
Evolutionary Analysis of Dipeptidyl Peptidase I


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
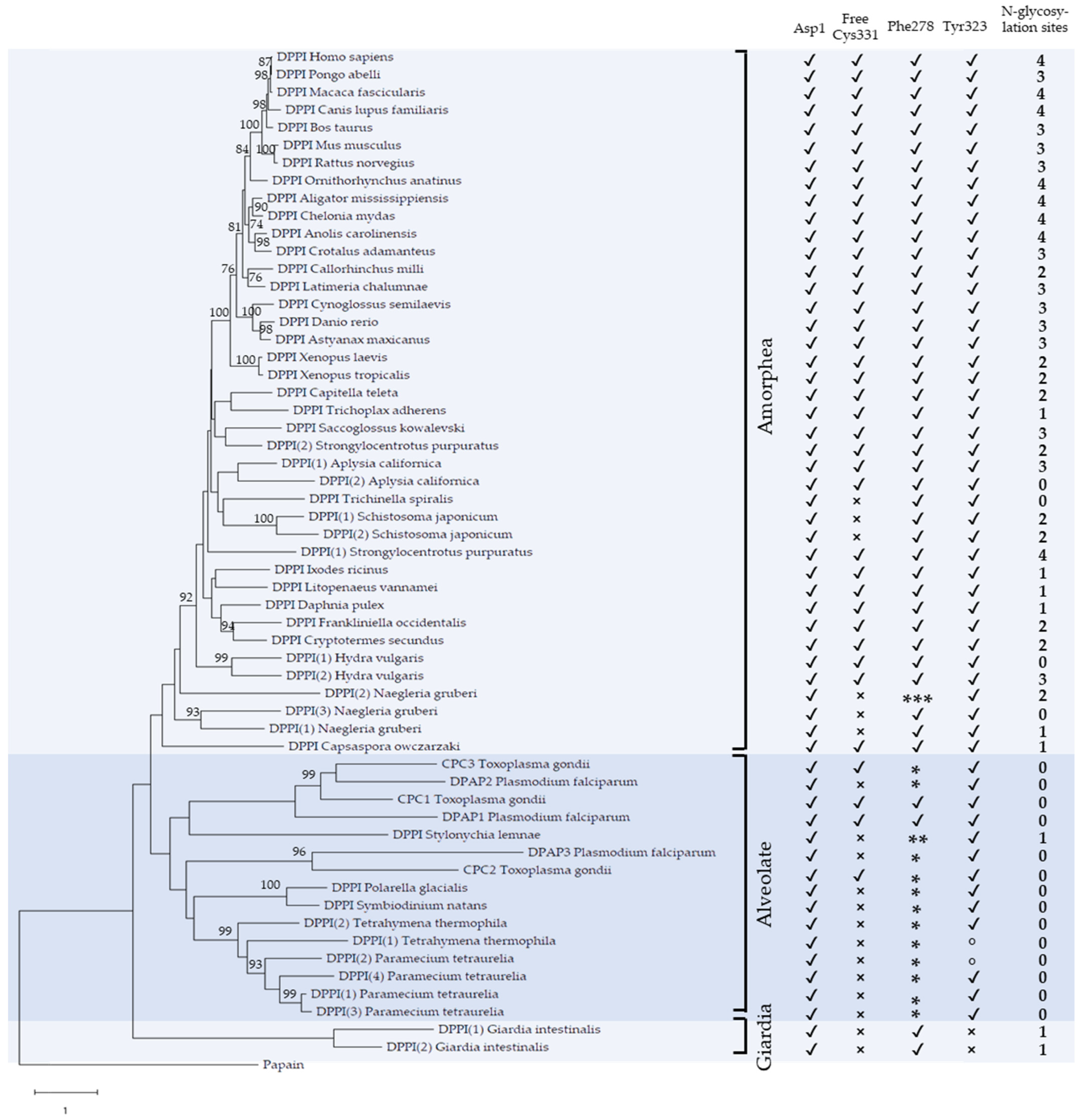
2.1. Phylogenetic Analysis
2.2. Primary and Tertiary Structure Conservation
2.3. Conservation of Functionally Important Structural Elements
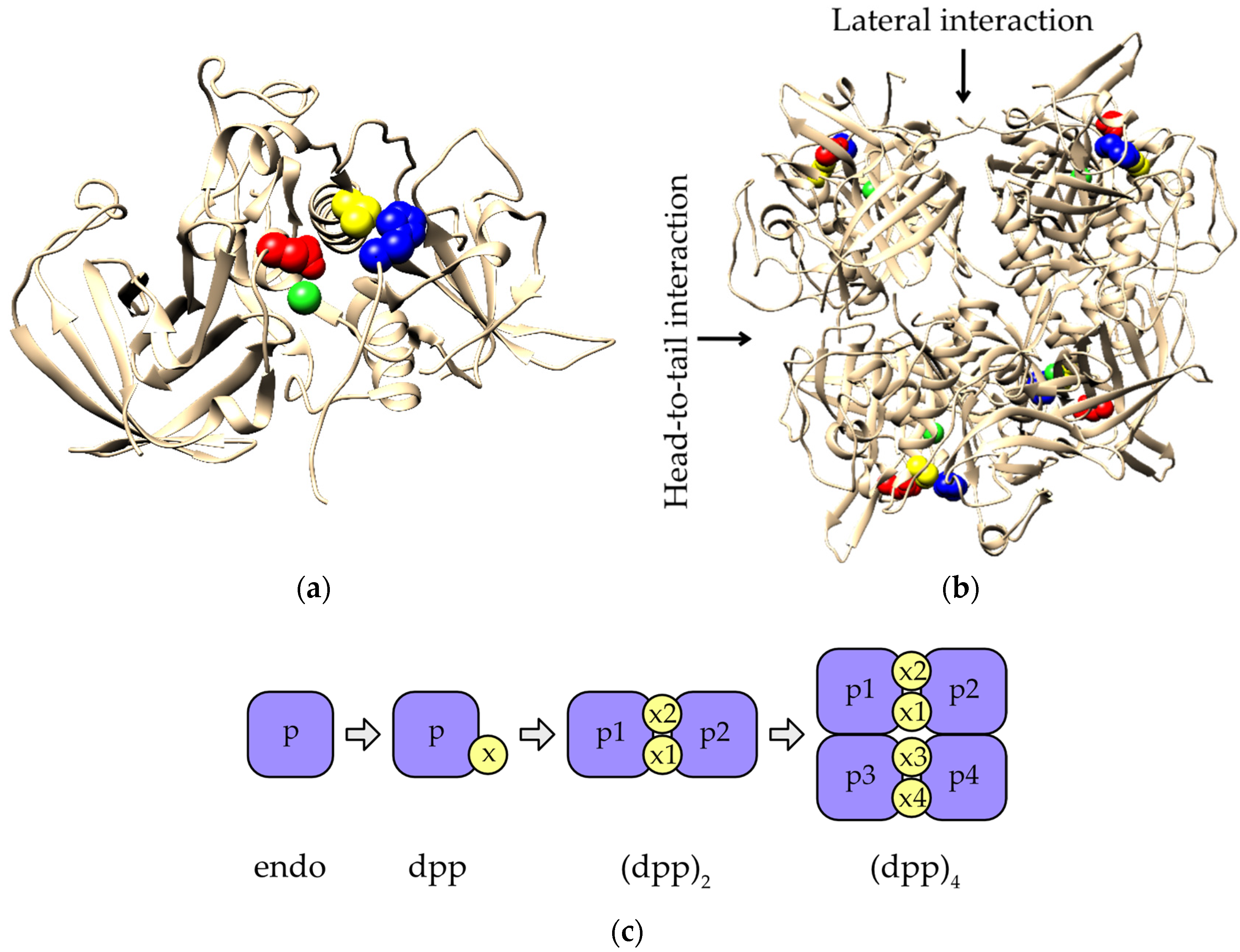
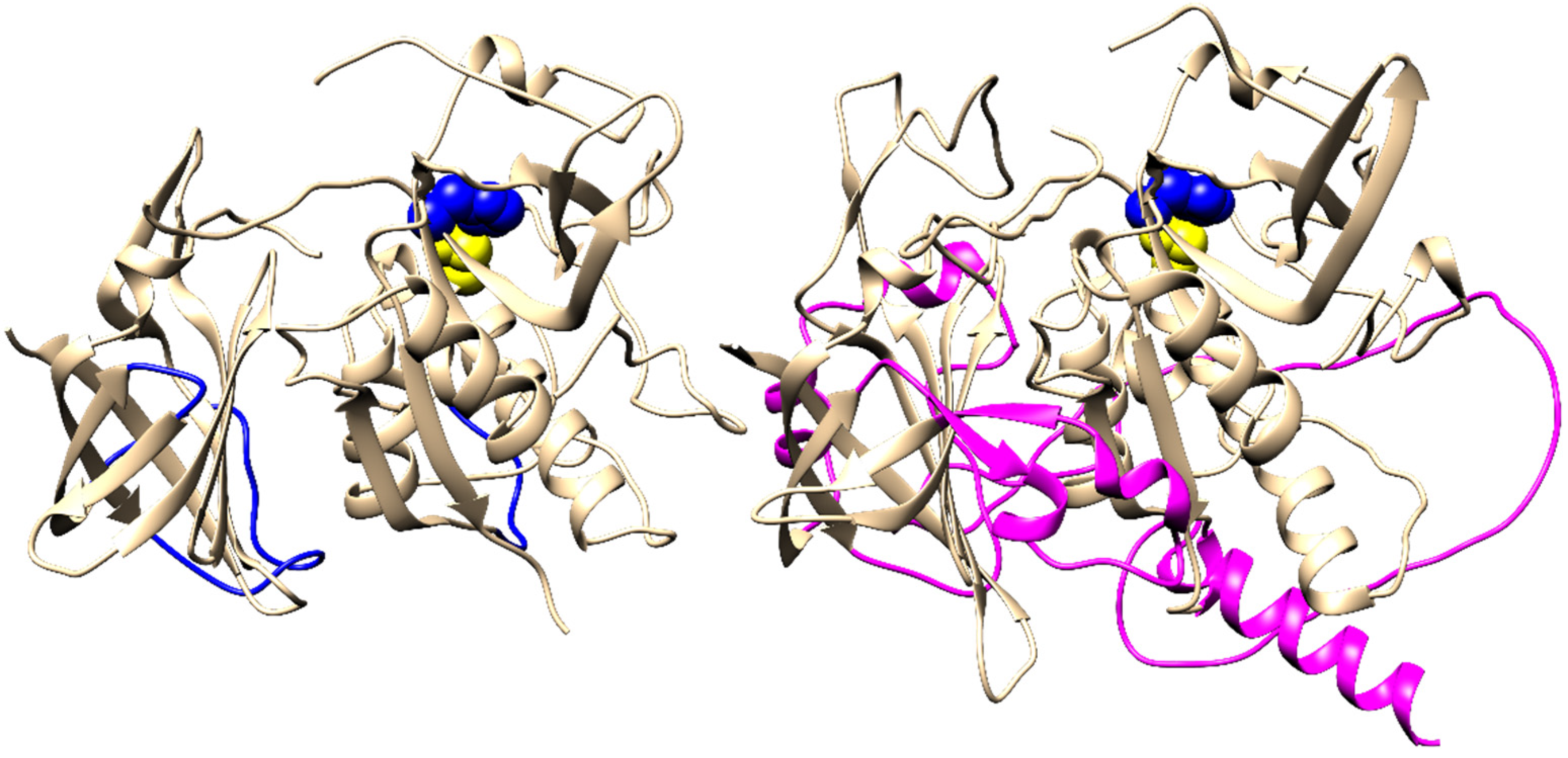
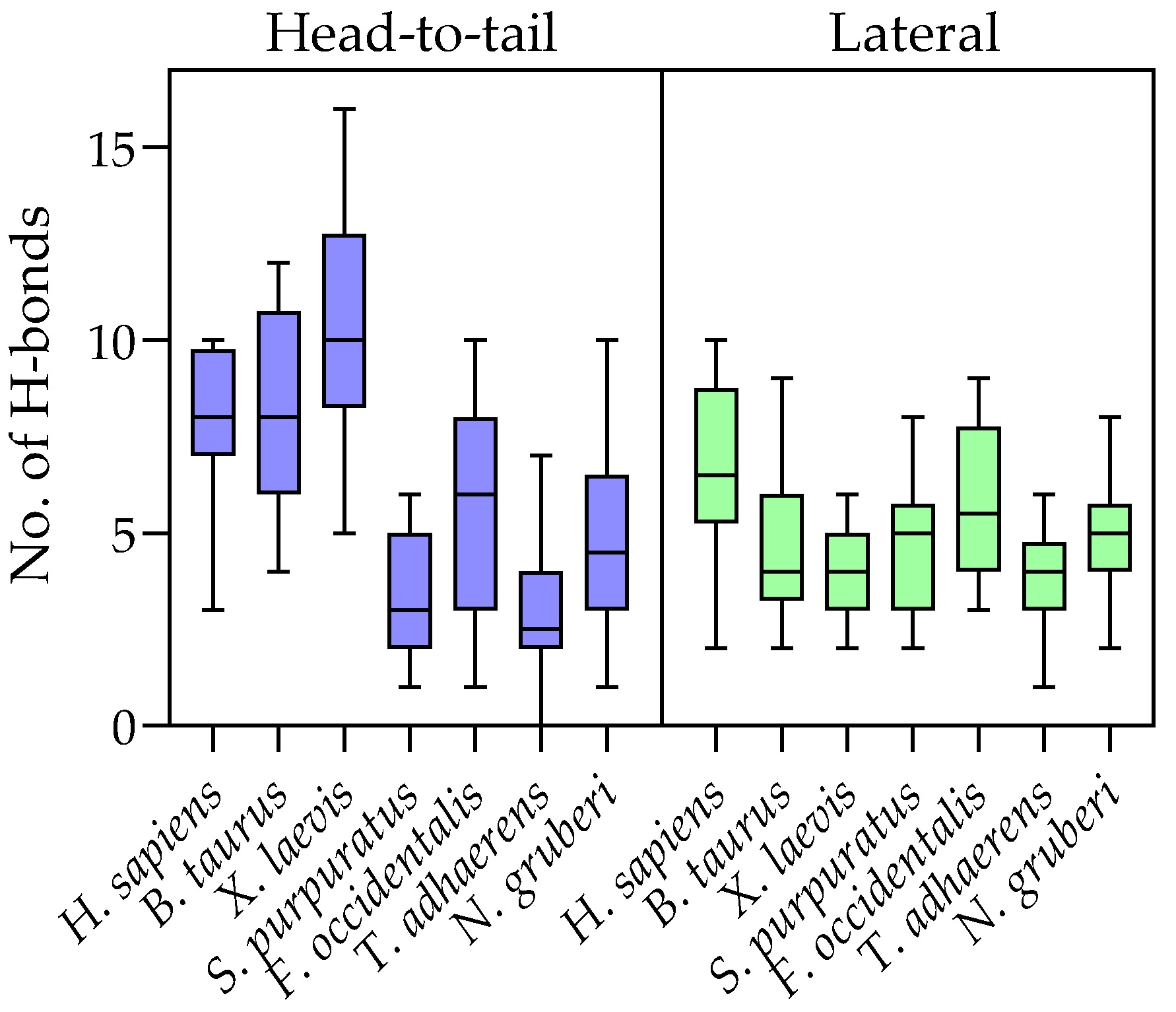
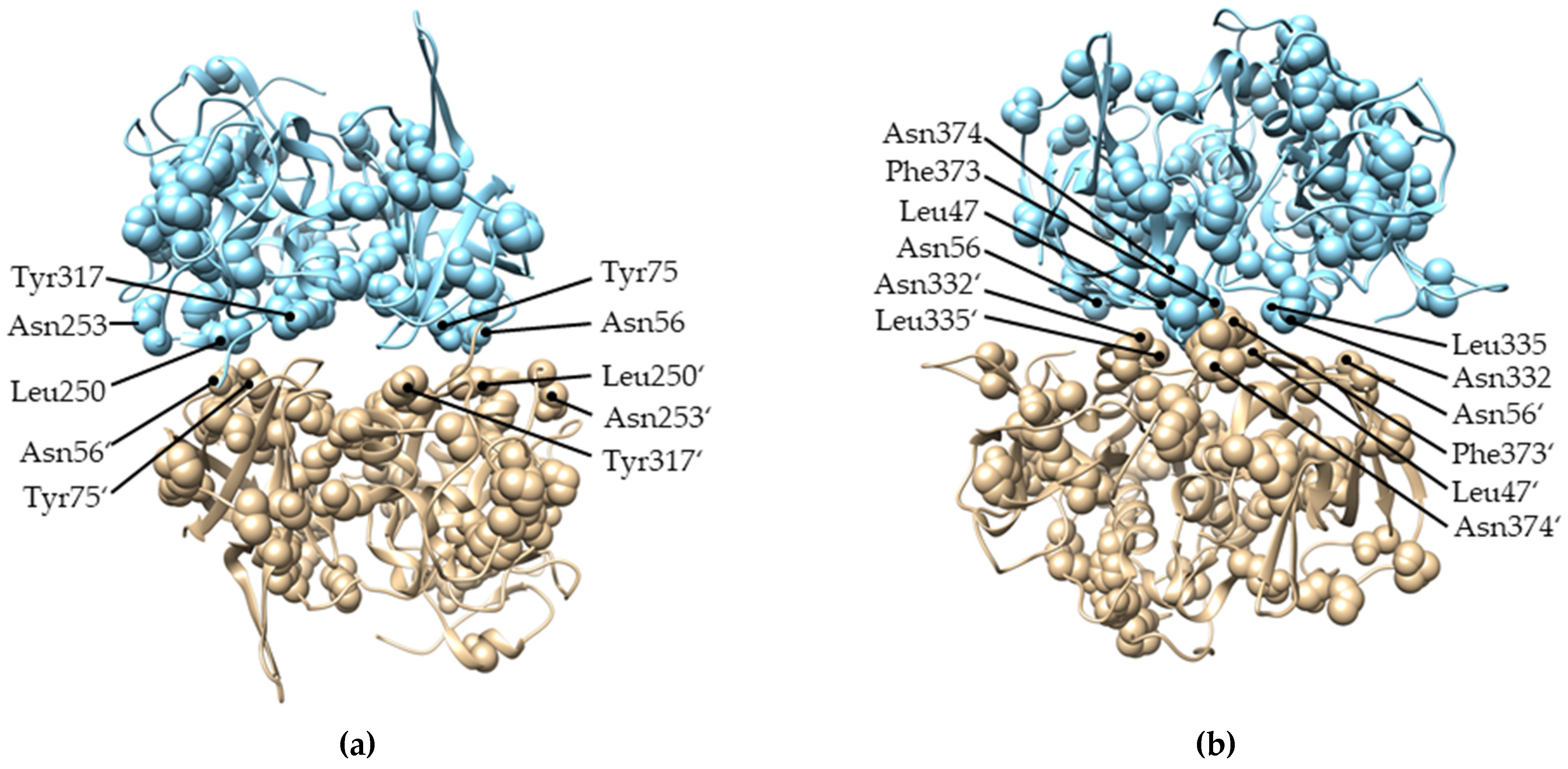
2.4. Evolution of Subunit Interfaces
3. Discussion
4. Materials and Methods
4.1. Sequence Retrieval
4.2. Phylogenetic Analysis
4.3. Functional Divergence Analysis
4.4. Three-Dimensional Representations and Structural Analyses
4.5. Homology Modeling with AlphaFold
4.6. Homology Modeling with Modeller
4.7. Statistical Analysis of N-Glycosylation Sites
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Novinec, M.; Lenarčič, B. Papain-like peptidases: Structure, function, and evolution. Biomol. Concepts 2013, 4, 287–308. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2018, 46, D624–D632. [Google Scholar] [CrossRef] [PubMed]
- Pham, C.T.N.; Ley, T.J. Dipeptidyl peptidase I is required for the processing and activation of granzymes A and B in vivo. Proc. Natl. Acad. Sci. USA 1999, 96, 8627–8632. [Google Scholar] [CrossRef] [PubMed]
