
Whole Genome Analysis of 335 New Bacterial Species from Human Microbiota Reveals a Huge Reservoir of Transferable Antibiotic Resistance Determinants



Abstract
:1. Introduction
2. Results
2.1. Prevalence and Description of ARGs Found in the New Bacterial Species from the Human Microbiota
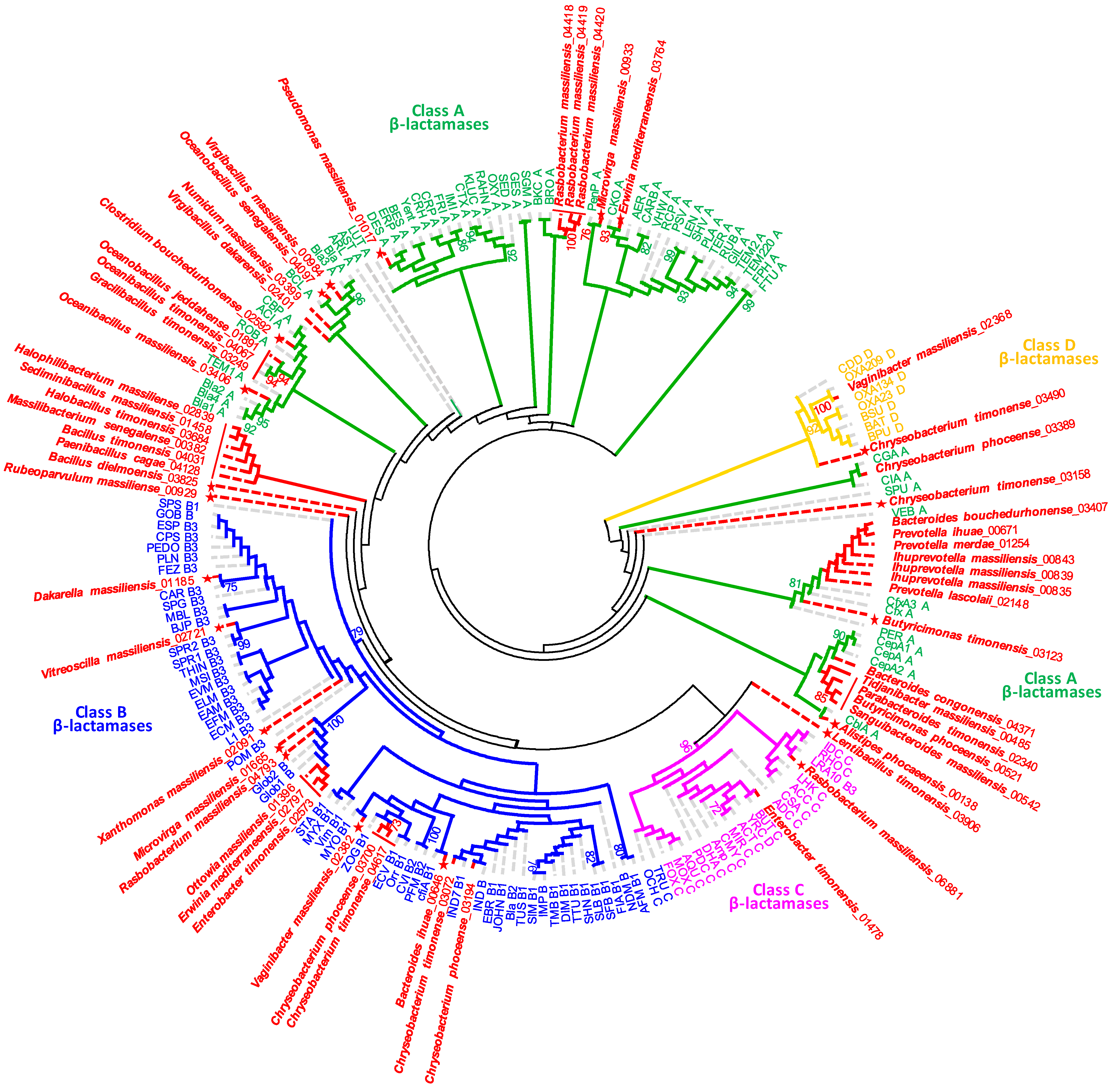
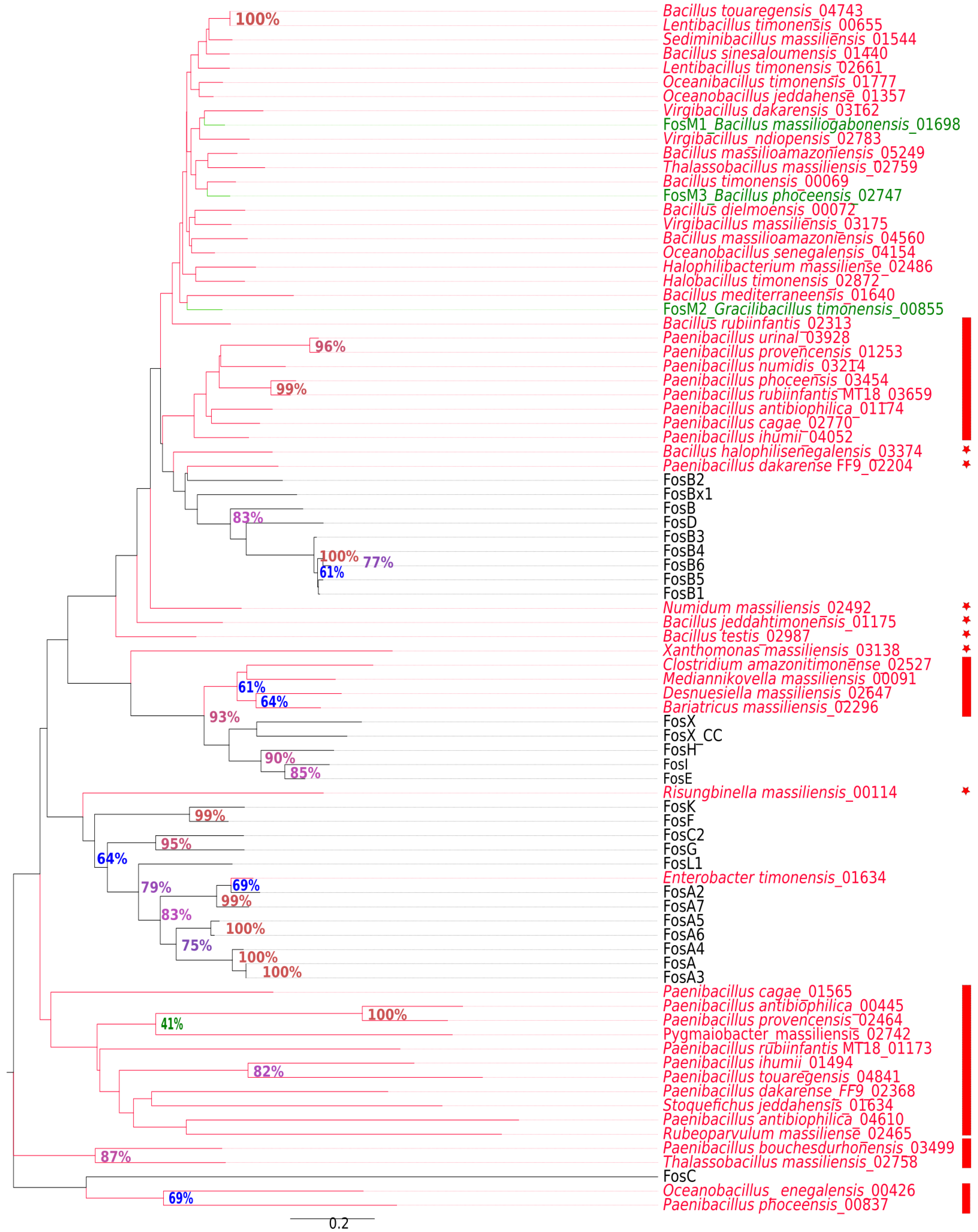
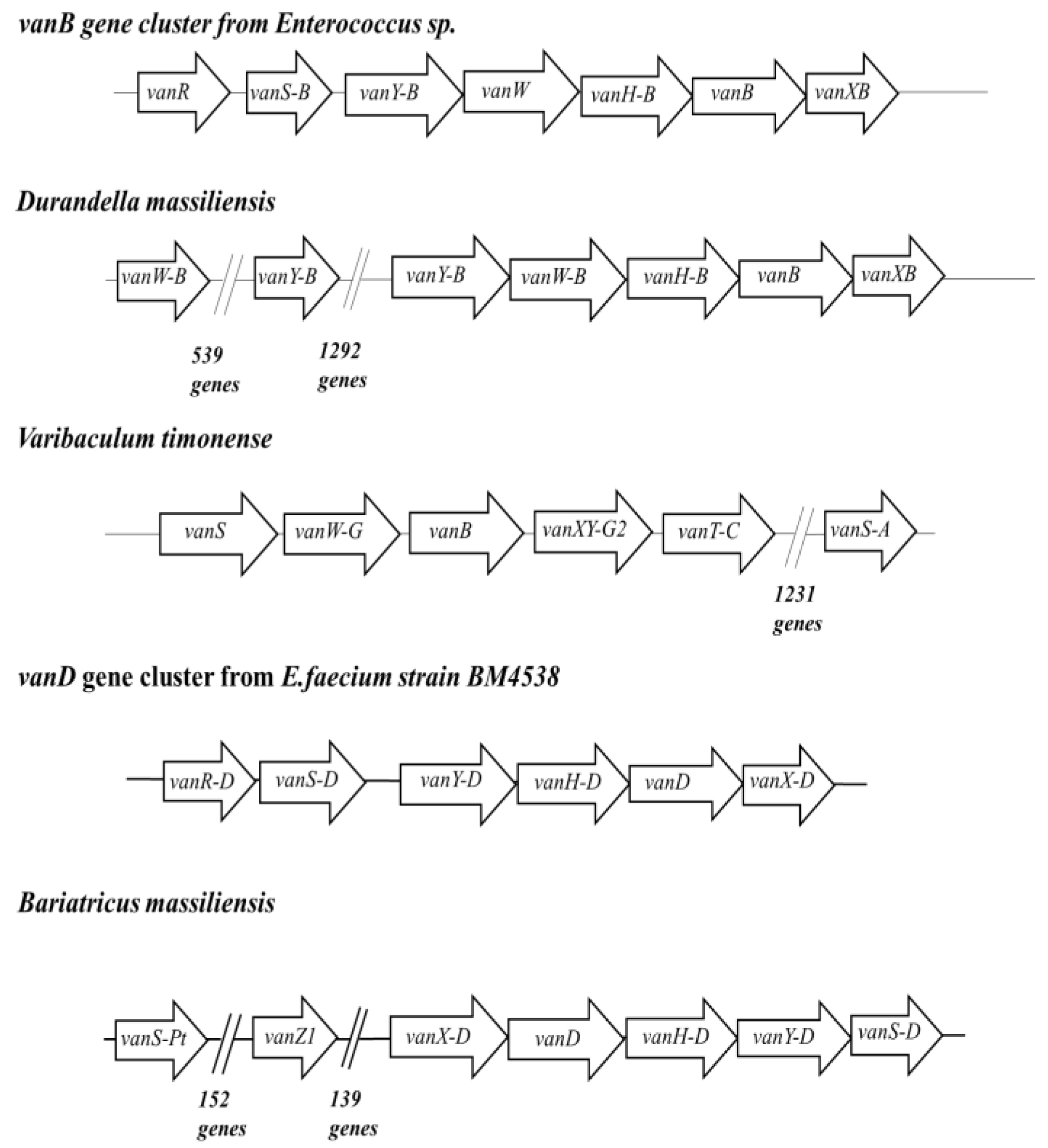
