
Upregulation of Cathepsin X in Glioblastoma: Interplay with γ-Enolase and the Effects of Selective Cathepsin X Inhibitors

 ,
,  , ,
, ,  , , , and
, , , and 
Abstract
:1. Introduction
2. Results
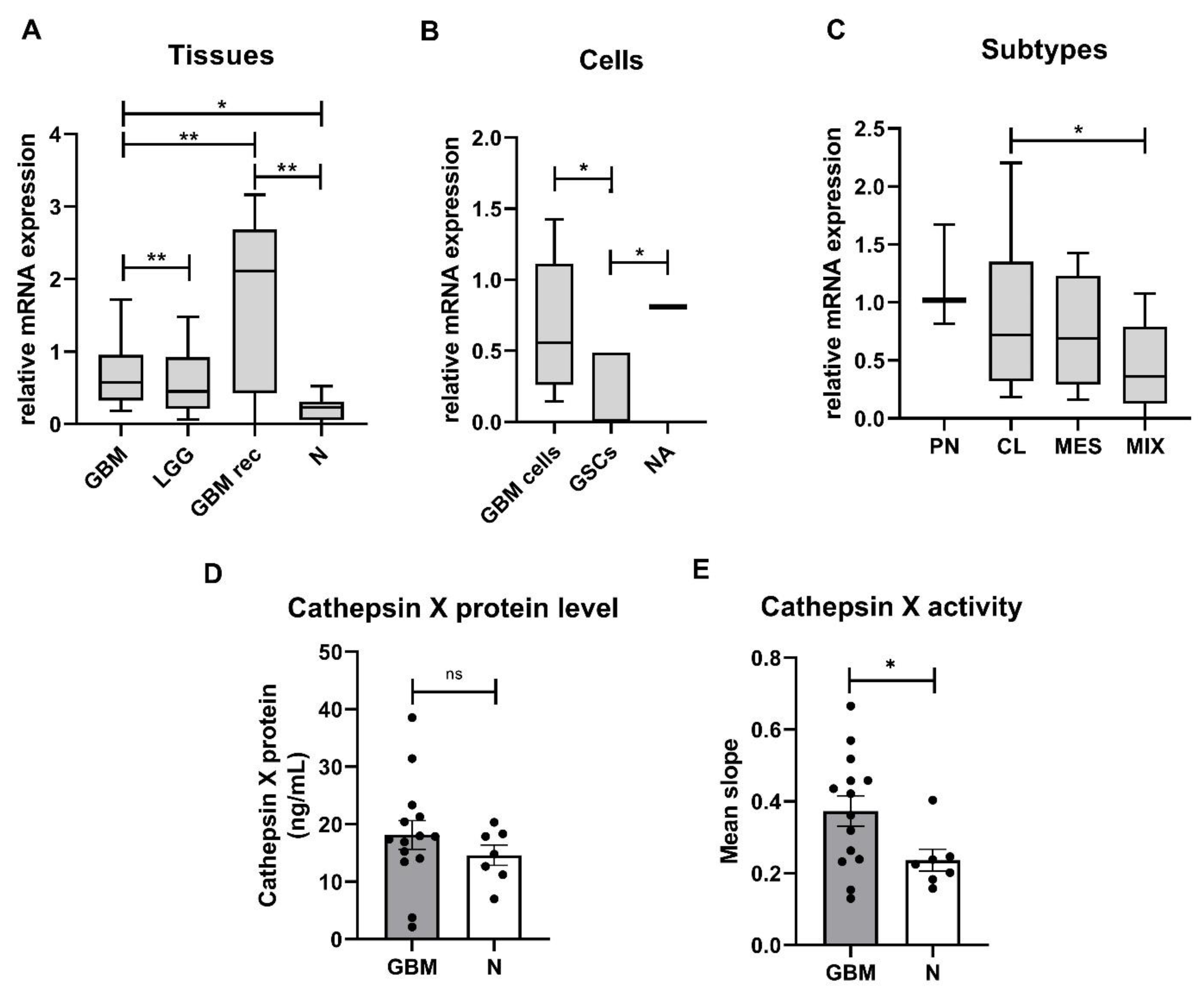
2.1. Expression and Enzymatic Activity of Cathepsin X Are Upregulated in GBM Tissues
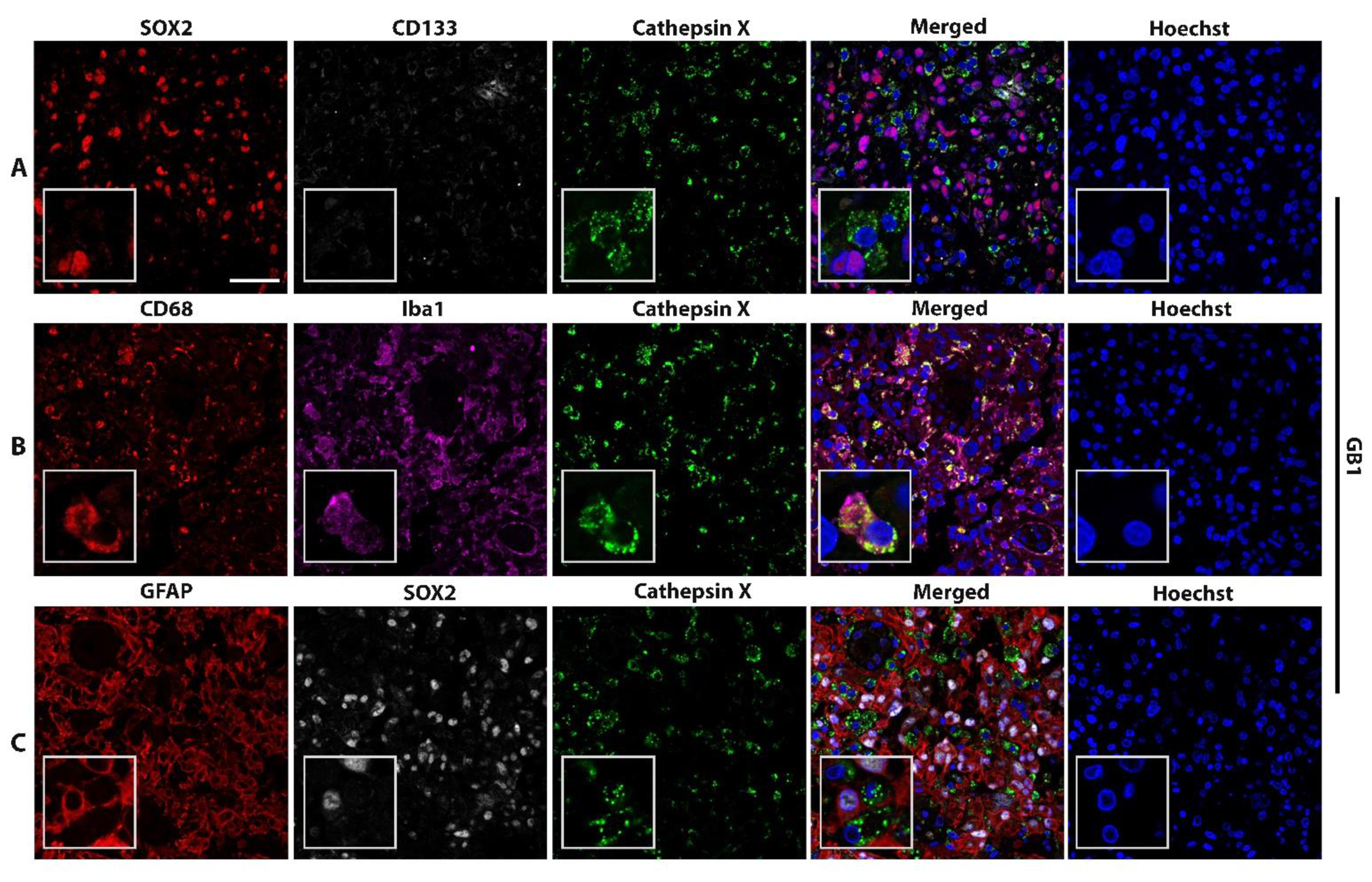
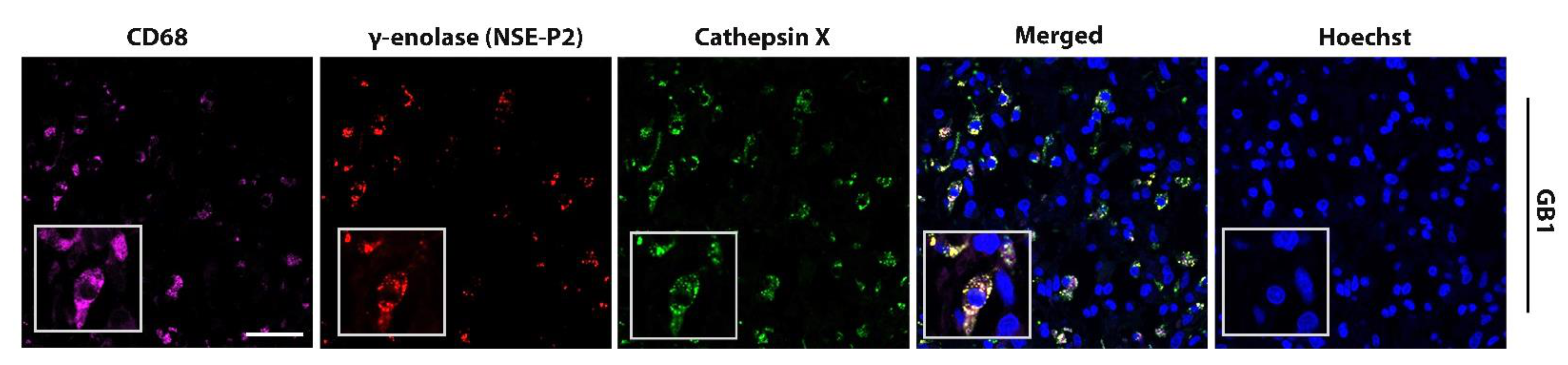
2.2. Macrophage-Specific and Microglia-Specific Localization of Cathepsin X in GBM Tissues
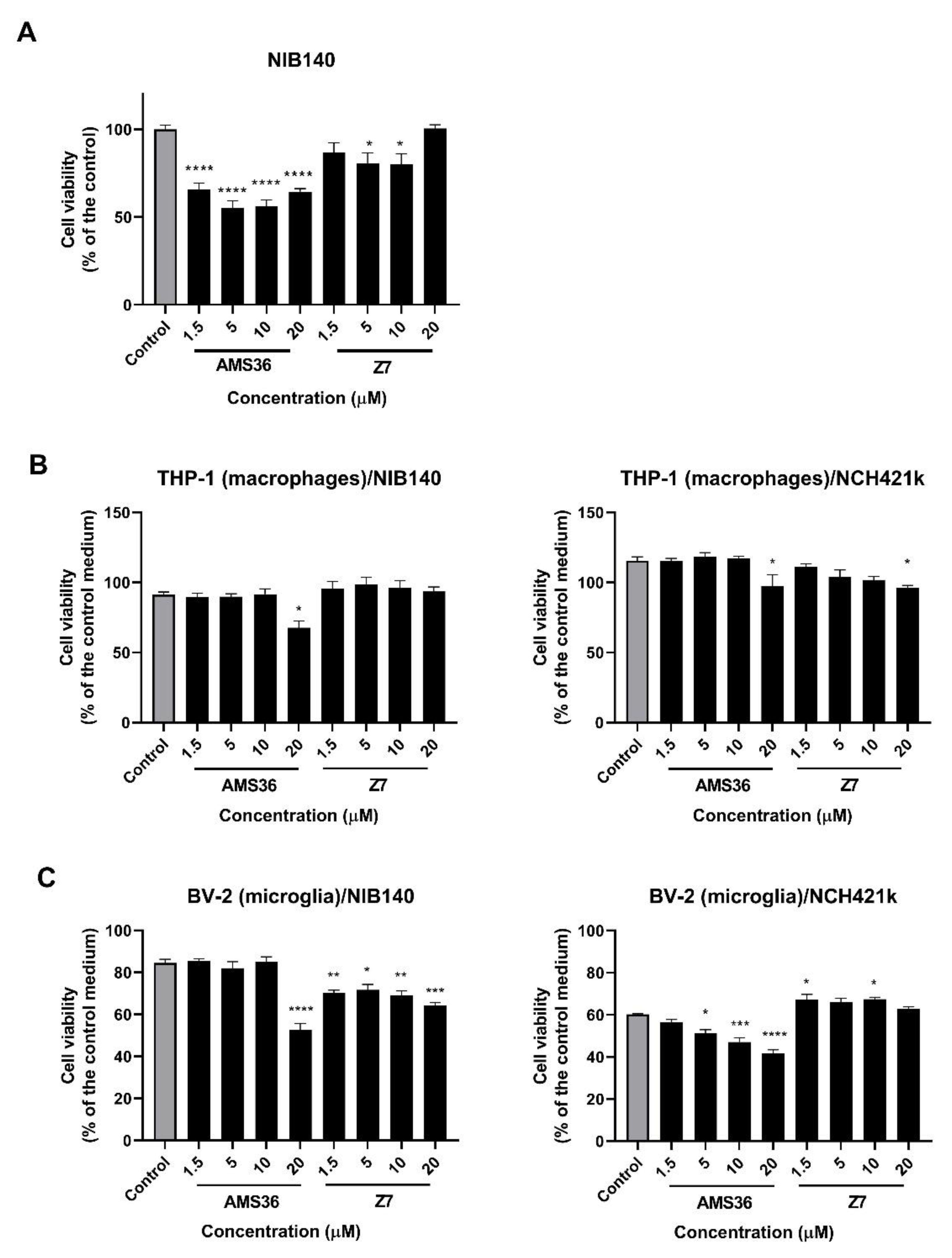
2.3. Cathepsin X Inhibition Decreases Viability of Primary Patient-Derived GBM Cells and GBM-Associated Cells
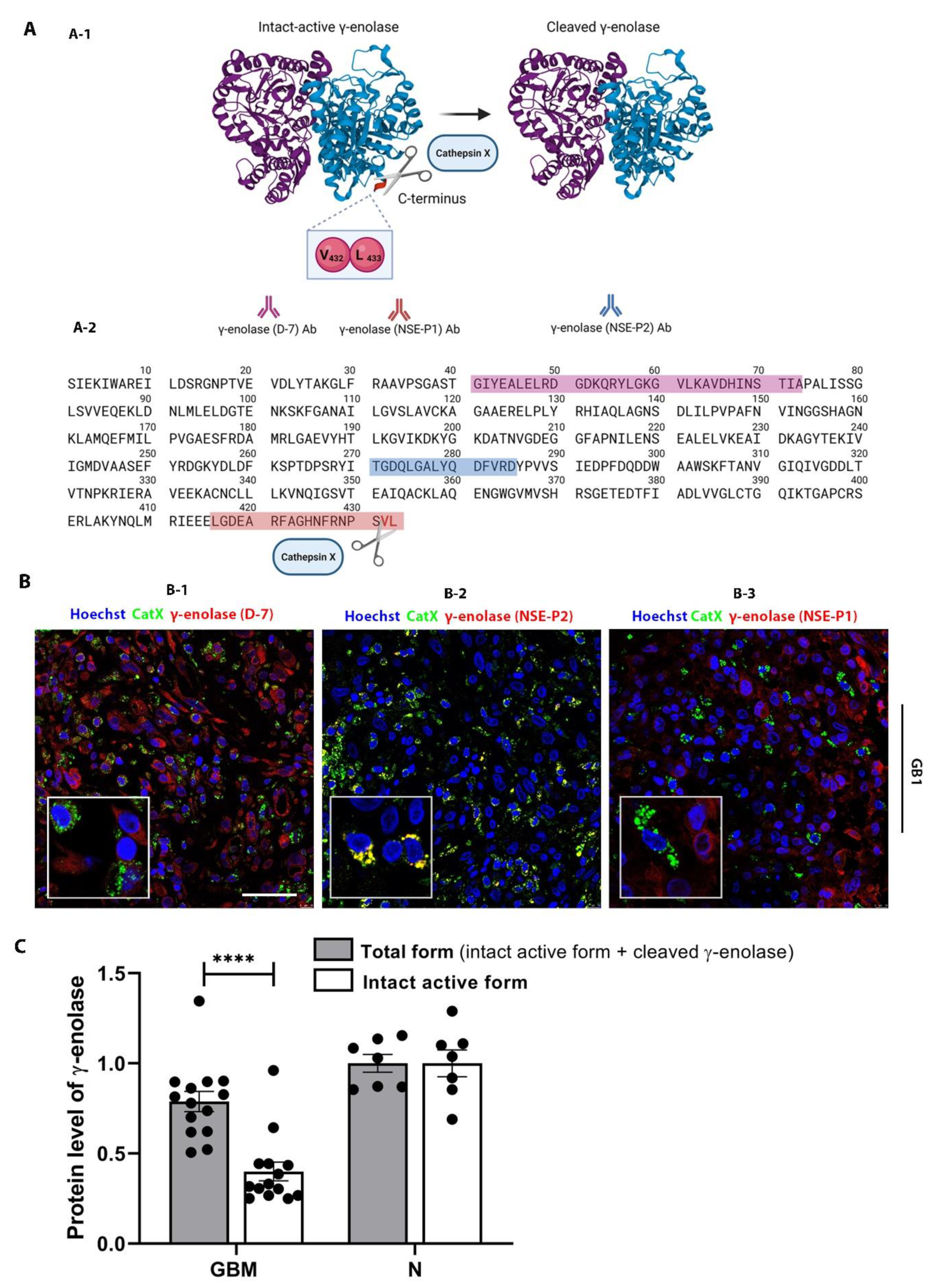
2.4. Interplay of Cathepsin X and γ-Enolase in GBM Tissues
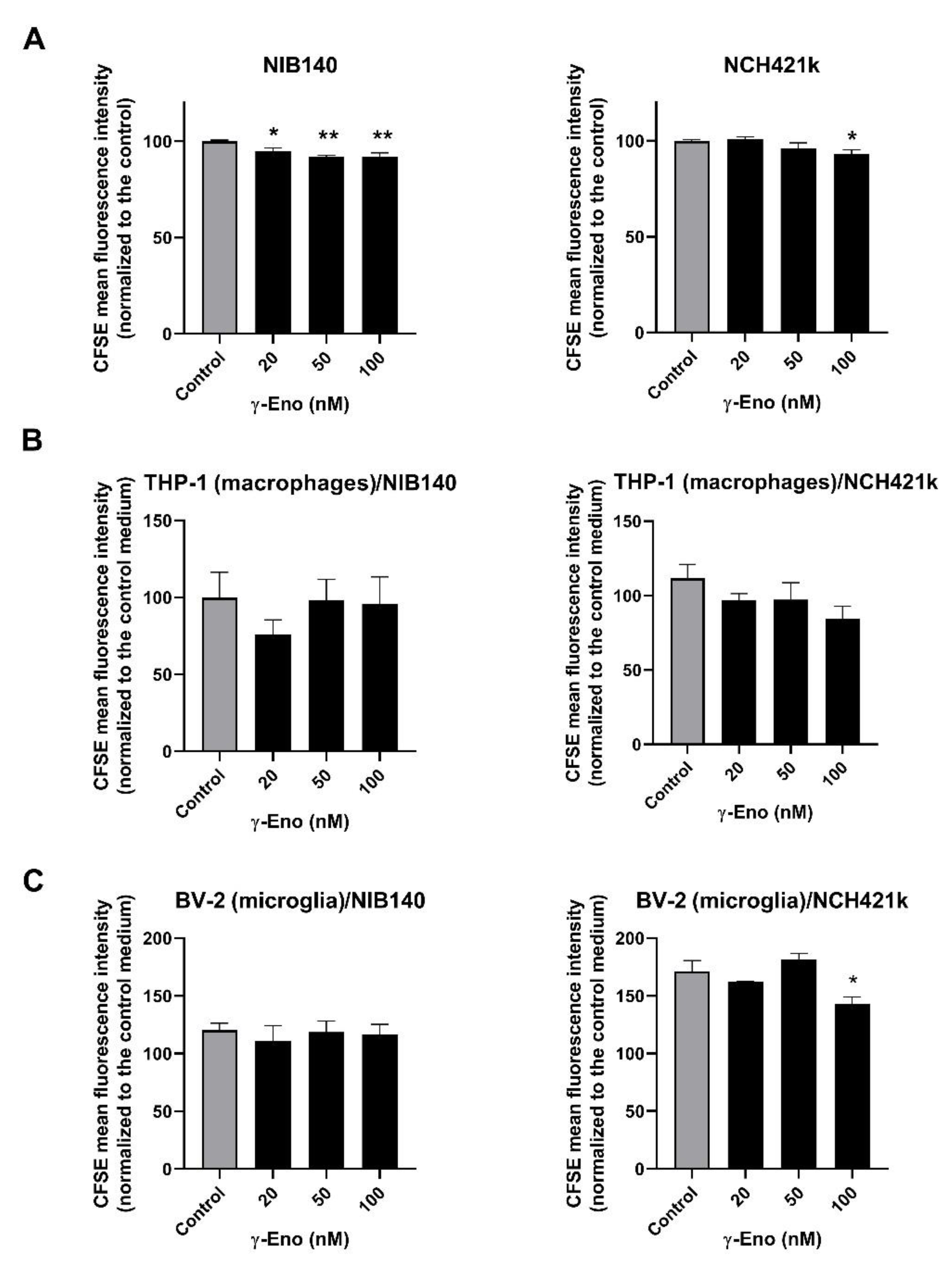
2.5. Proliferation of GBM Cells, GSCs, and GBM-Associated Cells Affected by the γ-Enolase Peptide
3. Discussion
4. Materials and Methods
4.1. Patient Samples
4.2. Establishment of Primary GBM Cells
4.3. GBM and Astrocyte Cell Cultures
4.4. Microglia and Macrophage Cell Cultures
4.5. Coculture Model
4.6. Real-Time Quantitative PCR

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Assay ID | Assay Type | # |
|---|---|---|---|
| GAPDH | Hs99999905_m1 | FAM-MGB, S(250rxns) | 4331182 |
| HPRT1 | Hs02800695_m1 | FAM-MGB, XS(75rxns) | 4453320 |
| COL1A2 | Hs01028956_m1 | FAM-MGB, XS(75rxns) | 4331182 |
| COL1A | Hs00164004_m1 | FAM-MGB, XS(75rxns) | 4331182 |
| TGFBI | Hs00998133_m1 | FAM-MGB, XS(75rxns) | 4331182 |