- Hart, T.C.; Hart, P.S.; Michalec, M.D.; Zhang, Y.; Firatli, E.; E Van Dyke, T.; Stabholz, A.; Zlorogorski, A.; Shapira, L.; A Soskolne, W. Haim-Munk syndrome and Papillon-Lefevre syndrome are allelic mutations in cathepsin C. J. Med Genet. 2000, 37, 88–94. [Google Scholar] [CrossRef]
- Korkmaz, B.; Caughey, G.H.; Chapple, I.; Gauthier, F.; Hirschfeld, J.; Jenne, D.E.; Kettritz, R.; Lalmanach, G.; Lamort, A.-S.; Lauritzen, C.; et al. Therapeutic targeting of cathepsin C: From pathophysiology to treatment. Pharmacol. Ther. 2018, 190, 202–236. [Google Scholar] [CrossRef]
- Korkmaz, B.; Lamort, A.-S.; Domain, R.; Beauvillain, C.; Gieldon, A.; Yildirim, A.; Stathopoulos, G.T.; Rhimi, M.; Jenne, D.E.; Kettritz, R. Cathepsin C inhibition as a potential treatment strategy in cancer. Biochem. Pharmacol. 2021, 194, 114803. [Google Scholar] [CrossRef]
- Turk, D.; Janjić, V.; Štern, I.; Podobnik, M.; Lamba, D.; Dahl, S.W.; Lauritzen, C.; Pedersen, J.; Turk, V.; Turk, B. Structure of human dipeptidyl peptidase I (cathepsin C): Exclusion domain added to an endopeptidase framework creates the machine for activation of granular serine proteases. EMBO J. 2001, 20, 6570–6582. [Google Scholar] [CrossRef]
