
The Role of the Lymphatic System in the Pathogenesis and Treatment of Inflammatory Bowel Disease

 ,
,
Abstract
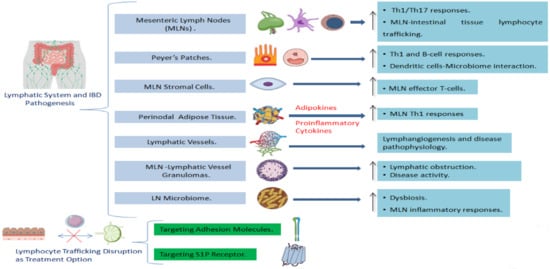
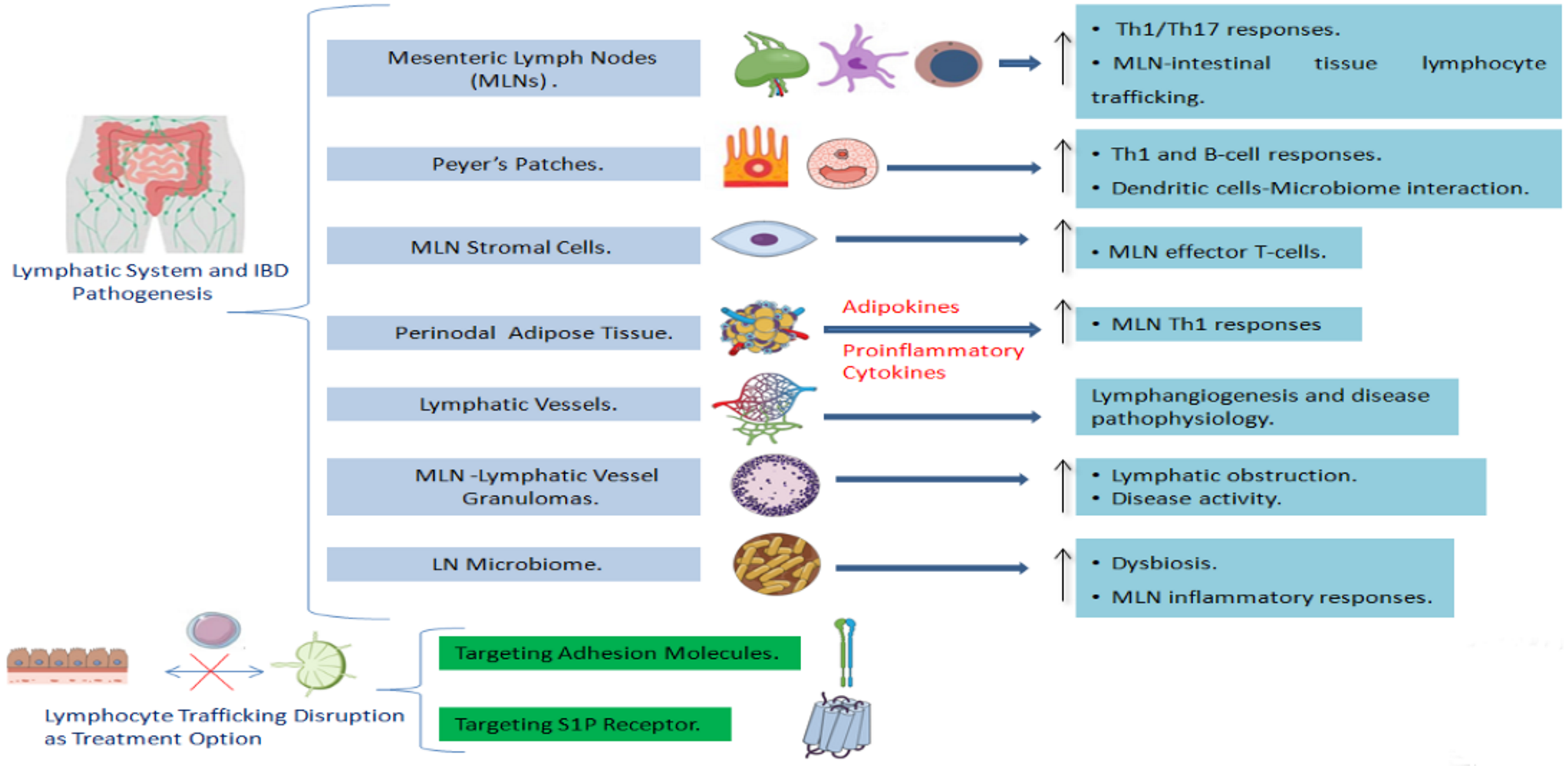{kind=link}
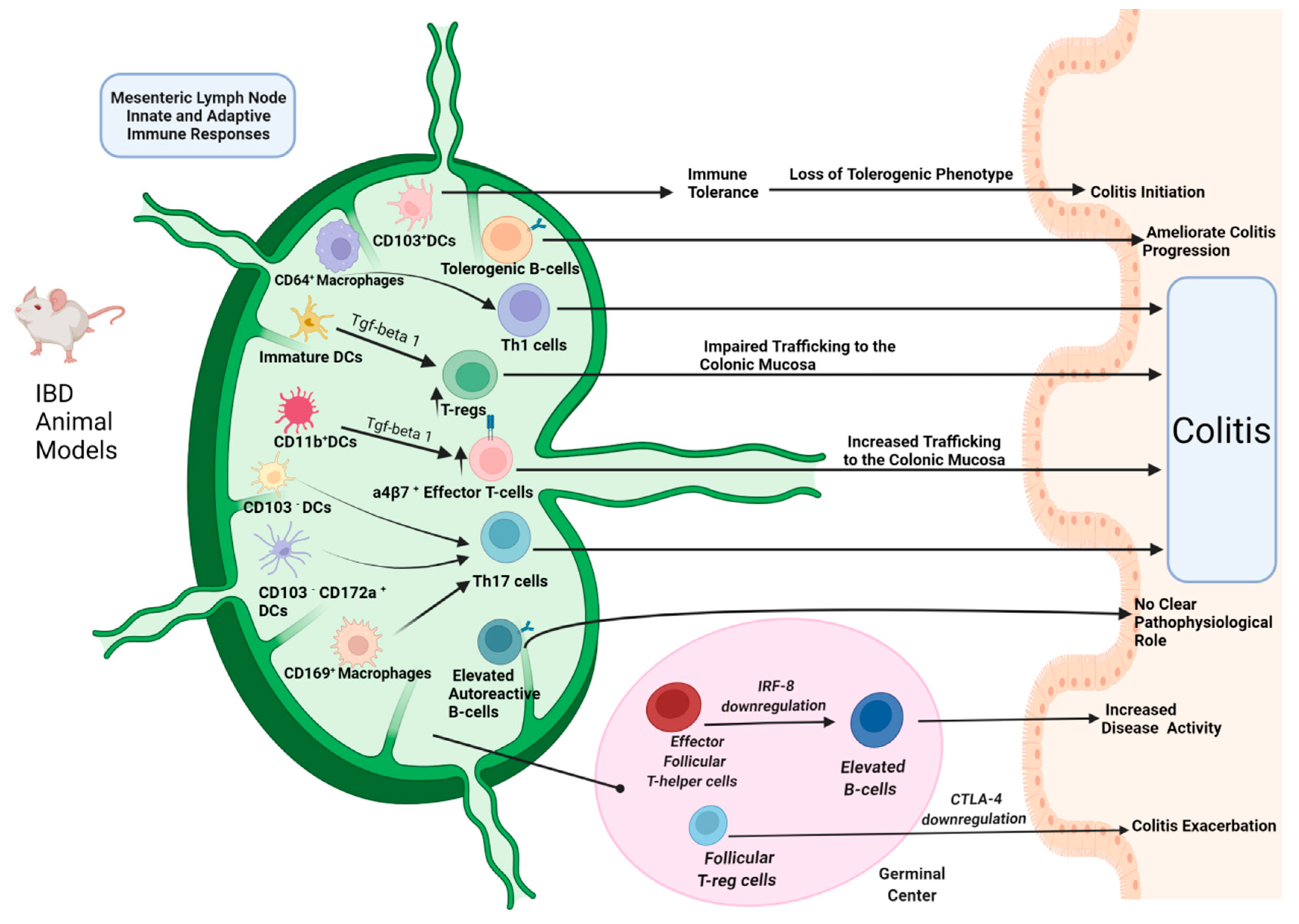{kind=link}
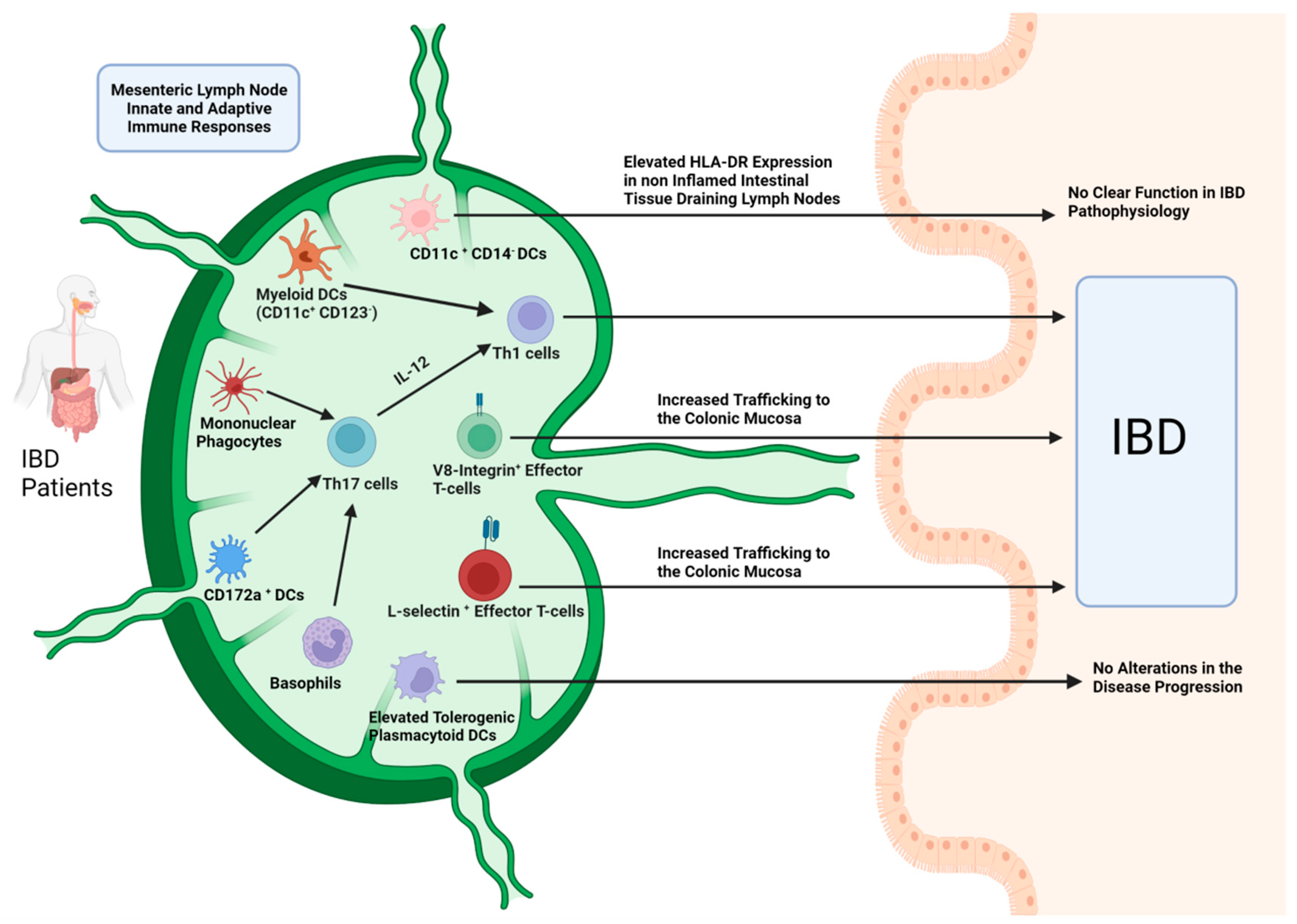{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. The Involvement of the Lymph Nodes in IBD
2.1.1. Innate Immune Responses
The Role of Mesenteric Lymph Node Dendritic Cells
The Role of Mesenteric Lymph Node Macrophages, Mononuclear Phagocytes and Basophils
2.1.2. Adaptive Immune Responses
Mesenteric Lymph Node T-Cell Subtypes Alterations
Mesenteric Lymph Node B-Cell Subtypes Alterations in IBD
2.2. The Involvement of Peyer’s Patches, and Colonic and Cecal Patches in IBD
2.3. The Involvement of the Lymphatic System Stromal Components in IBD
2.3.1. Lymph Node Stromal Cells and IBD
2.3.2. Adipose Tissue and Lymphatics in IBD: The Role of Perinodal Adipose Tissue
2.3.3. The Role of Lymphatic Vessels and Lymphatic Endothelium Alterations in IBD Pathogenesis
2.4. Granulomas in Mesenteric Lymph Nodes and Lymphatic Vessels as Indication of Crohn’s Disease
2.5. The Interaction between the Microbiome and the Lymphatic System in IBD
2.6. Lymphocyte Trafficking as a Potential Target for IBD Treatment
2.6.1. Adhesion Molecule Targeting as a Treatment Option
2.6.2. Targeting S1P Receptor as a Novel Therapeutic Approach
3. Discussion
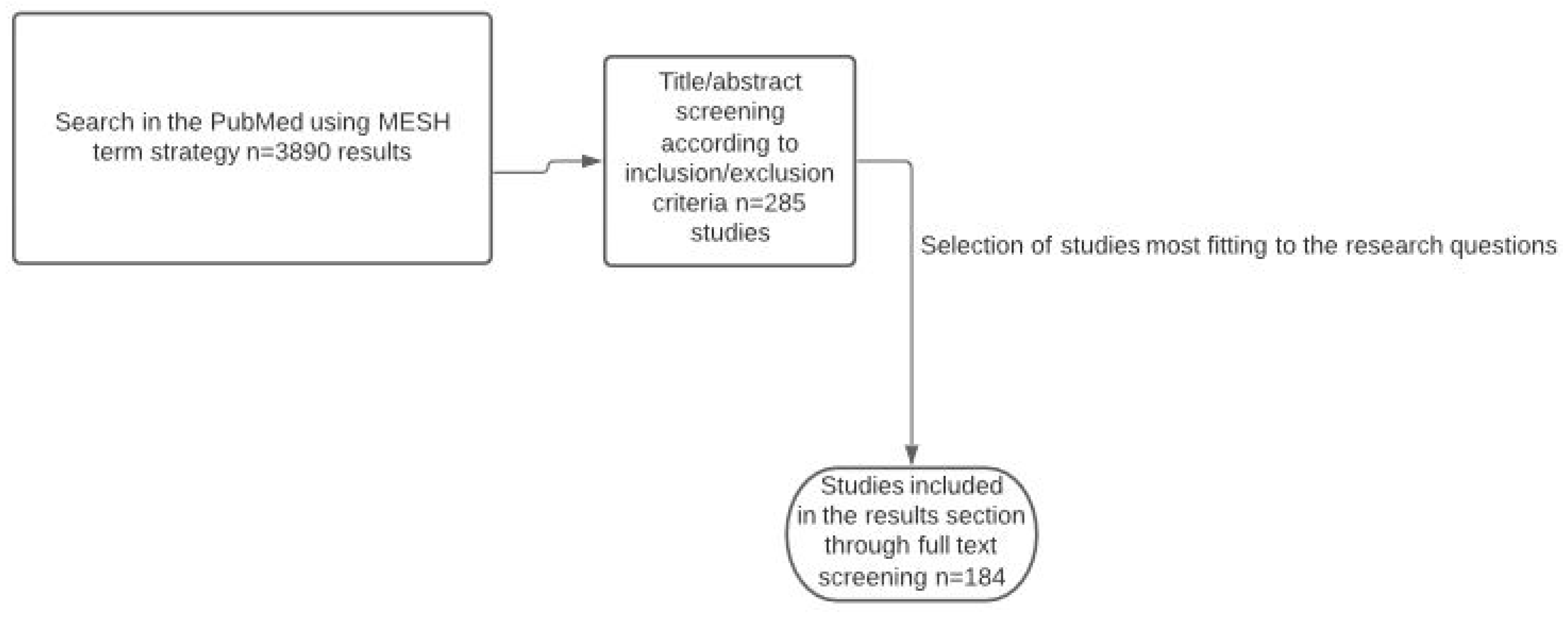
4. Materials and Methods
- What is the role of the adaptive and innate immune cells, as well as, stroma (including lymphatic endothelium) located in lymphoid tissues, regarding the pathogenesis of IBD (Crohn’s disease or ulcerative colitis)?
- What is the role of lymphatic obstruction in disease initiation and progression?
- Is there an interaction between the lymphoid tissues and the microbiome in relation to IBD pathogenesis?
- Which are the treatment options and targets interfering with lymphocyte trafficking in the GALT?
Inclusion and Exclusion Criteria
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
References
- Xavier, R.J.; Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Cook, M.G. The size and histological appearances of mesenteric lymph nodes in Crohn’s disease. Gut 1972, 13, 970–972. [Google Scholar] [CrossRef] [PubMed]
- Radmard, A.R.; Eftekhar Vaghefi, R.; Montazeri, S.A.; Naybandi Atashi, S.; Hashemi Taheri, A.P.; Haghighi, S.; Salehnia, A.; Dadgostar, M.; Malekzadeh, R. Mesenteric lymph nodes in MR enterography: Are they reliable followers of bowel in active Crohn’s disease? Eur. Radiol. 2018, 28, 4429–4437. [Google Scholar] [CrossRef]
- Gourtsoyianni, S.; Papanikolaou, N.; Amanakis, E.; Bourikas, L.; Roussomoustakaki, M.; Grammatikakis, J.; Gourtsoyiannis, N. Crohn’s disease lymphadenopathy: MR imaging findings. Eur. J. Radiol. 2009, 69, 425–428. [Google Scholar] [CrossRef] [PubMed]
- Maconi, G.; Di Sabatino, A.; Ardizzone, S.; Greco, S.; Colombo, E.; Russo, A.; Cassinotti, A.; Casini, V.; Corazza, G.R.; Bianchi Porro, G. Prevalence and clinical significance of sonographic detection of enlarged regional lymph nodes in Crohn’s disease. Scand. J. Gastroenterol. 2005, 40, 1328–1333. [Google Scholar] [CrossRef] [PubMed]
- Koboziev, I.; Karlsson, F.; Grisham, M.B. Gut-associated lymphoid tissue, T cell trafficking, and chronic intestinal inflammation. Ann. N. Y. Acad. Sci. 2010, 1207 (Suppl. 1), E86–E93. [Google Scholar] [CrossRef]
- Carlsen, H.S.; Baekkevold, E.S.; Johansen, F.E.; Haraldsen, G.; Brandtzaeg, P. B cell attracting chemokine 1 (CXCL13) and its receptor CXCR5 are expressed in normal and aberrant gut associated lymphoid tissue. Gut 2002, 51, 364–371. [Google Scholar] [CrossRef]
- Lenskaya, V.; Panji, P.; de Moll, E.H.; Christian, K.; Phelps, R.G. Oral lymphangiectasia and gastrointestinal Crohn disease. J. Cutan. Pathol. 2020, 47, 1080–1084. [Google Scholar] [CrossRef]
- Rahier, J.F.; De Beauce, S.; Dubuquoy, L.; Erdual, E.; Colombel, J.F.; Jouret-Mourin, A.; Geboes, K.; Desreumaux, P. Increased lymphatic vessel density and lymphangiogenesis in inflammatory bowel disease. Aliment Pharmacol. Ther. 2011, 34, 533–543. [Google Scholar] [CrossRef]
- Kovi, J.; Duong, H.D.; Hoang, C.T. Ultrastructure of intestinal lymphatics in Crohn’s disease. Am. J. Clin. Pathol. 1981, 76, 385–394. [Google Scholar] [CrossRef]
- Takesue, Y.; Ohge, H.; Uemura, K.; Imamura, Y.; Murakami, Y.; Yokoyama, T.; Kakehashi, M.; Sueda, T. Bacterial translocation in patients with Crohn’s disease undergoing surgery. Dis. Colon Rectum. 2002, 45, 1665–1671. [Google Scholar] [CrossRef]
- Diehl, G.E.; Longman, R.S.; Zhang, J.X.; Breart, B.; Galan, C.; Cuesta, A.; Schwab, S.R.; Littman, D.R. Microbiota restricts trafficking of bacteria to mesenteric lymph nodes by CX(3)CR1(hi) cells. Nature 2013, 494, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Baert, F.; Danese, S.; Krznarić, Ž.; Kobayashi, T.; Yao, X.; Chen, J.; Rosario, M.; Bhatia, S.; Kisfalvi, K.; et al. Efficacy and Safety of Vedolizumab Subcutaneous Formulation in a Randomized Trial of Patients with Ulcerative Colitis. Gastroenterology 2020, 158, 562–572.e12. [Google Scholar] [CrossRef] [PubMed]
- Feagan, B.G.; Sandborn, W.J.; Danese, S.; Wolf, D.C.; Liu, W.J.; Hua, S.Y.; Minton, N.; Olson, A.; D’Haens, G. Ozanimod induction therapy for patients with moderate to severe Crohn’s disease: A single-arm, phase 2, prospective observer-blinded endpoint study. Lancet Gastroenterol. Hepatol. 2020, 5, 819–828. [Google Scholar] [CrossRef]
- Stagg, A.J. Intestinal Dendritic Cells in Health and Gut Inflammation. Front. Immunol. 2018, 9, 2883. [Google Scholar] [CrossRef] [PubMed]
- Grainger, J.R.; Konkel, J.E.; Zangerle-Murray, T.; Shaw, T.N. Macrophages in gastrointestinal homeostasis and inflammation. Pflug. Arch. 2017, 469, 527–539. [Google Scholar] [CrossRef]
- Verstege, M.I.; ten Kate, F.J.; Reinartz, S.M.; van Drunen, C.M.; Slors, F.J.; Bemelman, W.A.; Vyth-Dreese, F.A.; te Velde, A.A. Dendritic cell populations in colon and mesenteric lymph nodes of patients with Crohn’s disease. J. Histochem. Cytochem. 2008, 56, 233–241. [Google Scholar] [CrossRef]
- Ishiguro, Y.; Sakuraba, H.; Yamagata, K.; Munakata, A. The presentation of haptenated proteins and activation of T cells in the mesenteric lymph nodes by dendritic cells in the TNBS colitis rat. Ann. N. Y. Acad. Sci. 2004, 1029, 346–347. [Google Scholar] [CrossRef]