| THBS1 | Hs00962908_m1 | FAM-MGB, XS(75rxns) | 4331182 |
| DAB2 | Hs01120074_m1 | FAM-MGB, XS(75rxns) | 4331182 |
| S100A4 | Hs00243202_m1 | FAM-MGB, XS(75rxns) | 4331182 |
| P2RX7 | Hs00175721_m1 | FAM-MGB, XS(75rxns) | 4331182 |
| STMN4 | Hs00229288_m1 | FAM-MGB, XS(75rxns) | 4331182 |
| SOX10 | Hs00366918_m1 | FAM-MGB, XS(75rxns) | 4331182 |
| ERBB3 | Hs00176538_m1 | FAM-MGB, XS(75rxns) | 4331182 |
| ACSBG1 | Hs00209500_m1 | FAM-MGB, XS(75rxns) | 4331182 |
| KCNF1 | Hs00266908_s1 | FAM-MGB, XS(75rxns) | 4331182 |
| OLIG2 | Hs00377820_m1 | FAM-MGB, XS(75rxns) | 4331182 |
| NOTCH1 | hs01062014_m1 | FAM-MGB, XS(75rxns) | 4331182 |
| NFKB1 | Hs00765730_m1 | FAM-MGB, XS(75rxns) | 4331182 |
| CD133 (PROM1) | Hs00195682_m1 | FAM-MGB, XS(75rxns) | 4331182 |
| SOX2 | Hs01053049_s1 | FAM-MGB, XS(75rxns) | 4331182 |
| OCT4 (POU5F1B) | Hs01596605_s1 | FAM-MGB, XS(75rxns) | 4331182 |
| CD15 (FUT4) | Hs01106466_s1 | FAM-MGB, XS(75rxns) | 4331182 |
| GFAP | Hs00909233_m1 | FAM-MGB, XS(75rxns) | 4331182 |
| TUB33 | Hs00801390_s1 | FAM-MGB, XS(75rxns) | 4331182 |
| Cathepsin X (CTSZ) | Hs00938366_m1 | FAM-MGB, XS(75rxns) | 4331182 |
4.7. Gene Expression Data Analyses
4.7.1. GBM Subtyping
4.7.2. Differentially Expressed Genes among the GBM Samples
4.7.3. Survival Analysis
4.8. Immunofluorescence
4.9. Protein Extraction from GBM Tissues and Nontumor Brain Tissues
4.10. Cathepsin X Activity
4.11. ELISAs
4.12. Cathepsin X Inhibitors and γ-Enolase C-Terminal Peptide
