieleman, D.P. Structures of Neat and Hydrated 1-Octanol from Computer Simulations. J. Am. Chem. Soc. 2002, 124, 15085–15093. [Google Scholar] [CrossRef]
- Marbeuf, A.; Brown, R. Molecular dynamics in n-alkanes: Premelting phenomena and rotator phases. J. Chem. Phys. 2006, 124, 054901. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temp. (K) | Dihedral Angle | ||
|---|---|---|---|
| C8-C7-C6-C5 | C7-C6-C5-C4 | C6-C5-C4-C3 | |
| 237 | 3 | 1 | 0 |
| 247 | 3 | 0 | 0 |
| 257 | 4 | 1 | 0 |
| 267 | 13 | 1 | 0 |
| 277 | 12 | 1 | 0 |
| 287 | 17 | 3 | 0 |
| 297 | 21 | 8 | 0 |
| 307 | 49 | 6 | 0 |
| 317 | 81 | 12 | 4 |
| 327 | 65 | 13 | 1 |
| 337 | 122 | 23 | 5 |
| 347 | 139 | 38 | 2 |
| 357 | 206 | 58 | 3 |
| 367 | 360 | 97 | 20 |
| 377 | 655 | 641 | 142 |
| 387 | 459 | 175 | 43 |
| 397 | 626 | 194 | 79 |
| 407 | 846 | 311 | 93 |
| 417 | 11,799 | 10,093 | 9972 |
| Temp. (K) | Dihedral Angle () | ||
|---|---|---|---|
| C8-C7-C6-C5 | C7-C6-C5-C4 | C6-C5-C4-C3 | |
| 150 | 178.77 ± 4.29 | 180.70 ± 3.48 | 180.05 ± 3.70 |
| 190 | 178.62 ± 4.80 | 180.85 ± 3.85 | 179.79 ± 4.22 |
| 230 | 179.00 ± 5.35 | 180.69 ± 4.16 | 179.98 ± 4.54 |
| 257 | 178.79 ± 5.69 | 180.87 ± 4.46 | 179.79 ± 4.90 |
| 270 | 178.51 ± 5.74 | 180.92 ± 4.58 | 179.93 ± 5.04 |
| 300 | 178.92 ± 6.07 | 180.65 ± 4.78 | 179.86 ± 5.13 |
| 350 | 178.89 ± 6.64 | 180.47 ± 5.21 | 180.32 ± 5.71 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pocheć, M.; Krupka, K.M.; Panek, J.J.; Orzechowski, K.; Jezierska, A. Inside out Approach to Rotator State in Hydrogen-Bonded System—Experimental and Theoretical Cross-Examination in n-Octanol. Int. J. Mol. Sci. 2022, 23, 2138. https://doi.org/10.3390/ijms23042138
Pocheć M, Krupka KM, Panek JJ, Orzechowski K, Jezierska A. Inside out Approach to Rotator State in Hydrogen-Bonded System—Experimental and Theoretical Cross-Examination in n-Octanol. International Journal of Molecular Sciences. 2022; 23(4):2138. https://doi.org/10.3390/ijms23042138
Chicago/Turabian StylePocheć, Michał, Katarzyna M. Krupka, Jarosław J. Panek, Kazimierz Orzechowski, and Aneta Jezierska. 2022. "Inside out Approach to Rotator State in Hydrogen-Bonded System—Experimental and Theoretical Cross-Examination in n-Octanol" International Journal of Molecular Sciences 23, no. 4: 2138. https://doi.org/10.3390/ijms23042138
APA StylePocheć, M., Krupka, K. M., Panek, J. J., Orzechowski, K., & Jezierska, A. (2022). Inside out Approach to Rotator State in Hydrogen-Bonded System—Experimental and Theoretical Cross-Examination in n-Octanol. International Journal of Molecular Sciences, 23(4), 2138. https://doi.org/10.3390/ijms23042138