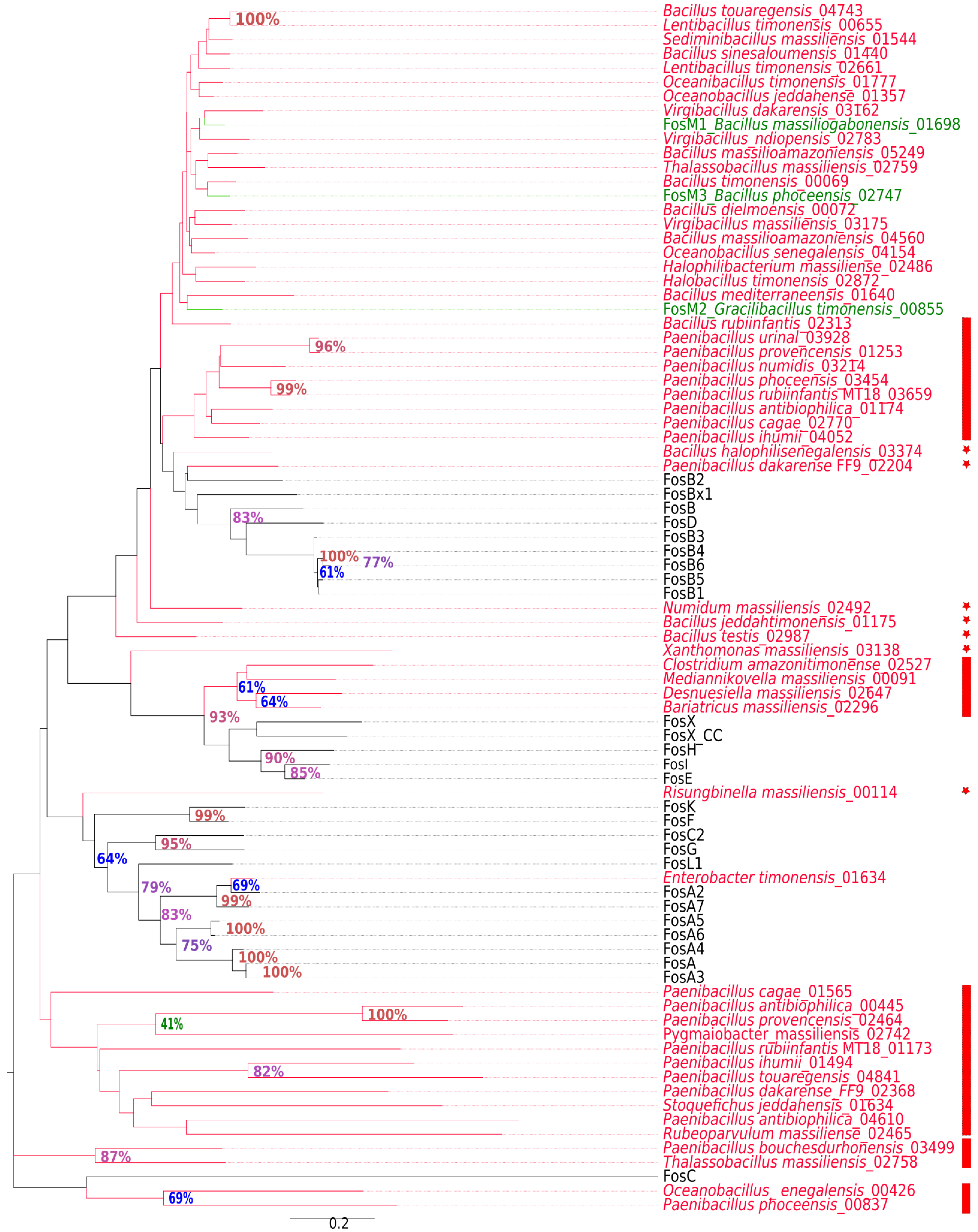
2.2. New Putative ARGs Conferring Resistance to β-Lactams, Fosfomycin and Vancomycin
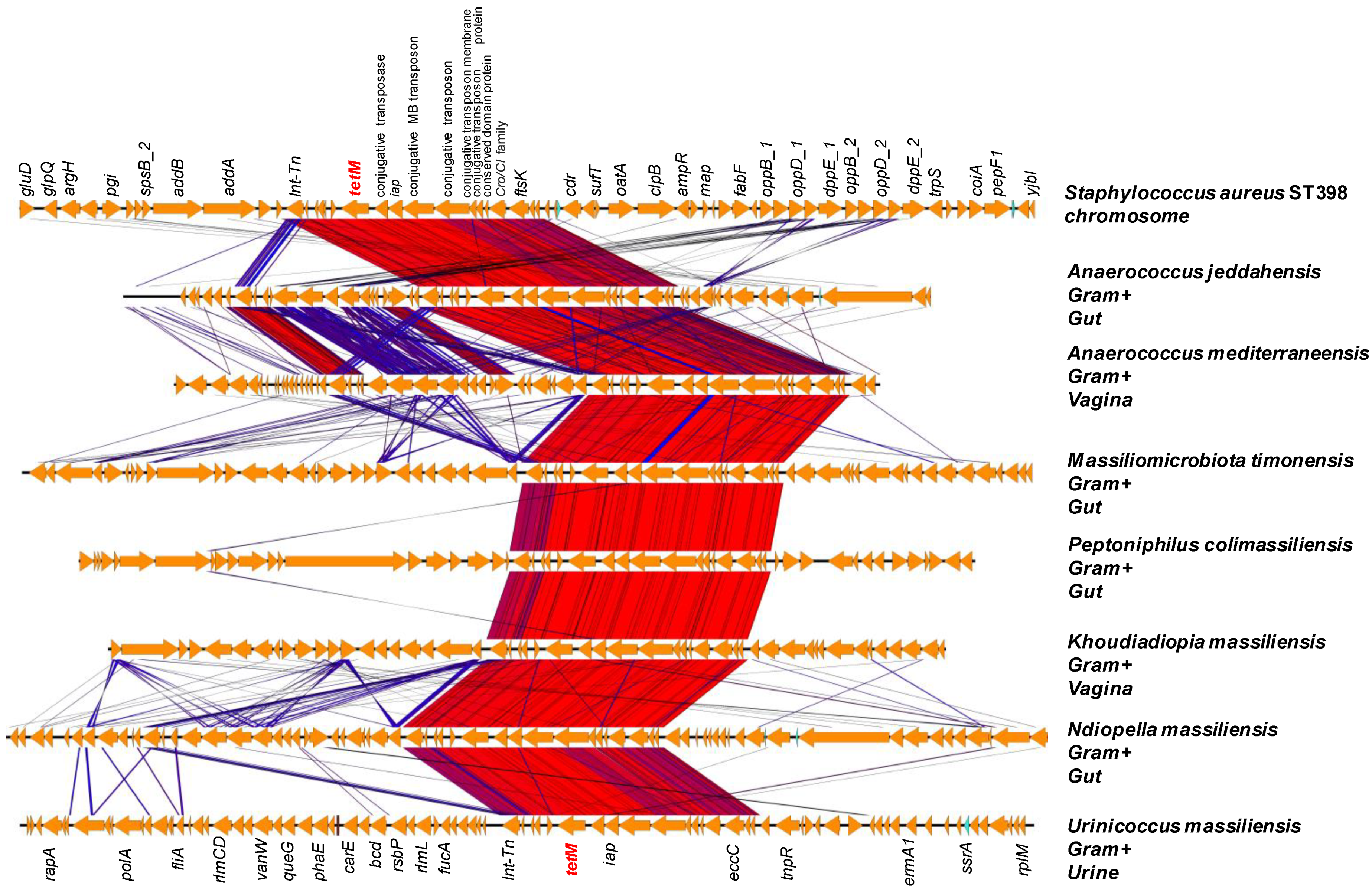
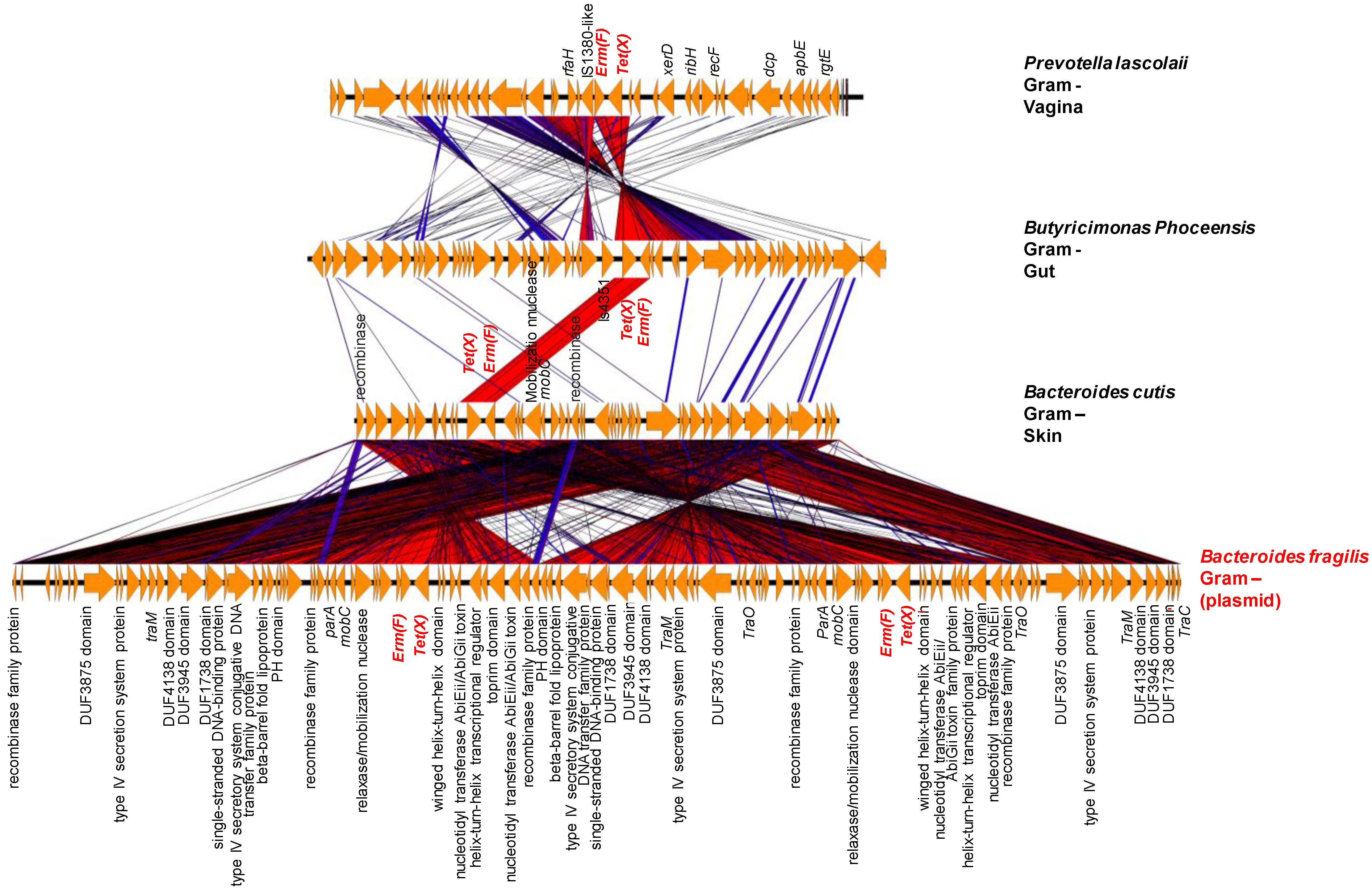
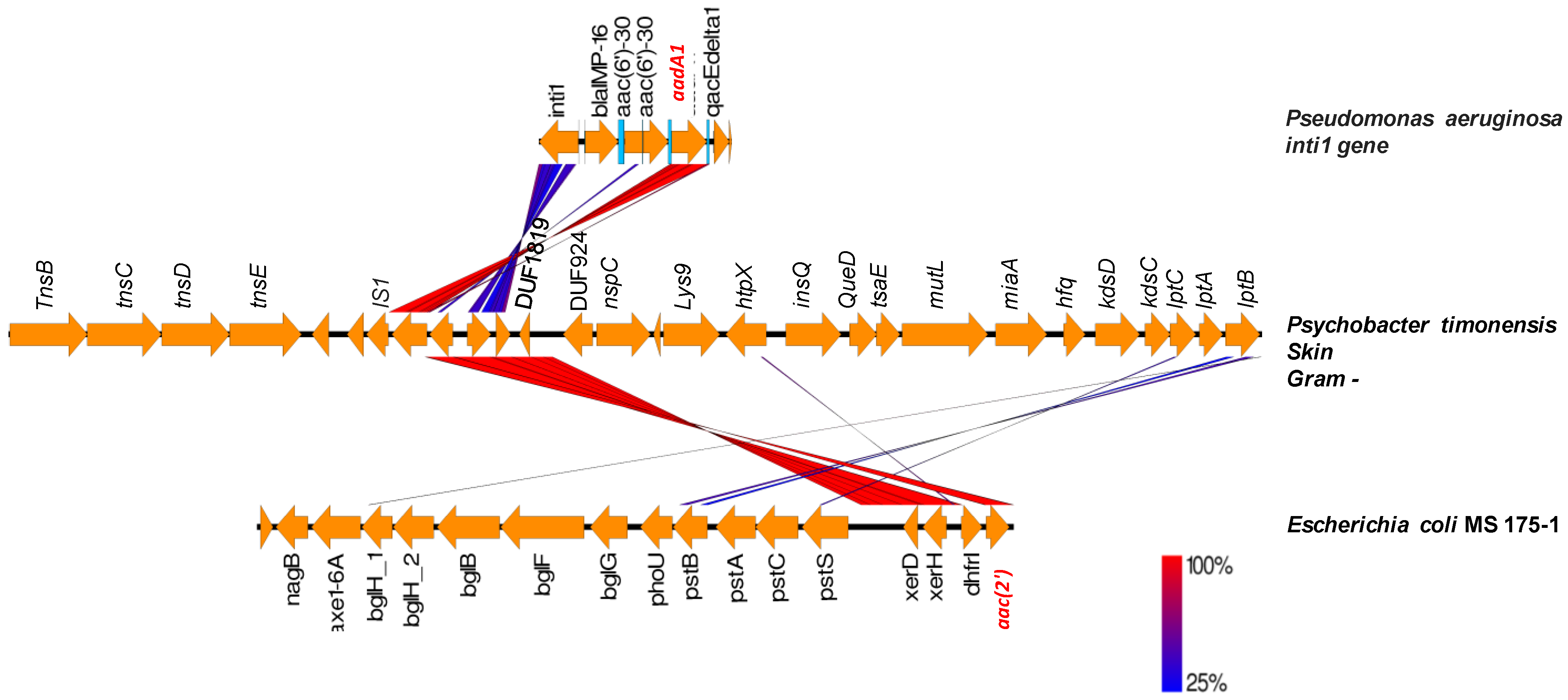
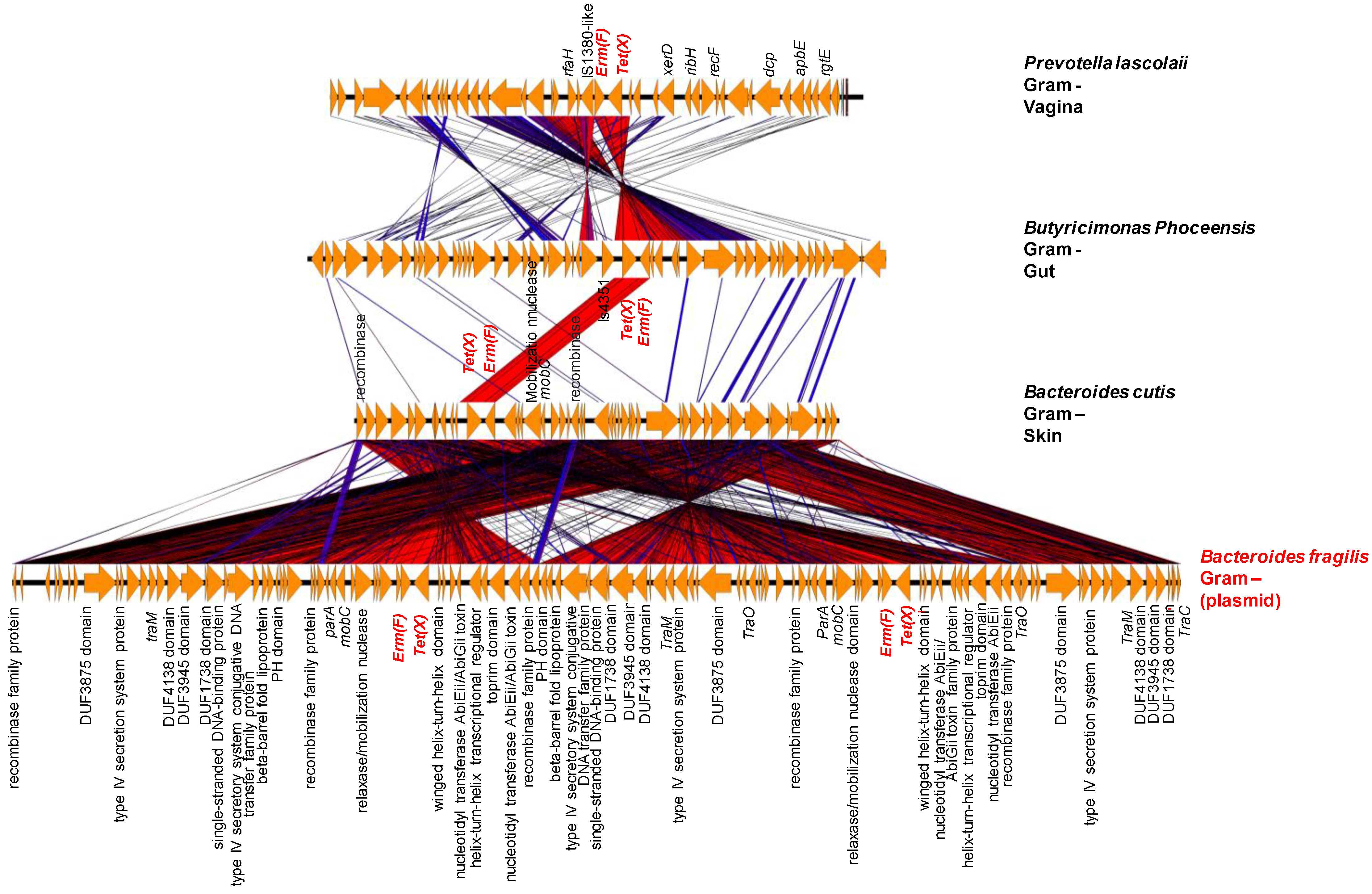
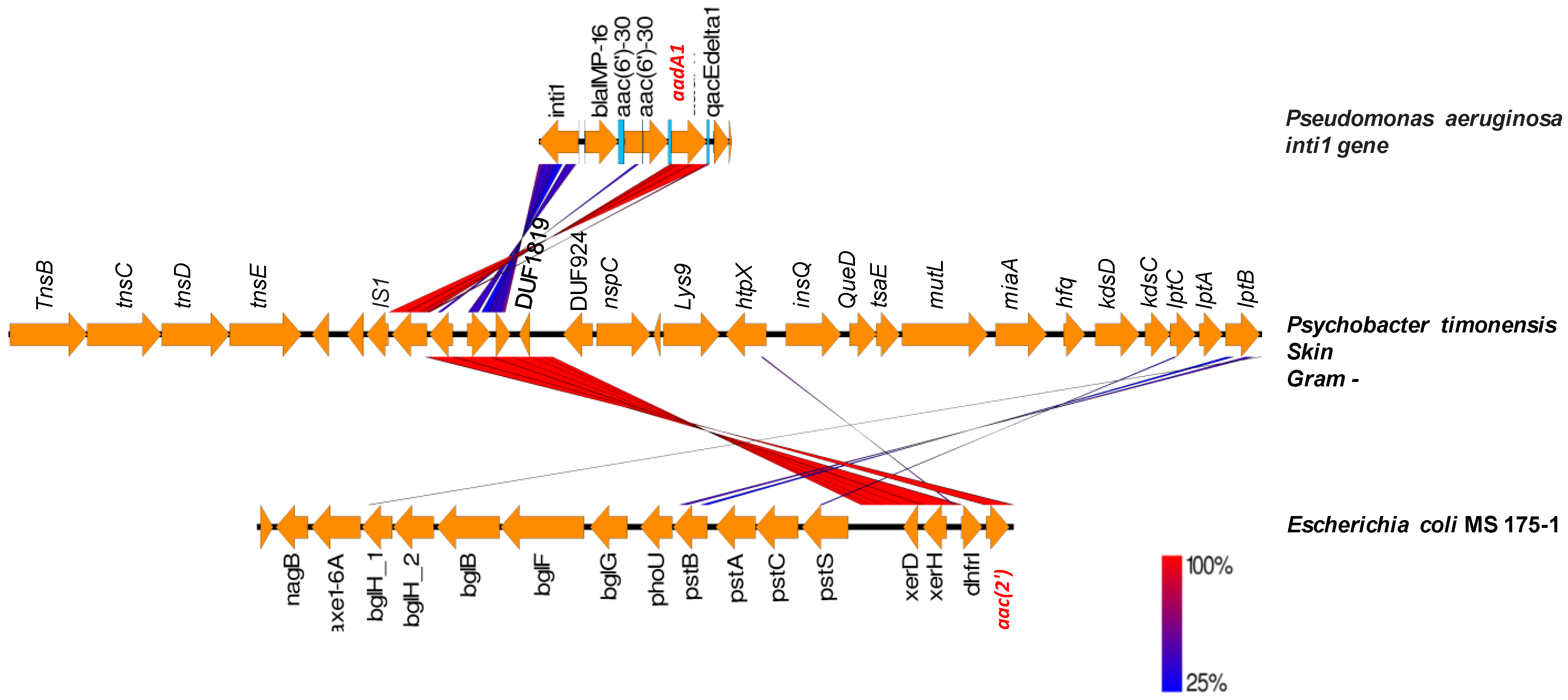
2.3. Origin of ARGs in the Human Microbiota
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Computational Method for Predicting ARGs
4.3. Phylogenetic Analysis
4.4. Bioinformatic Characterization of Conserved Protein Domains and Motifs
4.5. Detection of Mobile Genetic Elements Associated with ARGs and Determination of GC Content
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Adedeji, W.A. The treasure called antibiotics. Ann. Ib. Postgrad. Med. 2016, 14, 56–57. [Google Scholar] [PubMed]
- Davies, J.; Davies, D. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef] [Green Version]
- Holmes, A.H.; Moore, L.S.P.; Sundsfjord, A.; Steinbakk, M.; Regmi, S.; Karkey, A.; Guerin, P.J.; Piddock, L.J. Understanding the mechanisms and drivers of antimicrobial resistance. Lancet 2016, 387, 176–187. [Google Scholar] [CrossRef]
- Gullberg, E.; Cao, S.; Berg, O.G.; Ilbäck, C.; Sandegren, L.; Hughes, D.; Andersson, D.I. Selection of resistant bacteria at very low antibiotic concentrations. PLoS Pathog. 2011, 7, e1002158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. Global Action Plan on Antimicrobial Resistance. 2015. Available online: http://www.who.int/drugresistance/global_action_plan/en/ (accessed on 15 November 2021).