4.13. Cell Viability Assay
4.14. Cell Proliferation Assay
4.15. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro. Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Weller, M.; Lee, E.Q.; Alexander, B.M.; Barnholtz-Sloan, J.S.; Barthel, F.P.; Batchelor, T.T.; Bindra, R.S.; Chang, S.M.; Chiocca, E.A.; et al. Glioblastoma in adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro. Oncol. 2020, 22, 1073–1113. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Lah, T.T.; Novak, M.; Breznik, B. Brain malignancies: Glioblastoma and brain metastases. Semin. Cancer Biol. 2020, 60, 262–273. [Google Scholar] [CrossRef]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: A randomized clinical trial. J. Am. Med. Assoc. 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [Green Version]
- Teng, J.; Da Hora, C.C.; Kantar, R.S.; Nakano, I.; Wakimoto, H.; Batchelor, T.T.; Antonio Chiocca, E.; Badr, C.E.; Tannous, B.A. Dissecting inherent intratumor heterogeneity in patient-derived glioblastoma culture models. Neuro. Oncol. 2017, 19, 820–832. [Google Scholar] [CrossRef] [Green Version]
- Molina, E.S.; Pillat, M.M.; Moura-Neto, V.; Lah, T.T.; Ulrich, H. Glioblastoma stem-like cells: Approaches for isolation and characterization. J. Cancer Stem Cell Res. 2014, 2, e1007. [Google Scholar] [CrossRef]
- Lathia, J.D.; Mack, S.C.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [Green Version]