- Sakuraba, A.; Sato, T.; Kamada, N.; Kitazume, M.; Sugita, A.; Hibi, T. Th1/Th17 immune response is induced by mesenteric lymph node dendritic cells in Crohn’s disease. Gastroenterology 2009, 137, 1736–1745. [Google Scholar] [CrossRef]
- Kourepini, E.; Aggelakopoulou, M.; Alissafi, T.; Paschalidis, N.; Simoes, D.C.; Panoutsakopoulou, V. Osteopontin expression by CD103-dendritic cells drives intestinal inflammation. Proc. Natl. Acad. Sci. USA 2014, 111, E856–E865. [Google Scholar] [CrossRef]
- Fortin, G.; Raymond, M.; Van, V.Q.; Rubio, M.; Gautier, P.; Sarfati, M.; Franchimont, D. A role for CD47 in the development of experimental colitis mediated by SIRPalpha+CD103- dendritic cells. J. Exp. Med. 2009, 206, 1995–2011. [Google Scholar] [CrossRef]
- Baba, N.; Van, V.Q.; Wakahara, K.; Rubio, M.; Fortin, G.; Panzini, B.; Soucy, G.; Wassef, R.; Richard, C.; Tamaz, R.; et al. CD47 fusion protein targets CD172a+ cells in Crohn’s disease and dampens the production of IL-1β and TNF. J. Exp. Med. 2013, 210, 1251–1263. [Google Scholar] [CrossRef]
- Bsat, M.; Chapuy, L.; Baba, N.; Rubio, M.; Panzini, B.; Wassef, R.; Richard, C.; Soucy, G.; Mehta, H.; Sarfati, M. Differential accumulation and function of proinflammatory 6-sulfo LacNAc dendritic cells in lymph node and colon of Crohn’s versus ulcerative colitis patients. J. Leukoc. Biol. 2015, 98, 671–681. [Google Scholar] [CrossRef]
- Mann, E.R.; Bernardo, D.; Ng, S.C.; Rigby, R.J.; Al-Hassi, H.O.; Landy, J.; Peake, S.T.; Spranger, H.; English, N.R.; Thomas, L.V.; et al. Human gut dendritic cells drive aberrant gut-specific t-cell responses in ulcerative colitis, characterized by increased IL-4 production and loss of IL-22 and IFNγ. Inflamm. Bowel Dis. 2014, 20, 2299–2307. [Google Scholar] [CrossRef] [PubMed]
- Jaensson, E.; Uronen-Hansson, H.; Pabst, O.; Eksteen, B.; Tian, J.; Coombes, J.L.; Berg, P.L.; Davidsson, T.; Powrie, F.; Johansson-Lindbom, B.; et al. Small intestinal CD103+ dendritic cells display unique functional properties that are conserved between mice and humans. J. Exp. Med. 2008, 205, 2139–2149. [Google Scholar] [CrossRef] [PubMed]
- Laffont, S.; Siddiqui, K.R.; Powrie, F. Intestinal inflammation abrogates the tolerogenic properties of MLN CD103+ dendritic cells. Eur. J. Immunol. 2010, 40, 1877–1883. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, M.K.; Brynjólfsson, S.F.; Dige, A.; Uronen-Hansson, H.; Börjesson, L.G.; Bengtsson, J.L.; Gudjonsson, S.; Öhman, L.; Agnholt, J.; Sjövall, H.; et al. Macrophage and dendritic cell subsets in IBD: ALDH+ cells are reduced in colon tissue of patients with ulcerative colitis regardless of inflammation. Mucosal Immunol. 2016, 9, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, D.C.; Metzke, D.; Guckelberger, O.; Pascher, A.; Grötzinger, C.; Przesdzing, I.; Dörffel, Y.; Schmitz, J.; Thomas, S. Aberrant plasmacytoid dendritic cell distribution and function in patients with Crohn’s disease and ulcerative colitis. Clin. Exp. Immunol. 2011, 166, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Hostmann, A.; Kapp, K.; Beutner, M.; Ritz, J.P.; Loddenkemper, C.; Ignatius, R.; Duchmann, R.; Daum, S.; Gröne, J.; Hotz, H.; et al. Dendritic cells from human mesenteric lymph nodes in inflammatory and non-inflammatory bowel diseases: Subsets and function of plasmacytoid dendritic cells. Immunology 2013, 139, 100–108. [Google Scholar] [CrossRef]
- Do, J.S.; Visperas, A.; Freeman, M.L.; Iwakura, Y.; Oukka, M.; Min, B. Colitogenic effector T cells: Roles of gut-homing integrin, gut antigen specificity and γδ T cells. Immunol. Cell Biol. 2014, 92, 90–98. [Google Scholar] [CrossRef]
- Cai, Z.; Zhang, W.; Li, M.; Yue, Y.; Yang, F.; Yu, L.; Cao, X.; Wang, J. TGF-beta1 gene-modified, immature dendritic cells delay the development of inflammatory bowel disease by inducing CD4(+)Foxp3(+) regulatory T cells. Cell. Mol. Immunol. 2010, 7, 35–43, Erratum in Cell. Mol. Immunol. 2020, 17, 190–191. [Google Scholar] [CrossRef] [PubMed]
- Powrie, F.; Leach, M.W.; Mauze, S.; Menon, S.; Caddle, L.B.; Coffman, R.L. Inhibition of Th1 responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity 1994, 1, 553–562. [Google Scholar] [CrossRef]
- Yen, D.; Cheung, J.; Scheerens, H.; Poulet, F.; McClanahan, T.; McKenzie, B.; Kleinschek, M.A.; Owyang, A.; Mattson, J.; Blumenschein, W.; et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J. Clin. Investig. 2006, 116, 1310–1316. [Google Scholar] [CrossRef] [PubMed]
- Elson, C.O.; Cong, Y.; Weaver, C.T.; Schoeb, T.R.; McClanahan, T.K.; Fick, R.B.; Kastelein, R.A. Monoclonal anti-interleukin 23 reverses active colitis in a T cell-mediated model in mice. Gastroenterology 2007, 132, 2359–2370. [Google Scholar] [CrossRef] [PubMed]
- Feng, T.; Qin, H.; Wang, L.; Benveniste, E.N.; Elson, C.O.; Cong, Y. Th17 cells induce colitis and promote Th1 cell responses through IL-17 induction of innate IL-12 and IL-23 production. J. Immunol. 2011, 186, 6313–6318. [Google Scholar] [CrossRef] [PubMed]
- Tamoutounour, S.; Henri, S.; Lelouard, H.; de Bovis, B.; de Haar, C.; van der Woude, C.J.; Woltman, A.M.; Reyal, Y.; Bonnet, D.; Sichien, D.; et al. CD64 distinguishes macrophages from dendritic cells in the gut and reveals the Th1-inducing role of mesenteric lymph node macrophages during colitis. Eur. J. Immunol. 2012, 42, 3150–3166. [Google Scholar] [CrossRef] [PubMed]
- Chávez-Galán, L.; Olleros, M.L.; Vesin, D.; Garcia, I. Much More than M1 and M2 Macrophages, There are also CD169(+) and TCR(+) Macrophages. Front. Immunol. 2015, 6, 263. [Google Scholar] [CrossRef]
- Oetke, C.; Vinson, M.C.; Jones, C.; Crocker, P.R. Sialoadhesin-deficient mice exhibit subtle changes in B- and T-cell populations and reduced immunoglobulin M levels. Mol. Cell. Biol. 2006, 26, 1549–1557. [Google Scholar] [CrossRef]
- Li, Q.; Wang, D.; Hao, S.; Han, X.; Xia, Y.; Li, X.; Chen, Y.; Tanaka, M.; Qiu, C.H. CD169 Expressing Macrophage, a Key Subset in Mesenteric Lymph Nodes Promotes Mucosal Inflammation in Dextran Sulfate Sodium-Induced Colitis. Front. Immunol. 2017, 8, 669. [Google Scholar] [CrossRef]
- Chapuy, L.; Bsat, M.; Rubio, M.; Harvey, F.; Motta, V.; Schwenter, F.; Wassef, R.; Richard, C.; Deslandres, C.; Nguyen, B.N.; et al. Transcriptomic Analysis and High-dimensional Phenotypic Mapping of Mononuclear Phagocytes in Mesenteric Lymph Nodes Reveal Differences between Ulcerative Colitis and Crohn’s Disease. J. Crohns Colitis 2020, 14, 393–405. [Google Scholar] [CrossRef]
- Chapuy, L.; Bsat, M.; Mehta, H.; Rubio, M.; Wakahara, K.; Van, V.Q.; Baba, N.; Cheong, C.; Yun, T.J.; Panzini, B.; et al. Basophils increase in Crohn disease and ulcerative colitis and favor mesenteric lymph node memory TH17/TH1 response. J. Allergy Clin. Immunol. 2014, 134, 978–981.e1. [Google Scholar] [CrossRef] [PubMed]