- D’Costa, V.M.; King, C.E.; Kalan, L.; Morar, M.; Sung, W.W.; Schwarz, C.; Froese, D.; Zazula, G.; Calmels, F.; Debruyne, R.; et al. Antibiotic resistance is ancient. Nature 2011, 477, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Pishchany, G.; Kolter, R. On the possible ecological roles of antimicrobials. Mol. Microbiol. 2020, 113, 580–587. [Google Scholar] [CrossRef]
- Sana, T.G.; Lugo, K.A.; Monack, D.M. T6SS: The bacterial ‘fight club’ in the host gut. PLoS Pathog. 2017, 13, e1006325. [Google Scholar] [CrossRef] [Green Version]
- Fritz, S.; Rajaonison, A.; Chabrol, O.; Raoult, D.; Rolain, J.-M.; Merhej, V. Full-length title: NRPPUR database search and in vitro analysis identify an NRPS-PKS biosynthetic gene cluster with a potential antibiotic effect. BMC Bioinform. 2018, 19, 463. [Google Scholar] [CrossRef]
- Zipperer, A.; Konnerth, M.C.; Laux, C.; Berscheid, A.; Janek, D.; Weidenmaier, C.; Burian, M.; Schilling, N.A.; Slavetinsky, C.; Marschal, M.; et al. Human commensals producing a novel antibiotic impair pathogen colonization. Nature 2016, 535, 511–516. [Google Scholar] [CrossRef]
- Khabthani, S.; Rolain, J.-M.; Merhej, V. In Silico/In Vitro Strategies Leading to the Discovery of New Nonribosomal Peptide and Polyketide Antibiotics Active against Human Pathogens. Microorganisms 2021, 9, 2297. [Google Scholar] [CrossRef] [PubMed]
- Diene, S.M.; Pinault, L.; Baron, S.A.; Azza, S.; Armstrong, N.; Hadjadj, L.; Chabrière, E.; Rolain, J.M.; Pontarotti, P.; Raoult, D. A metallo-β-lactamase enzyme for internal detoxification of the antibiotic thienamycin. Sci. Rep. 2021, 11, 10062. [Google Scholar] [CrossRef] [PubMed]
- García, P.; Arca, P.; Evaristo Suárez, J. Product of fosC, a gene from Pseudomonas syringae, mediates fosfomycin resistance by using ATP as cosubstrate. Antimicrob. Agents Chemother. 1995, 39, 1569–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aminov, R.I.; Mackie, R.I. Evolution and ecology of antibiotic resistance genes. FEMS Microbiol. Lett. 2007, 271, 147–161. [Google Scholar] [CrossRef] [PubMed]
- von Wintersdorff, C.J.; Penders, J.; van Niekerk, J.M.; Mills, N.D.; Majumder, S.; van Alphen, L.B.; Savelkoul, P.H.; Wolffs, P.F. Dissemination of Antimicrobial Resistance in Microbial Ecosystems through Horizontal Gene Transfer. Front. Microbiol. 2016, 7, 173. [Google Scholar] [CrossRef] [Green Version]
- McInnes, R.S.; McCallum, G.E.; Lamberte, L.E.; van Schaik, W. Horizontal transfer of antibiotic resistance genes in the human gut microbiome. Curr. Opin. Microbiol. 2020, 53, 35–43. [Google Scholar] [CrossRef]
- Werren, J.H. Selfish genetic elements, genetic conflict, and evolutionary innovation. Proc. Natl. Acad. Sci. USA 2011, 108, 10863–10870. [Google Scholar] [CrossRef] [Green Version]
- Partridge, S.R.; Kwong, S.M.; Firth, N.; Jensen, S.O. Mobile Genetic Elements Associated with Antimicrobial Resistance. Clin. Microbiol. Rev. 2018, 31, e00088-17. [Google Scholar] [CrossRef] [Green Version]
- Smillie, C.S.; Smith, M.B.; Friedman, J.; Cordero, O.X.; David, L.A.; Alm, E.J. Ecology drives a global network of gene exchange connecting the human microbiome. Nature 2011, 480, 241–244. [Google Scholar] [CrossRef]
- van Schaik, W. The human gut resistome. Philos. Trans. R. Soc. B 2015, 370, 20140087. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Chen, X.; Skogerbø, G.; Zhang, P.; Chen, R.; He, S.; Huang, D.W. The human microbiome: A hot spot of microbial horizontal gene transfer. Genomics 2012, 100, 265–270. [Google Scholar] [CrossRef] [Green Version]
- Donia, M.S.; Cimermancic, P.; Schulze, C.J.; Wieland Brown, L.C.; Martin, J.; Mitreva, M.; Clardy, J.; Linington, R.G.; Fischbach, M.A. A systematic analysis of biosynthetic gene clusters in the human microbiome reveals a common family of antibiotics. Cell 2014, 158, 1402–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huddleston, J.R. Horizontal gene transfer in the human gastrointestinal tract: Potential spread of antibiotic resistance genes. Infect. Drug Resist. 2014, 7, 167–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rolain, J.-M. Food and human gut as reservoirs of transferable antibiotic resistance encoding genes. Front. Microbiol. 2013, 4, 173. [Google Scholar] [CrossRef] [Green Version]
- Olaitan, A.O.; Morand, S.; Rolain, J.-M. Emergence of colistin-resistant bacteria in humans without colistin usage: A new worry and cause for vigilance. Int. J. Antimicrob. Agents 2016, 47, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Clemente, J.C.; Pehrsson, E.C.; Blaser, M.J.; Sandhu, K.; Gao, Z.; Wang, B.; Magris, M.; Hidalgo, G.; Contreras, M.; Noya-Alarcón, Ó.; et al. The microbiome of uncontacted Amerindians. Sci. Adv. 2015, 1, e1500183. [Google Scholar] [CrossRef] [Green Version]
- Lagier, J.C.; Armougom, F.; Million, M.; Hugon, P.; Pagnier, I.; Robert, C.; Bittar, F.; Fournous, G.; Gimenez, G.; Maraninchi, M.; et al. Microbial culturomics: Paradigm shift in the human gut microbiome study. Clin. Microbiol. Infect. 2012, 18, 1185–1193. [Google Scholar] [CrossRef] [Green Version]
- Lagier, J.C.; Khelaifia, S.; Alou, M.T.; Ndongo, S.; Dione, N.; Hugon, P.; Caputo, A.; Cadoret, F.; Traore, S.I.; Seck, E.H.; et al. Culture of previously uncultured members of the human gut microbiota by culturomics. Nat. Microbiol. 2016, 1, 16203. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Derbyshire, M.K.; Gonzales, N.R.; Lu, S.; Chitsaz, F.; Geer, L.Y.; Geer, R.C.; He, J.; Gwadz, M.; Hurwitz, D.I.; et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015, 43, D222–D226. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Castañeda-García, A.; Blázquez, J.; Rodríguez-Rojas, A. Molecular mechanisms and clinical impact of acquired and intrinsic fosfomycin resistance. Antibiotics 2013, 2, 217–236. [Google Scholar] [CrossRef] [Green Version]
- Khabthani, S.; Hamel, M.; Baron, S.A.; Diene, S.M.; Rolain, J.-M.; Merhej, V. fosM, a New Family of Fosfomycin Resistance Genes Identified in Bacterial Species Isolated from Human Microbiota. Antimicrob. Agents Chemother. 2021, 65, e01712-20. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed]
- Yarygin, K.S.; Kovarsky, B.A.; Bibikova, T.S.; Melnikov, D.S.; Tyakht, A.V.; Alexeev, D.G. ResistoMap-online visualization of human gut microbiota antibiotic resistome. Bioinformatics 2017, 33, 2205–2206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, J.; Li, B.; Jiang, X.; Yang, Y.; Wells, G.F.; Zhang, T.; Li, X. Antibiotic resistome in a large-scale healthy human gut microbiota deciphered by metagenomic and network analyses. Environ. Microbiol. 2018, 20, 355–368. [Google Scholar] [CrossRef]