- Schechter, I.; Berger, A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967, 27, 157–162. [Google Scholar] [CrossRef]
- Rebernik, M.; Lenarčič, B.; Novinec, M. The catalytic domain of cathepsin C (dipeptidyl-peptidase I) alone is a fully functional endoprotease. Protein Expr. Purif. 2019, 157, 21–27. [Google Scholar] [CrossRef]
- Santilman, V.; Jadot, M.; Mainferme, F. Importance of the propeptide in the biosynthetic maturation of rat cathepsin C. Eur. J. Cell Biol. 2002, 81, 654–663. [Google Scholar] [CrossRef]
- Rebernik, M.; Snoj, T.; Klemenčič, M.; Novinec, M. Interplay between tetrameric structure, enzymatic activity and allosteric regulation of human dipeptidyl-peptidase I. Arch. Biochem. Biophys. 2019, 675, 108121. [Google Scholar] [CrossRef]
- Horn, M. Free-thiol Cys331 exposed during activation process is critical for native tetramer structure of cathepsin C (dipeptidyl peptidase I). Protein Sci. 2002, 11, 933–943. [Google Scholar] [CrossRef]
- Lamort, A.-S.; Hamon, Y.; Czaplewski, C.; Gieldon, A.; Seren, S.; Coquet, L.; Lecaille, F.; Lesner, A.; Lalmanach, G.; Gauthier, F.; et al. Processing and Maturation of Cathepsin C Zymogen: A Biochemical and Molecular Modeling Analysis. Int. J. Mol. Sci. 2019, 20, 4747. [Google Scholar] [CrossRef]