- Fell, J.M.; Walker-Smith, J.A.; Spencer, J.; MacDonald, T.T. The distribution of dividing T cells throughout the intestinal wall in inflammatory bowel disease (IBD). Clin. Exp. Immunol. 1996, 104, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Guillou, P.J.; Brennan, T.G.; Giles, G.R. Lymphocyte transformation in the mesenteric lymph nodes of patients with Crohn’s disease. Gut 1973, 14, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Bsat, M.; Chapuy, L.; Rubio, M.; Wassef, R.; Richard, C.; Schwenter, F.; Loungnarath, R.; Soucy, G.; Mehta, H.; Sarfati, M. Differential Pathogenic Th17 Profile in Mesenteric Lymph Nodes of Crohn’s Disease and Ulcerative Colitis Patients. Front. Immunol. 2019, 10, 1177. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Guo, R.; Luo, X.; Zhou, L. Ethyl pyruvate ameliorates experimental colitis in mice by inhibiting the HMGB1-Th17 and Th1/Tc1 responses. Int. Immunopharmacol. 2015, 29, 454–461. [Google Scholar] [CrossRef]
- Sasaoka, T.; Ito, M.; Yamashita, J.; Nakajima, K.; Tanaka, I.; Narita, M.; Hara, Y.; Hada, K.; Takahashi, M.; Ohno, Y.; et al. Treatment with IL-27 attenuates experimental colitis through the suppression of the development of IL-17-producing T helper cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G568–G576. [Google Scholar] [CrossRef]
- Bai, A.; Ma, A.G.; Yong, M.; Weiss, C.R.; Ma, Y.; Guan, Q.; Bernstein, C.N.; Peng, Z. AMPK agonist downregulates innate and adaptive immune responses in TNBS-induced murine acute and relapsing colitis. Biochem. Pharmacol. 2010, 80, 1708–1717. [Google Scholar] [CrossRef]
- Igaki, K.; Nakamura, Y.; Komoike, Y.; Uga, K.; Shibata, A.; Ishimura, Y.; Yamasaki, M.; Tsukimi, Y.; Tsuchimori, N. Pharmacological Evaluation of TAK-828F, a Novel Orally Available RORγt Inverse Agonist, on Murine Colitis Model. Inflammation 2019, 42, 91–102. [Google Scholar] [CrossRef]
- Guan, Q.; Ma, Y.; Hillman, C.L.; Qing, G.; Ma, A.G.; Weiss, C.R.; Zhou, G.; Bai, A.; Warrington, R.J.; Bernstein, C.N.; et al. Targeting IL-12/IL-23 by employing a p40 peptide-based vaccine ameliorates TNBS-induced acute and chronic murine colitis. Mol. Med. 2011, 17, 646–656. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, H.; Liu, L.; Li, H.; Shih, D.Q.; Zhang, X. 1,25-dihydroxyvitamin D3 regulates the development of chronic colitis by modulating both T helper (Th)1 and Th17 activation. APMIS 2015, 123, 490–501. [Google Scholar] [CrossRef]
- Hu, S.; Chen, M.; Wang, Y.; Wang, Z.; Pei, Y.; Fan, R.; Liu, X.; Wang, L.; Zhou, J.; Zheng, S.; et al. mTOR Inhibition Attenuates Dextran Sulfate Sodium-Induced Colitis by Suppressing T Cell Proliferation and Balancing TH1/TH17/Treg Profile. PLoS ONE 2016, 11, e0154564, Erratum in: PLoS ONE 2016, 11, e0159758. [Google Scholar] [CrossRef]
- Liu, X.J.; Yu, R.; Zou, K.F. Probiotic Mixture VSL#3 Alleviates Dextran Sulfate Sodium-induced Colitis in Mice by Downregulating T Follicular Helper Cells. Curr. Med. Sci. 2019, 39, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Qi, C.F.; Hu, Y.; Shan, Y.; Hsieh, Y.P.; Xu, F.; Lu, G.; Dai, J.; Gupta, M.; Cui, M.; et al. T follicular helper cells restricted by IRF8 contribute to T cell-mediated inflammation. J. Autoimmun. 2019, 96, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Chao, G.; Li, X.; Ji, Y.; Zhu, Y.; Li, N.; Zhang, N.; Feng, Z.; Niu, M. CTLA-4 regulates T follicular regulatory cell differentiation and participates in intestinal damage caused by spontaneous autoimmunity. Biochem. Biophys. Res. Commun. 2018, 505, 865–871. [Google Scholar] [CrossRef]
- Chao, G.; Li, X.; Ji, Y.; Zhu, Y.; Li, N.; Zhang, N.; Feng, Z.; Niu, M. MiR-155 controls follicular Treg cell-mediated humoral autoimmune intestinal injury by inhibiting CTLA-4 expression. Int. Immunopharmacol. 2019, 71, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Posnett, D.N.; Schmelkin, I.; Burton, D.A.; August, A.; McGrath, H.; Mayer, L.F. T cell antigen receptor V gene usage. Increases in V beta 8+ T cells in Crohn’s disease. J. Clin. Investig. 1990, 85, 1770–1776. [Google Scholar] [CrossRef]
- Szczepanik, M.; Gryglewski, A.; Bryniarski, K.; Stachura, J.; Ptak, W. Experimental inflammatory bowel disease—Role of T cells. J. Physiol. Pharmacol. 2000, 51, 333–346. [Google Scholar]
- Rudolphi, A.; Boll, G.; Poulsen, S.S.; Claesson, M.H.; Reimann, J. Gut-homing CD4+ T cell receptor alpha beta+ T cells in the pathogenesis of murine inflammatory bowel disease. Eur. J. Immunol. 1994, 24, 2803–2812. [Google Scholar] [CrossRef]
- Eberhardson, M.; Lindberg, K.; Karlsson, M.; Karlén, P.; Winqvist, O.; Thörn, M. The sentinel node technique is useful for studies of intestinal immunology in inflammatory bowel disease patients. Eur. J. Gastroenterol. Hepatol. 2008, 20, 985–988. [Google Scholar] [CrossRef]
- Papadakis, K.A.; Prehn, J.; Zhu, D.; Landers, C.; Gaiennie, J.; Fleshner, P.R.; Targan, S.R. Expression and regulation of the chemokine receptor CXCR3 on lymphocytes from normal and inflammatory bowel disease mucosa. Inflamm. Bowel Dis. 2004, 10, 778–788. [Google Scholar] [CrossRef]
- Hyun, J.G.; Lee, G.; Brown, J.B.; Grimm, G.R.; Tang, Y.; Mittal, N.; Dirisina, R.; Zhang, Z.; Fryer, J.P.; Weinstock, J.V.; et al. Anti-interferon-inducible chemokine, CXCL10, reduces colitis by impairing T helper-1 induction and recruitment in mice. Inflamm. Bowel Dis. 2005, 11, 799–805. [Google Scholar] [CrossRef]
- Schneider, M.A.; Meingassner, J.G.; Lipp, M.; Moore, H.D.; Rot, A. CCR7 is required for the in vivo function of CD4+ CD25+ regulatory T cells. J. Exp. Med. 2007, 204, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Richens, E.R.; Thorp, C.M.; Bland, P.W.; Gough, K.R. Peripheral blood and mesenteric lymph node lymphocytes in Crohn’s disease. Gut 1980, 21, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Meijer, C.J.; Bosman, F.T.; Lindeman, J. Evidence for predominant involvement of the B-cell system in the inflammatory process in Crohn’s disease. Scand. J. Gastroenterol. 1979, 14, 21–32. [Google Scholar] [CrossRef]
- Takagi, S.; Kinouchi, Y.; Chida, M.; Hiwatashi, N.; Noguchi, M.; Takahashi, S.; Shimosegawa, T. Strong telomerase activity of B lymphocyte from mesenteric lymph nodes of patients with inflammatory bowel disease. Dig. Dis. Sci. 2003, 48, 2091–2094. [Google Scholar] [CrossRef] [PubMed]