- Liu, L.; Feng, Y.; McNally, A.; Zong, Z. blaNDM-21, a new variant of blaNDM in an Escherichia coli clinical isolate carrying blaCTX-M-55 and rmtB. J. Antimicrob. Chemother. 2018, 73, 2336–2339. [Google Scholar] [CrossRef]
- Fu, Z.; Liu, Y.; Chen, C.; Guo, Y.; Ma, Y.; Yang, Y.; Hu, F.; Xu, X.; Wang, M. Characterization of Fosfomycin Resistance Gene, fosB, in Methicillin-Resistant Staphylococcus aureus Isolates. PLoS ONE 2016, 11, e0154829. [Google Scholar] [CrossRef] [Green Version]
- Sommer, M.O.A.; Dantas, G.; Church, G.M. Functional Characterization of the Antibiotic Resistance Reservoir in the Human Microflora. Science 2009, 325, 1128–1131. [Google Scholar] [CrossRef] [Green Version]
- Hadjadj, L.; Baron, S.A.; Diene, S.M.; Rolain, J.-M. How to discover new antibiotic resistance genes? Expert Rev. Mol. Diagn. 2019, 19, 349–362. [Google Scholar] [CrossRef]
- Privitera, G.; Dublanchet, A.; Sebald, M. Transfer of Multiple Antibiotic Resistance between Subspecies of Bacteroides fragilis. J. Infect. Dis. 1979, 139, 97–101. [Google Scholar] [CrossRef]
- Guiney, D.G.; Davis, C.E. Identification of a conjugative R plasmid in Bacteroides ochraceus capable of transfer to Escherichia coli. Nature 1978, 274, 181–182. [Google Scholar] [CrossRef]
- Wüst, J.; Hardegger, U. Transferable Resistance to Clindamycin, Erythromycin, and Tetracycline in Clostridium difficile. Antimicrob. Agents Chemother. 1983, 23, 784–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.J.; Markowitz, S.M.; Macrina, F.L. Transferable tetracycline resistance in Clostridium difficile. Antimicrob. Agents Chemother. 1981, 19, 997–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaffe, E.; Relman, D.A. Tracking microbial evolution in the human gut using Hi-C reveals extensive horizontal gene transfer, persistence and adaptation. Nat. Microbiol. 2020, 5, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Kieffer, N.; Nordmann, P.; Poirel, L. Moraxella species as potential sources of MCR-like polymyxin resistance determinants. Antimicrob. Agents Chemother. 2017, 61, e00129-17. [Google Scholar] [CrossRef] [Green Version]
- Khedher, M.B.; Baron, S.A.; Riziki, T.; Ruimy, R.; Raoult, D.; Diene, S.M.; Rolain, J.M. Massive analysis of 64,628 bacterial genomes to decipher water reservoir and origin of mobile colistin resistance genes: Is there another role for these enzymes? Sci. Rep. 2020, 10, 5970. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.Y.; Wang, Y.; Walsh, T.R.; Yi, L.X.; Zhang, R.; Spencer, J.; Doi, Y.; Tian, G.; Dong, B.; Huang, X.; et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: A microbiological and molecular biological study. Lancet Infect. Dis. 2016, 16, 161–168. [Google Scholar] [CrossRef]
- Toleman, M.A.; Spencer, J.; Jones, L.; Walsh, T.R. bla NDM-1 Is a Chimera Likely Constructed in Acinetobacter baumannii. Antimicrob. Agents Chemother. 2012, 56, 2773–2776. [Google Scholar] [CrossRef] [Green Version]
- Dortet, L.; Poirel, L.; Nordmann, P. Worldwide Dissemination of the NDM-Type carbapenemases in Gram-negative bacteria. BioMed Res. Int. 2014, 2014, 249856. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Feldgarden, M.; Brover, V.; Haft, D.H.; Prasad, A.B.; Slotta, D.J.; Tolstoy, I.; Tyson, G.H.; Zhao, S.; Hsu, C.H.; McDermott, P.F.; et al. Validating the AMRFinder Tool and Resistance Gene Database by Using Antimicrobial Resistance Genotype-Phenotype Correlations in a Collection of Isolates. Antimicrob. Agents Chemother. 2019, 63, e00483-19. [Google Scholar] [CrossRef] [Green Version]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef]
- Gupta, S.K.; Padmanabhan, B.R.; Diene, S.M.; Lopez-Rojas, R.; Kempf, M.; Landraud, L.; Rolain, J.-M. ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob. Agents Chemother. 2014, 58, 212–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sievers, F.; Higgins, D.G. Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 2018, 27, 135–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Carattoli, A.; Zankari, E.; García-Fernández, A.; Voldby Larsen, M.; Lund, O.; Villa, L.; Møller Aarestrup, F.; Hasman, H. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ATB | Genes Detected by BlastP | Diversity | Total |
|---|---|---|---|
| Aminoglycosides | aac(2′)-IIb,aac(3),aac(3)-If,aac(3)-Ih,aac(3)-IIe,aac(3)-IIIa,aac(3)-IIIb,aac(3)-VIII,aac(3)-Xa,aac(3)-XI,aac(6′),aac(6′)-34,aac(6′)-35,aac(6′)-Iad,aac(6′)-Iz,aacC7,aacC9,AAD(3′’),aad9,aadA1,aadA13,aadA31,aadD1,aadE,aadK,aadS,ant(3′’)-IIa,ant(3′’)-IIb,ant(3′’)-IIc,ant(4′)-Ic,ant(6),ant(6)-Ia,ant(6)-Ib,ant(6)-Ic,ant(9),aph,aph(2′’)-IIa,aph(3′)-Ia,aph(3′)-IIa,aph(3′)-IIc,aph(3′)-IIIa,aph(3′)-IVa,aph(3′)-Va,APH(6),aph(6)-Ia,aph(6)-Id,aph(9)-Ia,aac(2′). | 53 | 195 |
| Betalactams | bla,bla1,blaBCL-1,blaBKC,blaBKC-1,blaCAR-1,blaCBP,blaCIA-1,blaCIA-4,blaCKO-1,blaCMY-100,blaIND-4,blaIND-7,blaLRA-10_like,blaLUT-5,blaMYO-1,blaOXA-209,blaP,blaPOM,blaSPR-1,blaSPU-1,blaZOG-1,CblA,cepA,cepA-49,cfiA,cfxA,cfxA3,TEM. | 29 | 58 |
| Fosfomycin | fos,fos_related,fosA,fosA2,fosA-491618165,fosA7,fosA8,fosB,fosB2,fosB-38141535,fosBx1,fosX. | 12 | 58 |
| Glycopeptides | vanB,vanC1,vanD. | 3 | 51 |
| MLS | chrB_rRNA_meth,ereD,erm(31),erm(32),erm(35),erm(46),erm(49),erm(A),erm(B),erm(D),erm(F),erm(G),erm(X),erm(Y),ermk,llmA,lnu(A),lnu(AN2),lnu(C),lnu(D),lnu(G),lnuE,mph(B),mphI,mphK/ycbJ,mphL,mphM,mphO,myrA,rlmA,vgb(B),vgbC,cfr,cfr(B),cfr-Cb,clbC,vat,vat(B),vat(F),vat(H),vatI. | 41 | 270 |
| Nitroimidazole | nimA,nimB,nimD,nimE,nimJ. | 5 | 36 |
| Phenicol | cat,cat86,catA1,catA13,catA15,catA16,catA4,CatB,catB3,catP,catV,cpt. | 12 | 33 |
| Rifamycin | arr-269927220,arr-Ms,rphB,rphC,rphD. | 5 | 32 |
| Tetracyclines | otr(A),tet(32),tet(36),tet(M),tet(O),tet(Q),tet(T),tet(W),tet(X),tetB(P). | 10 | 156 |
| Total | 170 | 889 |
| Class | Conserved Motifs | Bacterial Species (ARGs ID) | |||
|---|---|---|---|---|---|
| A | STFK | SDN | EIDLN | KTG | Rubeoparvulum massiliensis (00929) |
| STFK | SDN | EPDLN | KSG | Gracilibacillus timonensis (03249), Oceanibacillus timonensis (04067) | |
| STFK | SDN | EPDLN | KTG | Rasbobactérium massiliensis (04418) | |
| STFK | SDN | ETDLN | KSG | Oceanobacillus jeddahense (01891) | |
| STFK | SDN | ETDLN | KTG | Microvirga massiliensis (00933) | |