- Majc, B.; Sever, T.; Zarić, M.; Breznik, B.; Turk, B.; Lah, T.T. Epithelial-to-mesenchymal transition as the driver of changing carcinoma and glioblastoma microenvironment. Biochim. Biophys. Acta - Mol. Cell Res. 2020, 1867, 118782. [Google Scholar] [CrossRef]
- Broekman, M.L.; Maas, S.L.N.; Abels, E.R.; Mempel, T.R.; Krichevsky, A.M.; Breakefield, X.O. Multidimensional communication in the microenvirons of glioblastoma. Nat. Rev. Neurol. 2018, 14, 482–495. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [Green Version]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef] [Green Version]
- Osuka, S.; Van Meir, E.G. Overcoming therapeutic resistance in glioblastoma: The way forward. J. Clin. Investig. 2017, 127, 415–426. [Google Scholar] [CrossRef] [Green Version]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.B.; Karpova, A.; Gritsenko, M.A.; Kyle, J.E.; Cao, S.; Li, Y.; Rykunov, D.; Colaprico, A.; Rothstein, J.H.; Hong, R.; et al. Proteogenomic and metabolomic characterization of human glioblastoma. Cancer Cell 2021, 39, 509–528. [Google Scholar] [CrossRef]
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M.; Chernikova, S.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Caneda, C.; et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep. 2017, 21, 1399–1410. [Google Scholar] [CrossRef] [Green Version]
- Glass, R.; Synowitz, M. CNS macrophages and peripheral myeloid cells in brain tumours. Acta Neuropathol. 2014, 128, 347–362. [Google Scholar] [CrossRef] [Green Version]