- Tabibian-Keissar, H.; Zuckerman, N.S.; Barak, M.; Dunn-Walters, D.K.; Steiman-Shimony, A.; Chowers, Y.; Ofek, E.; Rosenblatt, K.; Schiby, G.; Mehr, R.; et al. B-cell clonal diversification and gut-lymph node trafficking in ulcerative colitis revealed using lineage tree analysis. Eur. J. Immunol. 2008, 38, 2600–2609. [Google Scholar] [CrossRef]
- Mizoguchi, E.; Mizoguchi, A.; Chiba, C.; Niles, J.L.; Bhan, A.K. Antineutrophil cytoplasmic antibodies in T-cell receptor alpha-deficient mice with chronic colitis. Gastroenterology 1997, 113, 1828–1835. [Google Scholar] [CrossRef]
- Mizoguchi, A.; Mizoguchi, E.; Chiba, C.; Spiekermann, G.M.; Tonegawa, S.; Nagler-Anderson, C.; Bhan, A.K. Cytokine imbalance and autoantibody production in T cell receptor-alpha mutant mice with inflammatory bowel disease. J. Exp. Med. 1996, 183, 847–856. [Google Scholar] [CrossRef]
- Das, K.M.; Dasgupta, A.; Mandal, A.; Geng, X. Autoimmunity to cytoskeletal protein tropomyosin. A clue to the pathogenetic mechanism for ulcerative colitis. J. Immunol. 1993, 150, 2487–2493. [Google Scholar]
- Ebert, E.C.; Geng, X.; Lin, J.; Das, K.M. Autoantibodies against human tropomyosin isoform 5 in ulcerative colitis destroys colonic epithelial cells through antibody and complement-mediated lysis. Cell. Immunol. 2006, 244, 43–49. [Google Scholar] [CrossRef]
- Wei, B.; Velazquez, P.; Turovskaya, O.; Spricher, K.; Aranda, R.; Kronenberg, M.; Birnbaumer, L.; Braun, J. Mesenteric B cells centrally inhibit CD4+ T cell colitis through interaction with regulatory T cell subsets. Proc. Natl. Acad. Sci. USA 2005, 102, 2010–2015. [Google Scholar] [CrossRef]
- Saha, C.; Nigam, S.K.; Denker, B.M. Involvement of Galphai2 in the maintenance and biogenesis of epithelial cell tight junctions. J. Biol. Chem. 1998, 273, 21629–21633. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, A.; Mizoguchi, E.; Takedatsu, H.; Blumberg, R.S.; Bhan, A.K. Chronic intestinal inflammatory condition generates IL-10-producing regulatory B cell subset characterized by CD1d upregulation. Immunity 2002, 16, 219–230. [Google Scholar] [CrossRef]
- Takahashi, I.; Kiyono, H.; Hamada, S. CD4+ T-cell population mediates development of inflammatory bowel disease in T-cell receptor alpha chain-deficient mice. Gastroenterology 1997, 112, 1876–1886. [Google Scholar] [CrossRef]
- Takahashi, S.; Kawamura, T.; Kanda, Y.; Taniguchi, T.; Nishizawa, T.; Iiai, T.; Hatakeyama, K.; Abo, T. Multipotential acceptance of Peyer’s patches in the intestine for both thymus-derived T cells and extrathymic T cells in mice. Immunol. Cell Biol. 2005, 83, 504–510. [Google Scholar] [CrossRef]
- Salim, S.Y.; Silva, M.A.; Keita, A.V.; Larsson, M.; Andersson, P.; Magnusson, K.E.; Perdue, M.H.; Söderholm, J.D. CD83+CCR7− dendritic cells accumulate in the subepithelial dome and internalize translocated Escherichia coli HB101 in the Peyer’s patches of ileal Crohn’s disease. Am. J. Pathol. 2009, 174, 82–90. [Google Scholar] [CrossRef]
- Chassaing, B.; Etienne-Mesmin, L.; Bonnet, R.; Darfeuille-Michaud, A. Bile salts induce long polar fimbriae expression favouring Crohn’s disease-associated adherent-invasive Escherichia coli interaction with Peyer’s patches. Environ. Microbiol. 2013, 15, 355–371. [Google Scholar] [CrossRef]
- Larabi, A.; Salesse, L.; Cordonnier, C.; Etienne-Mesmin, L.; Barnich, N.; Dalmasso, G.; Nguyen, H.T.T. Differential miRNA-Gene Expression in M Cells in Response to Crohn’s Disease-Associated AIEC. Microorganisms 2020, 8, 1205. [Google Scholar] [CrossRef] [PubMed]
- Vazeille, E.; Chassaing, B.; Buisson, A.; Dubois, A.; de Vallée, A.; Billard, E.; Neut, C.; Bommelaer, G.; Colombel, J.F.; Barnich, N.; et al. GipA Factor Supports Colonization of Peyer’s Patches by Crohn’s Disease-associated Escherichia Coli. Inflamm. Bowel Dis. 2016, 22, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Kudo, T.; Nagata, S.; Aoyagi, Y.; Suzuki, R.; Matsuda, H.; Ohtsuka, Y.; Shimizu, T.; Okumura, K.; Yamashiro, Y. Polarized production of T-helper cell type 1 cells in Peyer’s patches in Crohn’s disease. Digestion 2004, 70, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Ohman, L.; Franzén, L.; Rudolph, U.; Birnbaumer, L.; Hörnquist, E.H. Regression of Peyer’s patches in G alpha i2 deficient mice prior to colitis is associated with reduced expression of Bcl-2 and increased apoptosis. Gut 2002, 51, 392–397. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ohman, L.; Aström, R.G.; Hultgren Hörnquist, E. Impaired B cell responses to orally administered antigens in lamina propria but not Peyer’s patches of Galphai2-deficient mice prior to colitis. Immunology 2005, 115, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Pararasa, C.; Zhang, N.; Tull, T.J.; Chong, M.H.A.; Siu, J.H.Y.; Guesdon, W.; Chavele, K.M.; Sanderson, J.D.; Langmead, L.; Kok, K.; et al. Reduced CD27-IgD- B Cells in Blood and Raised CD27-IgD- B Cells in Gut-Associated Lymphoid Tissue in Inflammatory Bowel Disease. Front. Immunol. 2019, 10, 361. [Google Scholar] [CrossRef] [PubMed]
- Qian, T.; Hong, J.; Wang, L.; Wang, Z.; Lu, Z.; Li, Y.; Liu, R.; Chu, Y. Regulation of CD11b by HIF-1α and the STAT3 signaling pathway contributes to the immunosuppressive function of B cells in inflammatory bowel disease. Mol. Immunol. 2019, 111, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, S.; Gu, S.; Yamashita, A. Correlation of rectum-associated lymph nodules with the development of experimentally induced acute colonic inflammation in rats. J. Gastroenterol. Hepatol. 2001, 16, 1360–1367. [Google Scholar] [CrossRef]
- Watabe, T.; Nagaishi, T.; Tsugawa, N.; Kojima, Y.; Jose, N.; Hosoya, A.; Onizawa, M.; Nemoto, Y.; Oshima, S.; Nakamura, T.; et al. B cell activation in the cecal patches during the development of an experimental colitis model. Biochem. Biophys. Res. Commun. 2018, 496, 367–373. [Google Scholar] [CrossRef]
- Dohi, T.; Fujihashi, K.; Rennert, P.D.; Iwatani, K.; Kiyono, H.; McGhee, J.R. Hapten-induced colitis is associated with colonic patch hypertrophy and T helper cell 2-type responses. J. Exp. Med. 1999, 189, 1169–1180. [Google Scholar] [CrossRef]
- Gardenbroek, T.J.; Pinkney, T.D.; Sahami, S.; Morton, D.G.; Buskens, C.J.; Ponsioen, C.Y.; Tanis, P.J.; Löwenberg, M.; van den Brink, G.R.; Broeders, I.A.; et al. The ACCURE-trial: The effect of appendectomy on the clinical course of ulcerative colitis, a randomised international multicenter trial (NTR2883) and the ACCURE-UK trial: A randomised external pilot trial (ISRCTN56523019). BMC Surg. 2015, 15, 30, Erratum in BMC Surg. 2016, 16, 1. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, F.C.; Liblau, R.S.; von Boehmer, H.; Pittet, M.J.; Lee, J.W.; Turley, S.J.; Khazaie, K. Direct presentation of antigen by lymph node stromal cells protects against CD8 T-cell-mediated intestinal autoimmunity. Gastroenterology 2008, 134, 1028–1037. [Google Scholar] [CrossRef]