| STFK | SDN | ETELN | KTG | Pseudomonas massiliensis (01017) | |
| STFK | SDN | EVELN | KSG | Lentibacillus timonensis (03906) | |
| STFK | SDS | X | KTG | Rabobacterium massiliensis (04420) | |
| STHK | SDN | EPALN | KTG | Virgibacillus massiliensis (00984) | |
| STHK | SDN | EPELN | KSG | Virgibacillus dakarensis (02401) | |
| STHK | SDN | EPELN | KTG | Numidum massiliensis (03399), Oceanobacillus senegalensis (04097) | |
| STHK | SDN | KSG | Erwinia mediterraneensis (03764) | ||
| STVK | SDS | X | KTG | Rasbobacterium massiliensis (04419) | |
| STYK | SDN | EPDLN | KSG | Bacillus timonensis (04031) | |
| STYK | SDN | EPDLN | KSG | Massilibacterium senegalense (00382), Oceanbacillus massiliensis (03406) | |
| STYK | SDN | EPELN | KSG | Halobacillus timonensis (03684) | |
| STYK | SDN | EPELN | KSG | Paenibacillus cagae (04128) | |
| STYK | SDN | EPNLN | KSG | Halophilibacterium massiliense (02839) | |
| STYK | SDN | ETELN | KSG | Bacillus dielmoensis (03825), Clostridium bouchedurhonensis (02592) | |
| STYK | X | ETELN | KSG | Sediminibacillus massiliensis (01458) | |
| SVFK | SDN | X | KTG | Alistipes phocaeensis (00138), Bacteroides bouchedurhonensis (03407), Bacteroides congolense (04371), Butyricimonas phoceensis (00521), Butyricimonas timonensis (03123), Chryseobacterium phoceense (03389), Ihuprevotella massiliensis (00839), Ihuprevotella massiliensis (00843), Ihuprevotella massiliensis (00835), Parabacteroides timonensis (02340), Prevotella ihumii (00671), Prevotella lascolaii (02148), Prevotella merdae (01254), Sanguibacter massiliensis (00542), Tidjanibacter massiliensis (00485) | |
| SVFK | SDN | X | X | Chryseobacterium timonensis (03158), Chryseobacterium timonense (03490) | |
| B | HARLDQ | Vitreoscilla massiliensis (02721) | |||
| B1 | HXHXD | Bacteroides ihumii (00646), Chryseobacterium phoceensis (03194), Chryseobacterium phoceensis (03700), Chryseobacterium timonensis (03072), Chryseobacterium timonensis (04617), Vaginibacter massiliensis (02382) | |||
| B3 | HAHADH | Xanthomonas massiliensis (02091) | |||
| HGHFDH | Dakarella massiliensis (01185) | ||||
| HXHXDH | Enterobacter timonensis (02573), Erwinia mediterraneensi (02797), Microvirga massiliensis (01665), Ottowia massiliensis (01396), Rasbobacterium massiliensis (04793) | ||||
| C | SVSK | YAN | KTG | Enterobacter timonensis (01478) | |
| SVSK | YSN | KTG | Rasbobacterium massiliensis (06881) | ||
| D | STFK | SCV | X | KTG | Vaginibacter massiliensis (02368) |
| Genome | Gene | ARGs | Closely Related Species | [Dif] | Transposase |
|---|---|---|---|---|---|
| Anaerococcus jeddahense | 01594 | tet(M) | Staphylococcus aureus | 5 | 01589 |
| Massiliomicrobiota timonensis | 00843 | tet(M) | 0 | 0838 | |
| Peptoniphilus colimassiliensis | 00489 | tet(M) | 14 | 00484 | |
| Khoudiadiopia massiliensis | 00250 | tet(M) | 5 | 00245 | |
| Ndiopella massiliensis | 00481 | tet(M) | 11 | 00476 | |
| Urinicoccus massiliensis | 01024 | tet(M) | 0 | 01019 | |
| Polynesia massiliensis | 06038 | tet(M) | 11 | 06033 | |
| Anaerococcus mediterraneense | 01722 | tet(M) | 1 | 01727 | |
| Anaerococcus mediannikovii | 00814 | ant | Streptococcus mitis | 5 | 00817_00819 |
| Anaerococcus mediannikovii | 00808 | erm | Streptococus pyogenes | 3 | 00817_00819 |
| Anaerococcus mediannikovii | 00812 | aph | Plasmid in Enterococcus faecalis, plasmid in K. oxytoca | 11 | 00817_00819 |
| Intestinibacillus timonensis | 00006 | aph | 8 | ||
| Peptoniphilus colimassiliensis | 00331 | aph | 5 | 00324_00326 | |
| Peptoniphilus colimassiliensis | 00335 | erm | S. aureus, S. pyogenes,S. parasanguis | 20 | 00324_00326 |
| Peptoniphilus raoultii | 01230 | erm | 5 | 01231_01233 | |
| Urinacoccus massiliensis | 01047 | erm | 6 | 01037 | |
| Clostridium bouchedurhonense | 00004 | erm | Enterococcus faecium/Staphylococcus epidermidis, plasmid in S. pyogenes, plasmid in streptococcus sanguis, plasmid in Lactobacillus reuteri, Clostridioides difficile | 13 | |
| Emergencia timonensis | 01552 | erm | 16 | 01558 | |
| Negativibacillus massiliensis | 02069 | erm | 16 | 02058 | |
| Intestinibacillus timonensis | 01053 | erm | 20 | ||
| Sanamassiliae timonensis | 00487 | erm | 16 | 00477 | |
| Neobittarella massiliensis | 00011 | erm | 24 | ||
| Psychrobacter timonensis | 02207 | aac | Escherichia coli | 5 | 02201 |
| Desnuesiella massiliensis | 01710 | ant | S. aureus | 5 | 01706 |
| Malibacterium massiliense | 00202 | tet | Lawsonia intracellularis | 10 | |
| Phocibacillus massiliensis | 01825 * | tet | 5 | ||
| Clostridium phoceensis | 03063 * | tet | 11 | ||
| Megasphaera vaginalis | 01258 | tet | Clostridiodes difficile | 14 | |
| Emergencia timonensis | 03403 | tet | Vibrio sp., E. faecalis Clostridium septicum | 12 | 03398 |
| Bariatricus massiliensis | 00013 * | aad | Plasmid in Campylobacter jejuni | 9 | |
| Neglecta timonensis | 01846 | erm | Lysinibacillus sphaericus,Bacteroides thetaiotaomicron,Bacterioides ovatus | 26 | 01848_01850 |
| Megamonas massiliensis | 00045 | lnu | Streptococcus agalactiae | 2 | |
| Megamonas massiliensis | 00845 | lnu | 2 | ||
| Megamonas massiliensis | 01002 | lnu | 2 | ||
| Megamonas massiliensis | 01006 | lnu | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khabthani, S.; Rolain, J.-M.; Merhej, V. Whole Genome Analysis of 335 New Bacterial Species from Human Microbiota Reveals a Huge Reservoir of Transferable Antibiotic Resistance Determinants. Int. J. Mol. Sci. 2022, 23, 2137. https://doi.org/10.3390/ijms23042137
Khabthani S, Rolain J-M, Merhej V. Whole Genome Analysis of 335 New Bacterial Species from Human Microbiota Reveals a Huge Reservoir of Transferable Antibiotic Resistance Determinants. International Journal of Molecular Sciences. 2022; 23(4):2137. https://doi.org/10.3390/ijms23042137
Chicago/Turabian StyleKhabthani, Sami, Jean-Marc Rolain, and Vicky Merhej. 2022. "Whole Genome Analysis of 335 New Bacterial Species from Human Microbiota Reveals a Huge Reservoir of Transferable Antibiotic Resistance Determinants" International Journal of Molecular Sciences 23, no. 4: 2137. https://doi.org/10.3390/ijms23042137
APA StyleKhabthani, S., Rolain, J.-M., & Merhej, V. (2022). Whole Genome Analysis of 335 New Bacterial Species from Human Microbiota Reveals a Huge Reservoir of Transferable Antibiotic Resistance Determinants. International Journal of Molecular Sciences, 23(4), 2137. https://doi.org/10.3390/ijms23042137