- Morantz, R.A.; Wood, G.W.; Foster, M.; Clark, M.; Gollahon, K. Macrophages in experimental and human brain tumors. Part 2: Studies of the macrophage content of human brain tumors. J. Neurosurg. 1979, 50, 305–311. [Google Scholar] [CrossRef]
- Zhou, W.; Ke, S.Q.; Huang, Z.; Flavahan, W.; Fang, X.; Paul, J.; Wu, L.; Sloan, A.E.; McLendon, R.E.; Li, X.; et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat. Cell Biol. 2015, 17, 170–182. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and molecular identity of tumor-associated macrophages in glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef] [Green Version]
- Kos, J.; Vižin, T.; Pečar Fonović, U.; Pišlar, A. Intracellular signaling by cathepsin X: Molecular mechanisms and diagnostic and therapeutic opportunities in cancer. Semin. Cancer Biol. 2015. [Google Scholar] [CrossRef]
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim. Biophys. Acta 2012, 1824, 68–88. [Google Scholar] [CrossRef] [Green Version]
- Brix, K.; Dunkhorst, A.; Mayer, K.; Jordans, S. Cysteine cathepsins: Cellular roadmap to different functions. Biochimie 2008, 90, 194–207. [Google Scholar] [CrossRef]
- Olson, O.C.; Joyce, J.A. Cysteine cathepsin proteases: Regulators of cancer progression and therapeutic response. Nat. Publ. Gr. 2015. [Google Scholar] [CrossRef]
- Habič, A.; Novak, M.; Majc, B.; Lah Turnšek, T.; Breznik, B. Proteases Regulate Cancer Stem Cell Properties and Remodel Their Microenvironment. J. Histochem. Cytochem. 2021. [Google Scholar] [CrossRef]