- Basic, M.; Peppermüller, P.P.; Bolsega, S.; Bleich, A.; Bornemann, M.; Bode, U.; Buettner, M. Lymph Node Stromal Cells From Different Draining Areas Distinctly Regulate the Development of Chronic Intestinal Inflammation. Front. Immunol. 2021, 11, 549473. [Google Scholar] [CrossRef]
- Westcott, E.D.; Mattacks, C.A.; Windsor, A.C.; Knight, S.C.; Pond, C.M. Perinodal adipose tissue and fatty acid composition of lymphoid tissues in patients with and without Crohn’s disease and their implications for the etiology and treatment of CD. Ann. N. Y. Acad. Sci. 2006, 1072, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Westcott, E.; Windsor, A.; Mattacks, C.; Pond, C.; Knight, S. Fatty acid compositions of lipids in mesenteric adipose tissue and lymphoid cells in patients with and without Crohn’s disease and their therapeutic implications. Inflamm. Bowel Dis. 2005, 11, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Gambero, A.; Maróstica, M.; Abdalla Saad, M.J.; Pedrazzoli, J., Jr. Mesenteric adipose tissue alterations resulting from experimental reactivated colitis. Inflamm. Bowel Dis. 2007, 13, 1357–1364. [Google Scholar] [CrossRef] [PubMed]
- Acedo, S.C.; Gotardo, E.M.; Lacerda, J.M.; de Oliveira, C.C.; de Oliveira Carvalho, P.; Gambero, A. Perinodal adipose tissue and mesenteric lymph node activation during reactivated TNBS-colitis in rats. Dig. Dis. Sci. 2011, 56, 2545–2552. [Google Scholar] [CrossRef]
- Schwager, S.; Detmar, M. Inflammation and Lymphatic Function. Front. Immunol. 2019, 10, 308. [Google Scholar] [CrossRef] [PubMed]
- Pedica, F.; Ligorio, C.; Tonelli, P.; Bartolini, S.; Baccarini, P. Lymphangiogenesis in Crohn’s disease: An immunohistochemical study using monoclonal antibody D2-40. Virchows Arch. 2008, 452, 57–63. [Google Scholar] [CrossRef]
- Geleff, S.; Schoppmann, S.F.; Oberhuber, G. Increase in podoplanin-expressing intestinal lymphatic vessels in inflammatory bowel disease. Virchows Arch. 2003, 442, 231–237. [Google Scholar] [CrossRef]
- Fogt, F.; Pascha, T.L.; Zhang, P.J.; Gausas, R.E.; Rahemtulla, A.; Zimmerman, R.L. Proliferation of D2-40-expressing intestinal lymphatic vessels in the lamina propria in inflammatory bowel disease. Int. J. Mol. Med. 2004, 13, 211–214. [Google Scholar] [CrossRef]
- Rehal, S.; Stephens, M.; Roizes, S.; Liao, S.; von der Weid, P.Y. Acute small intestinal inflammation results in persistent lymphatic alterations. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 314, G408–G417. [Google Scholar] [CrossRef]
- Rehal, S.; von der Weid, P.Y. Experimental ileitis alters prostaglandin biosynthesis in mesenteric lymphatic and blood vessels. Prostaglandins Other Lipid Mediat. 2015, 116, 37–48. [Google Scholar] [CrossRef]
- Becker, F.; Potepalov, S.; Shehzahdi, R.; Bernas, M.; Witte, M.; Abreo, F.; Traylor, J.; Orr, W.A.; Tsunoda, I.; Alexander, J.S. Downregulation of FoxC2 Increased Susceptibility to Experimental Colitis: Influence of Lymphatic Drainage Function? Inflamm. Bowel Dis. 2015, 21, 1282–1296. [Google Scholar] [CrossRef] [PubMed]
- Mathias, R.; von der Weid, P.Y. Involvement of the NO-cGMP-K(ATP) channel pathway in the mesenteric lymphatic pump dysfunction observed in the guinea pig model of TNBS-induced ileitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G623–G634. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wu, T.F.; Carati, C.J.; Macnaughton, W.K.; von der Weid, P.Y. Contractile activity of lymphatic vessels is altered in the TNBS model of guinea pig ileitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 291, G566–G574. [Google Scholar] [CrossRef] [PubMed]
- Horjus Talabur Horje, C.S.; Smids, C.; Meijer, J.W.; Groenen, M.J.; Rijnders, M.K.; van Lochem, E.G.; Wahab, P.J. High endothelial venules associated with T cell subsets in the inflamed gut of newly diagnosed inflammatory bowel disease patients. Clin. Exp. Immunol. 2017, 188, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Low, S.; Hirakawa, J.; Hoshino, H.; Uchimura, K.; Kawashima, H.; Kobayashi, M. Role of MAdCAM-1-Expressing High Endothelial Venule-Like Vessels in Colitis Induced in Mice Lacking Sulfotransferases Catalyzing L-Selectin Ligand Biosynthesis. J. Histochem. Cytochem. 2018, 66, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Suzawa, K.; Kobayashi, M.; Sakai, Y.; Hoshino, H.; Watanabe, M.; Harada, O.; Ohtani, H.; Fukuda, M.; Nakayama, J. Preferential induction of peripheral lymph node addressin on high endothelial venule-like vessels in the active phase of ulcerative colitis. Am. J. Gastroenterol. 2007, 102, 1499–1509. [Google Scholar] [CrossRef]
- Jurisic, G.; Sundberg, J.P.; Bleich, A.; Leiter, E.H.; Broman, K.W.; Buechler, G.; Alley, L.; Vestweber, D.; Detmar, M. Quantitative lymphatic vessel trait analysis suggests Vcam1 as candidate modifier gene of inflammatory bowel disease. Genes Immun. 2010, 11, 219–231. [Google Scholar] [CrossRef]
- Ungaro, F.; Garlatti, V.; Massimino, L.; Spinelli, A.; Carvello, M.; Sacchi, M.; Spanò, S.; Colasante, G.; Valassina, N.; Vetrano, S.; et al. mTOR-Dependent Stimulation of IL20RA Orchestrates Immune Cell Trafficking through Lymphatic Endothelium in Patients with Crohn’s Disease. Cells 2019, 8, 924. [Google Scholar] [CrossRef]
- Li, Y.; Ge, Y.; Gong, J.; Zhu, W.; Cao, L.; Guo, Z.; Gu, L.; Li, J. Mesenteric Lymphatic Vessel Density Is Associated with Disease Behavior and Postoperative Recurrence in Crohn’s Disease. J. Gastrointest. Surg. 2018, 22, 2125–2132. [Google Scholar] [CrossRef]
- Dickerson, L.K.; De Freitas, S.; Pozo, M.E.; Safar, B. Letter to the Editor: Mesenteric Lymphatic Vessel Density Is Associated with Disease Behavior and Postoperative Recurrence in Crohn's Disease. J. Gastrointest. Surg. 2019, 23, 181–182. [Google Scholar] [CrossRef]
- Rahier, J.F.; Dubuquoy, L.; Colombel, J.F.; Jouret-Mourin, A.; Delos, M.; Ferrante, M.; Sokol, H.; Hertogh, G.D.; Salleron, J.; Geboes, K.; et al. Decreased lymphatic vessel density is associated with postoperative endoscopic recurrence in Crohn’s disease. Inflamm. Bowel Dis. 2013, 19, 2084–2090. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Li, Y.; Cao, L.; Cai, X.; Ge, Y.; Zhu, W. Decreased Expression of Prox1 Is Associated with Postoperative Recurrence in Crohn’s Disease. J. Crohns Colitis 2018, 12, 1210–1218. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Higashiyama, M.; Hozumi, H.; Sato, S.; Furuhashi, H.; Takajo, T.; Maruta, K.; Yasutake, Y.; Narimatsu, K.; Yoshikawa, K.; et al. Platelet interaction with lymphatics aggravates intestinal inflammation by suppressing lymphangiogenesis. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G276–G285. [Google Scholar] [CrossRef]