- Klemba, M.; Gluzman, I.; Goldberg, D.E. A Plasmodium falciparum Dipeptidyl Aminopeptidase I Participates in Vacuolar Hemoglobin Degradation. J. Biol. Chem. 2004, 279, 43000–43007. [Google Scholar] [CrossRef]
- Arastu-Kapur, S.; Ponder, E.L.; Fonović, U.P.; Yeoh, S.; Yuan, F.; Fonovic, M.; Grainger, M.; I Phillips, C.; Powers, J.C.; Bogyo, M. Identification of proteases that regulate erythrocyte rupture by the malaria parasite Plasmodium falciparum. Nat. Chem. Biol. 2008, 4, 203–213. [Google Scholar] [CrossRef]
- Wang, F.; Krai, P.; Deu, E.; Bibb, B.; Lauritzen, C.; Pedersen, J.; Bogyo, M.; Klemba, M. Biochemical characterization of Plasmodium falciparum dipeptidyl aminopeptidase 1. Mol. Biochem. Parasitol. 2011, 175, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Que, X.; Engel, J.C.; Ferguson, D.; Wunderlich, A.; Tomavo, S.; Reed, S.L. Cathepsin Cs Are Key for the Intracellular Survival of the Protozoan Parasite, Toxoplasma gondii. J. Biol. Chem. 2007, 282, 4994–5003. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Zou, Y.; Su, Z.; Huang, W.; Zhou, Z.; Arendsee, Z.; Zeng, Y. An Update of DIVERGE Software for Functional Divergence Analysis of Protein Family. Mol. Biol. Evol. 2013, 30, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Gu, X. Statistical methods for testing functional divergence after gene duplication. Mol. Biol. Evol. 1999, 16, 1664–1674. [Google Scholar] [CrossRef]