- Levicar, N.; Strojnik, T.; Kos, J.; Dewey, R.A.; Pilkington, G.J.; Lah T., T. Lysosomal enzymes, cathepsins in brain tumour invasion. J. Neurooncol. 2002, 58, 21–32. [Google Scholar] [CrossRef]
- Rudzińska, M.; Parodi, A.; Soond, S.M.; Vinarov, A.Z.; Korolev, D.O.; Morozov, A.O.; Daglioglu, C.; Tutar, Y.; Zamyatnin, A.A. The role of cysteine cathepsins in cancer progression and drug resistance. Int. J. Mol. Sci. 2019, 20, 3602. [Google Scholar] [CrossRef]
- Benavides, F.; Perez, C.; Blando, J.; Contreras, O.; Shen, J.; Coussens, L.M.; Fischer, S.M.; Kusewitt, D.F.; Digiovanni, J.; Conti, C.J. Protective role of cathepsin L in mouse skin carcinogenesis. Mol. Carcinog. 2012, 51, 352–361. [Google Scholar] [CrossRef] [Green Version]
- Dennemärker, J.; Lohmüller, T.; Mayerle, J.; Tacke, M.; Lerch, M.M.; Coussens, L.M.; Peters, C.; Reinheckel, T. Deficiency for the cysteine protease cathepsin L promotes tumor progression in mouse epidermis. Oncogene 2009, 29, 1611–1621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Otín, C.; Matrisian, L.M. Emerging roles of proteases in tumour suppression. Nat. Rev. Cancer 2007, 7, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Lah T., T.; Durán Alonso, M.B.; Van Noorden, C.J.F. Antiprotease therapy in cancer: Hot or not? Expert Opin. Biol. Ther. 2006, 6, 257–279. [Google Scholar] [CrossRef] [PubMed]
- Breznik, B.; Limbaeck Stokin, C.; Kos, J.; Khurshed, M.; Hira, V.V.V.; Bošnjak, R.; Lah T., T.; Van Noorden, C.J.F. Cysteine cathepsins B, X and K expression in peri-arteriolar glioblastoma stem cell niches. J. Mol. Histol. 2018, 49, 481–497. [Google Scholar] [CrossRef] [Green Version]