- Bandinelli, F.; Milia, A.F.; Manetti, M.; Lastraioli, E.; D’Amico, M.; Tonelli, P.; Fazi, M.; Arcangeli, A.; Matucci-Cerinic, M.; Ibba-Manneschi, L. Lymphatic endothelial progenitor cells and vascular endothelial growth factor-C in spondyloarthritis and Crohn’s disease: Two overlapping diseases? Clin. Exp. Rheumatol. 2015, 33, 195–200. [Google Scholar]
- Tacconi, C.; Schwager, S.; Cousin, N.; Bajic, D.; Sesartic, M.; Sundberg, J.P.; Neri, D.; Detmar, M. Antibody-Mediated Delivery of VEGFC Ameliorates Experimental Chronic Colitis. ACS Pharmacol. Transl. Sci. 2019, 2, 342–352. [Google Scholar] [CrossRef]
- D’Alessio, S.; Tacconi, C.; Danese, S. Targeting lymphatics in inflammatory bowel disease. Oncotarget 2015, 6, 34047–34048. [Google Scholar] [CrossRef]
- Hosono, K.; Kojo, K.; Narumiya, S.; Majima, M.; Ito, Y. Prostaglandin E receptor EP4 stimulates lymphangiogenesis to promote mucosal healing during DSS-induced colitis. Biomed. Pharmacother. 2020, 128, 110264. [Google Scholar] [CrossRef]
- Jurisic, G.; Sundberg, J.P.; Detmar, M. Blockade of VEGF receptor-3 aggravates inflammatory bowel disease and lymphatic vessel enlargement. Inflamm. Bowel Dis. 2013, 19, 1983–1989. [Google Scholar] [CrossRef]
- Davis, R.B.; Kechele, D.O.; Blakeney, E.S.; Pawlak, J.B.; Caron, K.M. Lymphatic deletion of calcitonin receptor-like receptor exacerbates intestinal inflammation. JCI Insight 2017, 2, e92465. [Google Scholar] [CrossRef]
- Lee, A.S.; Sung, M.J.; Kim, W.; Jung, Y.J. COMP-angiopoietin-1 ameliorates inflammation-induced lymphangiogenesis in dextran sulfate sodium (DSS)-induced colitis model. J. Mol. Med. 2018, 96, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S.; Hur, H.J.; Sung, M.J. The Effect of Artemisinin on Inflammation-Associated Lymphangiogenesis in Experimental Acute Colitis. Int. J. Mol. Sci. 2020, 21, 8068. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Zhao, J.; Qin, L.; Qiao, M. Promoting inflammatory lymphangiogenesis by vascular endothelial growth factor-C (VEGF-C) aggravated intestinal inflammation in mice with experimental acute colitis. Braz. J. Med. Biol. Res. 2016, 49, e4738. [Google Scholar] [CrossRef]
- Sura, R.; Colombel, J.F.; Van Kruiningen, H.J. Lymphatics, tertiary lymphoid organs and the granulomas of Crohn’s disease: An immunohistochemical study. Aliment Pharmacol. Ther. 2011, 33, 930–939. [Google Scholar] [CrossRef]
- Li, Y.; Stocchi, L.; Liu, X.; Rui, Y.; Liu, G.; Remzi, F.H.; Shen, B. Presence of Granulomas in Mesenteric Lymph Nodes Is Associated with Postoperative Recurrence in Crohn’s Disease. Inflamm. Bowel Dis. 2015, 21, 2613–2618. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Yuan, L.; Li, Y.; Shen, B.; Xie, H.; Liu, X. Association of granulomas in mesenteric lymph nodes in Crohn’s disease with younger age and transmural inflammation. J. Gastroenterol. Hepatol. 2017, 32, 1463–1468. [Google Scholar] [CrossRef]
- Geboes, K.; van den Oord, J.; De Wolf-Peeters, C.; Desmet, V.; Rutgeerts, P.; Janssens, J.; Vantrappen, G.; Penninckx, F.; Kerremans, R. The cellular composition of granulomas in mesenteric lymph nodes from patients with Crohn’s disease. Virchows Arch. A Pathol. Anat. Histopathol. 1986, 409, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Kodama, M.; Kobayashi, D.; Abe, K.; Sahara, R.; Yamana, T.; Furukawa, S.; Yao, T.; Tamura, T.; Okano, S. Epithelioid Cell Granulomas in Crohn’s Disease Are Differentially Associated with Blood Vessels and Lymphatic Vessels: A Sequential Double Immunostaining Study. J. Histochem. Cytochem. 2020, 68, 553–560. [Google Scholar] [CrossRef]
- Cui, Y.; Lu, S.Y.; Xu, J.; Peng, Y.S.; Miao, Q.; Wang, X.Q.; Chen, X.Y.; Ran, Z.H. Microscopic features of small bowel mucosa of patients with Crohn’s disease. BMC Gastroenterol. 2019, 19, 232. [Google Scholar] [CrossRef]
- Ishida, M.; Iwai, M.; Yoshida, K.; Kagotani, A.; Okabe, H. Metastatic Crohn’s disease accompanying granulomatous vasculitis and lymphangitis in the vulva. Int. J. Clin. Exp. Pathol. 2013, 6, 2263–2266. [Google Scholar]
- O’Brien, C.L.; Pavli, P.; Gordon, D.M.; Allison, G.E. Detection of bacterial DNA in lymph nodes of Crohn’s disease patients using high throughput sequencing. Gut 2014, 63, 1596–1606. [Google Scholar] [CrossRef]
- Fiorucci, S.; Distrutti, E.; Mencarelli, A.; Barbanti, M.; Palazzini, E.; Morelli, A. Inhibition of intestinal bacterial translocation with rifaximin modulates lamina propria monocytic cells reactivity and protects against inflammation in a rodent model of colitis. Digestion 2002, 66, 246–256. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, M.G.; Coffey, J.C.; McDermott, K.; Cotter, P.D.; Cabrera-Rubio, R.; Kiely, P.A.; Dunne, C.P. The Human Mesenteric Lymph Node Microbiome Differentiates Between Crohn’s Disease and Ulcerative Colitis. J. Crohns Colitis. 2019, 13, 58–66. [Google Scholar] [CrossRef]
- Feagan, B.G.; Sandborn, W.J.; D’Haens, G.; Lee, S.D.; Allez, M.; Fedorak, R.N.; Seidler, U.; Vermeire, S.; Lawrance, I.C.; Maroney, A.C.; et al. Randomised clinical trial: Vercirnon, an oral CCR9 antagonist, vs. placebo as induction therapy in active Crohn’s disease. Aliment Pharmacol. Ther. 2015, 42, 1170–1181. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Colombel, J.F.; Ghosh, S.; Sands, B.E.; Dryden, G.; Hébuterne, X.; Leong, R.W.; Bressler, B.; Ullman, T.; Lakatos, P.L.; et al. Eldelumab [Anti-IP-10] Induction Therapy for Ulcerative Colitis: A Randomised, Placebo-Controlled, Phase 2b Study. J. Crohns Colitis. 2016, 10, 418–428. [Google Scholar] [CrossRef]
- Sans, M.; Panés, J.; Ardite, E.; Elizalde, J.I.; Arce, Y.; Elena, M.; Palacín, A.; Fernández–Checa, J.C.; Anderson, D.C.; Lobb, R.; et al. VCAM-1 and ICAM-1 mediate leukocyte-endothelial cell adhesion in rat experimental colitis. Gastroenterology 1999, 116, 874–883. [Google Scholar] [CrossRef]
- Soriano, A.; Salas, A.; Salas, A.; Sans, M.; Gironella, M.; Elena, M.; Anderson, D.C.; Piqué, J.M.; Panés, J. VCAM-1, but not ICAM-1 or MAdCAM-1, immunoblockade ameliorates DSS-induced colitis in mice. Lab. Investig. J. Tech. Methods Pathol. 2000, 80, 1541–1551. [Google Scholar] [CrossRef]