- Gu, X. Maximum-Likelihood Approach for Gene Family Evolution Under Functional Divergence. Mol. Biol. Evol. 2001, 18, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Gorter, J.; Gruber, M. Cathepsin C: An allosteric enzyme. Biochim. Biophys. Acta (BBA) Enzym. 1970, 198, 546–555. [Google Scholar] [CrossRef]
- Cigic, B.; Pain, R.H. Location of the binding site for chloride ion activation of cathepsin C. JBIC J. Biol. Inorg. Chem. 1999, 264, 944–951. [Google Scholar] [CrossRef]
- Kastritis, P.; Bonvin, A.M.J.J. On the binding affinity of macromolecular interactions: Daring to ask why proteins interact. J. R. Soc. Interface 2013, 10, 20120835. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef]
- Burki, F.; Roger, A.J.; Brown, M.W.; Simpson, A.G. The New Tree of Eukaryotes. Trends Ecol. Evol. 2020, 35, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.; Cambra, I.; González-Melendi, P.; Santamaria, M.E.; Díaz, I. C1A cysteine-proteases and their inhibitors in plants. Physiol. Plant. 2012, 145, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, V.E.; Iribarren, P.A.; Niemirowicz, G.T.; Cazzulo, J.J. Update on relevant trypanosome peptidases: Validated targets and future challenges. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2020, 1869, 140577. [Google Scholar] [CrossRef]
- Camejo, A.; Gold, D.A.; Lu, D.; McFetridge, K.; Julien, L.; Yang, N.; Jensen, K.D.; Saeij, J.P. Identification of three novel Toxoplasma gondii rhoptry proteins. Int. J. Parasitol. 2013, 44, 147–160. [Google Scholar] [CrossRef]
- Vries, L.E.; Sanchez, M.I.; Groborz, K.; Kuppens, L.; Poreba, M.; Lehmann, C.; Nevins, N.; Withers-Martinez, C.; Hirst, D.J.; Yuan, F.; et al. Characterization ofP. falciparumdipeptidyl aminopeptidase 3 specificity identifies differences in amino acid preferences between peptide-based substrates and covalent inhibitors. FEBS J. 2019, 286, 3998–4023. [Google Scholar] [CrossRef]
- O’Farrell, P.A.; Gonzalez, F.; Zheng, W.; Johnston, S.A.; Joshua-Tor, L. Crystal structure of human bleomycin hydrolase, a self-compartmentalizing cysteine protease. Structure 1999, 7, 619–627. [Google Scholar] [CrossRef]
- Dolenc, I.; Štefe, I.; Turk, D.; Taler-Verčič, A.; Turk, B.; Turk, V.; Stoka, V. Human cathepsin X/Z is a biologically active homodimer. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2020, 1869, 140567. [Google Scholar] [CrossRef]