- Kos, J.; Sekirnik, A.; Premzl, A.; Bergant Zavašnik, V.; Langerholc, T.; Turk, B.; Werle, B.; Golouh, R.; Repnik, U.; Jeras, M.; et al. Carboxypeptidases cathepsins X and B display distinct protein profile in human cells and tissues. Exp. Cell Res. 2005, 306, 103–113. [Google Scholar] [CrossRef]
- Dolenc, I.; Štefe, I.; Turk, D.; Taler-Verčič, A.; Turk, B.; Turk, V.; Stoka, V. Human cathepsin X/Z is a biologically active homodimer. Biochim. Biophys. Acta - Proteins Proteomics 2021, 1869, 140567. [Google Scholar] [CrossRef]
- Pišlar, A.; Kos, J. Cysteine cathepsins in neurological disorders. Mol. Neurobiol. 2014, 49, 1017–1030. [Google Scholar] [CrossRef]
- Wendt, W.; Zhu, X.R.; Lübbert, H.; Stichel, C.C. Differential expression of cathepsin X in aging and pathological central nervous system of mice. Exp. Neurol. 2007, 204, 525–540. [Google Scholar] [CrossRef]
- Stichel, C.C.; Luebbert, H. Inflammatory processes in the aging mouse brain: Participation of dendritic cells and T-cells. Neurobiol. Aging 2007, 28, 1507–1521. [Google Scholar] [CrossRef]
- Hafner, A.; Glavan, G.; Obermajer, N.; Živin, M.; Schliebs, R.; Kos, J. Neuroprotective role of γ-enolase in microglia in a mouse model of Alzheimer’s disease is regulated by cathepsin X. Aging Cell 2013, 12, 604–614. [Google Scholar] [CrossRef]