- Gironella, M.; Mollà, M.; Salas, A.; Soriano, A.; Sans, M.; Closa, D.; Engel, P.; Salas, A.; Piqué, J.M.; Panés, J. The role of P-selectin in experimental colitis as determined by antibody immunoblockade and genetically deficient mice. J. Leukoc. Biol. 2002, 72, 56–64. [Google Scholar] [PubMed]
- Dalmasso, G.; Cottrez, F.; Imbert, V.; Lagadec, P.; Peyron, J.F.; Rampal, P.; Czerucka, D.; Groux, H.; Foussat, A.; Brun, V. Saccharomyces boulardii inhibits inflammatory bowel disease by trapping T cells in mesenteric lymph nodes. Gastroenterology 2006, 131, 1812–1825, Erratum in Gastroenterology 2007, 132, 1637. [Google Scholar] [CrossRef]
- Bellaguarda, E.; Keyashian, K.; Pekow, J.; Rubin, D.T.; Cohen, R.D.; Sakuraba, A. Prevalence of Antibodies against JC Virus in Patients with Refractory Crohn’s Disease and Effects of Natalizumab Therapy. Clin. Gastroenterol. Hepatol. 2015, 13, 1919–1925. [Google Scholar] [CrossRef]
- Feagan, B.G.; Rutgeerts, P.; Sands, B.E.; Hanauer, S.; Colombel, J.F.; Sandborn, W.J.; Van Assche, G.; Axler, J.; Kim, H.J.; Danese, S.; et al. Vedolizumab as induction and maintenance therapy for ulcerative colitis. N. Engl. J. Med. 2013, 369, 699–710. [Google Scholar] [CrossRef]
- Danese, S.; Sandborn, W.J.; Colombel, J.F.; Vermeire, S.; Glover, S.C.; Rimola, J.; Siegelman, J.; Jones, S.; Bornstein, J.D.; Feagan, B.G. Endoscopic, Radiologic, and Histologic Healing with Vedolizumab in Patients with Active Crohn’s Disease. Gastroenterology 2019, 157, 1007–1018.e7. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Feagan, B.G.; Rutgeerts, P.; Hanauer, S.; Colombel, J.F.; Sands, B.E.; Lukas, M.; Fedorak, R.N.; Lee, S.; Bressler, B.; et al. Vedolizumab as induction and maintenance therapy for Crohn’s disease. N. Engl. J. Med. 2013, 369, 711–721. [Google Scholar] [CrossRef]
- Loftus, E.V., Jr.; Feagan, B.G.; Panaccione, R.; Colombel, J.F.; Sandborn, W.J.; Sands, B.E.; Danese, S.; D’Haens, G.; Rubin, D.T.; Shafran, I.; et al. Long-term safety of vedolizumab for inflammatory bowel disease. Aliment Pharmacol. Ther. 2020, 52, 1353–1365. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Kaser, A. Vedolizumab, a humanized mAb against the α4β7 integrin for the potential treatment of ulcerative colitis and Crohn’s disease. Curr. Opin. Investig. Drugs 2010, 11, 1295–1304. [Google Scholar] [PubMed]
- Zeissig, S.; Rosati, E.; Dowds, C.M.; Aden, K.; Bethge, J.; Schulte, B.; Pan, W.H.; Mishra, N.; Zuhayra, M.; Marx, M.; et al. Vedolizumab is associated with changes in innate rather than adaptive immunity in patients with inflammatory bowel disease. Gut 2019, 68, 25–39. [Google Scholar] [CrossRef]
- Lichnog, C.; Klabunde, S.; Becker, E.; Fuh, F.; Tripal, P.; Atreya, R.; Klenske, E.; Erickson, R.; Chiu, H.; Reed, C.; et al. Cellular Mechanisms of Etrolizumab Treatment in Inflammatory Bowel Disease. Front. Pharmacol. 2019, 10, 39. [Google Scholar] [CrossRef]
- Zundler, S.; Schillinger, D.; Fischer, A.; Atreya, R.; López-Posadas, R.; Watson, A.; Neufert, C.; Atreya, I.; Neurath, M.F. Blockade of αEβ7 integrin suppresses accumulation of CD8+ and Th9 lymphocytes from patients with IBD in the inflamed gut in vivo. Gut 2017, 66, 1936–1948. [Google Scholar] [CrossRef]
- Available online: ClinicalTrials.gov/NCT02403323 (accessed on 20 November 2021).
- Zelman-Toister, E.; Bakos, E.; Cohen, S.; Zigmond, E.; Shezen, E.; Grabovsky, V.; Sagiv, A.; Hart, G.; Kaushansky, N.; Ben-Nun, A.; et al. CD151 Regulates T-Cell Migration in Health and Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2016, 22, 257–267. [Google Scholar] [CrossRef]
- Sugiura, T.; Kageyama, S.; Andou, A.; Miyazawa, T.; Ejima, C.; Nakayama, A.; Dohi, T.; Eda, H. Oral treatment with a novel small molecule alpha 4 integrin antagonist, AJM300, prevents the development of experimental colitis in mice. J. Crohns Colitis 2013, 7, e533–e542. [Google Scholar] [CrossRef]
- Yoshimura, N.; Watanabe, M.; Motoya, S.; Tominaga, K.; Matsuoka, K.; Iwakiri, R.; Watanabe, K.; Hibi, T.; AJM300 Study Group. Safety and Efficacy of AJM300, an Oral Antagonist of α4 Integrin, in Induction Therapy for Patients with Active Ulcerative Colitis. Gastroenterology 2015, 149, 1775–1783.e2. [Google Scholar] [CrossRef] [PubMed]
- Lucaciu, L.A.; Seicean, R.; Seicean, A. Small molecule drugs in the treatment of inflammatory bowel diseases: Which one, when and why?—A systematic review. Eur. J. Gastroenterol. Hepatol. 2020, 32, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Philpott, J.R.; Miner, P.B., Jr. Antisense inhibition of ICAM-1 expression as therapy provides insight into basic inflammatory pathways through early experiences in IBD. Expert Opin. Biol. Ther. 2008, 8, 1627–1632. [Google Scholar] [CrossRef] [PubMed]
- Greuter, T.; Biedermann, L.; Rogler, G.; Sauter, B.; Seibold, F. Alicaforsen, an antisense inhibitor of ICAM-1, as treatment for chronic refractory pouchitis after proctocolectomy: A case series. United Eur. Gastroenterol. J. 2016, 4, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Greuter, T.; Vavricka, S.R.; Biedermann, L.; Pilz, J.; Borovicka, J.; Seibold, F.; Sauter, B.; Rogler, G. Alicaforsen, an Antisense Inhibitor of Intercellular Adhesion Molecule-1, in the Treatment for Left-Sided Ulcerative Colitis and Ulcerative Proctitis. Dig. Dis. 2018, 36, 123–129. [Google Scholar] [CrossRef]
- Miner, P.B., Jr.; Wedel, M.K.; Xia, S.; Baker, B.F. Safety and efficacy of two dose formulations of alicaforsen enema compared with mesalazine enema for treatment of mild to moderate left-sided ulcerative colitis: A randomized, double-blind, active-controlled trial. Aliment Pharmacol. Ther. 2006, 23, 1403–1413, Erratum in Aliment Pharmacol. Ther. 2006, 24, 1268. [Google Scholar] [CrossRef]
- van Deventer, S.J.; Tami, J.A.; Wedel, M.K. A randomised, controlled, double blind, escalating dose study of alicaforsen enema in active ulcerative colitis. Gut 2004, 53, 1646–1651. [Google Scholar] [CrossRef][Green Version]
- van Deventer, S.J.; Wedel, M.K.; Baker, B.F.; Xia, S.; Chuang, E.; Miner, P.B., Jr. A phase II dose ranging, double-blind, placebo-controlled study of alicaforsen enema in subjects with acute exacerbation of mild to moderate left-sided ulcerative colitis. Aliment Pharmacol. Ther. 2006, 23, 1415–1425. [Google Scholar] [CrossRef]
- Miner, P.; Wedel, M.; Bane, B.; Bradley, J. An enema formulation of alicaforsen, an antisense inhibitor of intercellular adhesion molecule-1, in the treatment of chronic, unremitting pouchitis. Aliment Pharmacol. Ther. 2004, 19, 281–286. [Google Scholar] [CrossRef]
- Available online: ClinicalTrials.gov/NCT02525523 (accessed on 21 November 2021).