- Dolenc, I.; Turk, B.; Pungercic, G.; Ritonja, A.; Turk, V. Oligomeric Structure and Substrate Induced Inhibition of Human Cathepsin C. J. Biol. Chem. 1995, 270, 21626–21631. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.; Kim, B.-H.; Grishin, N.V. PROMALS3D: A tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 2008, 36, 2295–2300. [Google Scholar] [CrossRef] [PubMed]
- Le, S.Q.; Gascuel, O. An Improved General Amino Acid Replacement Matrix. Mol. Biol. Evol. 2008, 25, 1307–1320. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varda, N.; Novinec, M. Evolutionary Analysis of Dipeptidyl Peptidase I. Int. J. Mol. Sci. 2022, 23, 1852. https://doi.org/10.3390/ijms23031852
Varda N, Novinec M. Evolutionary Analysis of Dipeptidyl Peptidase I. International Journal of Molecular Sciences. 2022; 23(3):1852. https://doi.org/10.3390/ijms23031852
Chicago/Turabian StyleVarda, Nina, and Marko Novinec. 2022. "Evolutionary Analysis of Dipeptidyl Peptidase I" International Journal of Molecular Sciences 23, no. 3: 1852. https://doi.org/10.3390/ijms23031852
APA StyleVarda, N., & Novinec, M. (2022). Evolutionary Analysis of Dipeptidyl Peptidase I. International Journal of Molecular Sciences, 23(3), 1852. https://doi.org/10.3390/ijms23031852