- Yacyshyn, B.R.; Bowen-Yacyshyn, M.B.; Jewell, L.; Tami, J.A.; Bennett, C.F.; Kisner, D.L.; Shanahan, W.R., Jr. A placebo-controlled trial of ICAM-1 antisense oligonucleotide in the treatment of Crohn’s disease. Gastroenterology 1998, 114, 1133–1142, Erratum in Gastroenterology 2001, 121, 747. [Google Scholar] [CrossRef]
- Yacyshyn, B.R.; Barish, C.; Goff, J.; Dalke, D.; Gaspari, M.; Yu, R.; Tami, J.; Dorr, F.A.; Sewell, K.L. Dose ranging pharmacokinetic trial of high-dose alicaforsen (intercellular adhesion molecule-1 antisense oligodeoxynucleotide) (ISIS 2302) in active Crohn’s disease. Aliment Pharmacol. Ther. 2002, 16, 1761–1770. [Google Scholar] [CrossRef]
- Yacyshyn, B.R.; Schievella, A.; Sewell, K.L.; Tami, J.A. Gene polymorphisms and serological markers of patients with active Crohn’s disease in a clinical trial of antisense to ICAM-1. Clin. Exp. Immunol. 2005, 141, 141–147. [Google Scholar] [CrossRef]
- Yacyshyn, B.; Chey, W.Y.; Wedel, M.K.; Yu, R.Z.; Paul, D.; Chuang, E. A randomized, double-masked, placebo-controlled study of alicaforsen, an antisense inhibitor of intercellular adhesion molecule 1, for the treatment of subjects with active Crohn’s disease. Clin. Gastroenterol. Hepatol. 2007, 5, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.; Nikolaus, S.; Malchow, H.; Kruis, W.; Lochs, H.; Raedler, A.; Hahn, E.G.; Krummenerl, T.; Steinmann, G.; German ICAM-1 Study Group. Absence of efficacy of subcutaneous antisense ICAM-1 treatment of chronic active Crohn’s disease. Gastroenterology 2001, 120, 1339–1346. [Google Scholar] [CrossRef]
- Halpert, G.; Eitan, T.; Voronov, E.; Apte, R.N.; Rath-Wolfson, L.; Albeck, M.; Kalechman, Y.; Sredni, B. Multifunctional activity of a small tellurium redox immunomodulator compound, AS101, on dextran sodium sulfate-induced murine colitis. J. Biol. Chem. 2014, 289, 17215–17227. [Google Scholar] [CrossRef] [PubMed]
- Matloubian, M.; Lo, C.G.; Cinamon, G.; Lesneski, M.J.; Xu, Y.; Brinkmann, V.; Allende, M.L.; Proia, R.L.; Cyster, J.G. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 2004, 427, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Davis, M.D.; Heise, C.E.; Albert, R.; Cottens, S.; Hof, R.; Bruns, C.; Prieschl, E.; Baumruker, T.; Hiestand, P.; et al. The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J. Biol. Chem. 2002, 277, 21453–21457. [Google Scholar] [CrossRef] [PubMed]
- Mehling, M.; Brinkmann, V.; Antel, J.; Bar-Or, A.; Goebels, N.; Vedrine, C.; Kristofic, C.; Kuhle, J.; Lindberg, R.L.; Kappos, L. FTY720 therapy exerts differential effects on T cell subsets in multiple sclerosis. Neurology 2008, 71, 1261–1267. [Google Scholar] [CrossRef]
- Mizushima, T.; Ito, T.; Kishi, D.; Kai, Y.; Tamagawa, H.; Nezu, R.; Kiyono, H.; Matsuda, H. Therapeutic effects of a new lymphocyte homing reagent FTY720 in interleukin-10 gene-deficient mice with colitis. Inflamm. Bowel Dis. 2004, 10, 182–192. [Google Scholar] [CrossRef]
- Deguchi, Y.; Andoh, A.; Yagi, Y.; Bamba, S.; Inatomi, O.; Tsujikawa, T.; Fujiyama, Y. The S1P receptor modulator FTY720 prevents the development of experimental colitis in mice. Oncol. Rep. 2006, 16, 699–703. [Google Scholar] [CrossRef]
- Daniel, C.; Sartory, N.; Zahn, N.; Geisslinger, G.; Radeke, H.H.; Stein, J.M. FTY720 ameliorates Th1-mediated colitis in mice by directly affecting the functional activity of CD4+CD25+ regulatory T cells. J. Immunol. 2007, 178, 2458–2468. [Google Scholar] [CrossRef]
- Daniel, C.; Sartory, N.A.; Zahn, N.; Schmidt, R.; Geisslinger, G.; Radeke, H.H.; Stein, J.M. FTY720 ameliorates oxazolone colitis in mice by directly affecting T helper type 2 functions. Mol. Immunol. 2007, 44, 3305–3316. [Google Scholar] [CrossRef] [PubMed]
- Karuppuchamy, T.; Behrens, E.H.; González-Cabrera, P.; Sarkisyan, G.; Gima, L.; Boyer, J.D.; Bamias, G.; Jedlicka, P.; Veny, M.; Clark, D.; et al. Sphingosine-1-phosphate receptor-1 (S1P1) is expressed by lymphocytes, dendritic cells, and endothelium and modulated during inflammatory bowel disease. Mucosal Immunol. 2017, 10, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Karuppuchamy, T.; Tyler, C.J.; Lundborg, L.R.; Pérez-Jeldres, T.; Kimball, A.K.; Clambey, E.T.; Jedlicka, P.; Rivera-Nieves, J. Sphingosine-1-Phosphate Lyase Inhibition Alters the S1P Gradient and Ameliorates Crohn’s-Like Ileitis by Suppressing Thymocyte Maturation. Inflamm. Bowel Dis. 2020, 26, 216–228. [Google Scholar] [CrossRef] [PubMed]
- Scott, F.L.; Clemons, B.; Brooks, J.; Brahmachary, E.; Powell, R.; Dedman, H.; Desale, H.G.; Timony, G.A.; Martinborough, E.; Rosen, H.; et al. Ozanimod (RPC1063) is a potent sphingosine-1-phosphate receptor-1 (S1P1 ) and receptor-5 (S1P5 ) agonist with autoimmune disease-modifying activity. Br. J. Pharmacol. 2016, 173, 1778–1792. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Feagan, B.G.; D’Haens, G.; Wolf, D.C.; Jovanovic, I.; Hanauer, S.B.; Ghosh, S.; Petersen, A.; Hua, S.Y.; Lee, J.H.; et al. Ozanimod as Induction and Maintenance Therapy for Ulcerative Colitis. N. Engl. J. Med. 2021, 385, 1280–1291. [Google Scholar] [CrossRef]
- Al-Shamma, H.; Lehmann-Bruinsma, K.; Carroll, C.; Solomon, M.; Komori, H.K.; Peyrin-Biroulet, L.; Adams, J. The Selective Sphingosine 1-Phosphate Receptor Modulator Etrasimod Regulates Lymphocyte Trafficking and Alleviates Experimental Colitis. J. Pharmacol. Exp. Ther. 2019, 369, 311–317. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Peyrin-Biroulet, L.; Zhang, J.; Chiorean, M.; Vermeire, S.; Lee, S.D.; Kühbacher, T.; Yacyshyn, B.; Cabell, C.H.; Naik, S.U.; et al. Efficacy and Safety of Etrasimod in a Phase 2 Randomized Trial of Patients with Ulcerative Colitis. Gastroenterology 2020, 158, 550–561. [Google Scholar] [CrossRef]
- Shimano, K.; Maeda, Y.; Kataoka, H.; Murase, M.; Mochizuki, S.; Utsumi, H.; Oshita, K.; Sugahara, K. Amiselimod (MT-1303), a novel sphingosine 1-phosphate receptor-1 functional antagonist, inhibits progress of chronic colitis induced by transfer of CD4+CD45RBhigh T cells. PLoS ONE 2019, 14, e0226154. [Google Scholar] [CrossRef]
- D’Haens, G.; Danese, S.; Davies, M.; Watanabe, M.; Hibi, T. A Phase II, Multicentre, Randomised, Double-Blind, Placebo-Controlled Study to Evaluate Safety, Tolerability and Efficacy of Amiselimod in Patients with Moderate to Severe Active Crohn’s Disease. J. Crohns Colitis 2021, jjab201. [Google Scholar] [CrossRef]
- Available online: ClinicalTrials.gov/NCT03464097 (accessed on 30 November 2021).
- Available online: ClinicalTrials.gov/NCT04173273 (accessed on 30 November 2021).
- de Hair, M.J.; Zijlstra, I.A.; Boumans, M.J.; van de Sande, M.G.; Maas, M.; Gerlag, D.M.; Tak, P.P. Hunting for the pathogenesis of rheumatoid arthritis: Core-needle biopsy of inguinal lymph nodes as a new research tool. Ann. Rheum. Dis. 2012, 71, 1911–1912. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nikolakis, D.; de Voogd, F.A.E.; Pruijt, M.J.; Grootjans, J.; van de Sande, M.G.; D’Haens, G.R. The Role of the Lymphatic System in the Pathogenesis and Treatment of Inflammatory Bowel Disease. Int. J. Mol. Sci. 2022, 23, 1854. https://doi.org/10.3390/ijms23031854
Nikolakis D, de Voogd FAE, Pruijt MJ, Grootjans J, van de Sande MG, D’Haens GR. The Role of the Lymphatic System in the Pathogenesis and Treatment of Inflammatory Bowel Disease. International Journal of Molecular Sciences. 2022; 23(3):1854. https://doi.org/10.3390/ijms23031854
Chicago/Turabian StyleNikolakis, Dimitrios, Floris A. E. de Voogd, Maarten J. Pruijt, Joep Grootjans, Marleen G. van de Sande, and Geert R. D’Haens. 2022. "The Role of the Lymphatic System in the Pathogenesis and Treatment of Inflammatory Bowel Disease" International Journal of Molecular Sciences 23, no. 3: 1854. https://doi.org/10.3390/ijms23031854
APA StyleNikolakis, D., de Voogd, F. A. E., Pruijt, M. J., Grootjans, J., van de Sande, M. G., & D’Haens, G. R. (2022). The Role of the Lymphatic System in the Pathogenesis and Treatment of Inflammatory Bowel Disease. International Journal of Molecular Sciences, 23(3), 1854. https://doi.org/10.3390/ijms23031854