- Pišlar, A.; Tratnjek, L.; Glavan, G.; Zidar, N.; Živin, M.; Kos, J. Neuroinflammation-Induced Upregulation of Glial Cathepsin X Expression and Activity in vivo. Front. Mol. Neurosci. 2020, 13, 1–17. [Google Scholar] [CrossRef]
- Obermajer, N.; Doljak, B.; Jamnik, P.; Pečar Fonović, U.; Kos, J. Cathepsin X cleaves the C-terminal dipeptide of alpha- and gamma-enolase and impairs survival and neuritogenesis of neuronal cells. Int. J. Biochem. Cell Biol. 2009, 41, 1685–1696. [Google Scholar] [CrossRef]
- Haque, A.; Polcyn, R.; Matzelle, D.; Banik, N.L. New insights into the role of neuron-specific enolase in neuro-inflammation, neurodegeneration, and neuroprotection. Brain Sci. 2018, 8, 33. [Google Scholar] [CrossRef] [Green Version]
- Hafner, A.; Obermajer, N.; Kos, J. γ-Enolase C-terminal peptide promotes cell survival and neurite outgrowth by activation of the PI3K/Akt and MAPK/ERK signalling pathways. Biochem. J. 2012, 443, 439–450. [Google Scholar] [CrossRef]
- Takei, N.; Kondo, J.; Nagaike, K.; Ohsawa, K.; Kato, K.; Kohsaka, S. Neuronal survival factor from bovine brain is identical to neuron-specific enolase. J. Neurochem. 1991, 57, 1178–1184. [Google Scholar] [CrossRef]
- Vizin, T.; Kos, J. Gamma-enolase: A well-known tumour marker, with a less-known role in cancer. Radiol. Oncol. 2015, 49, 217. [Google Scholar] [CrossRef] [Green Version]
- Behnan, J.; Stangeland, B.; Hosainey, S.A.M.; Joel, M.; Olsen, T.K.; Micci, F.; Glover, J.C.; Isakson, P.; Brinchmann, J.E. Differential propagation of stroma and cancer stem cells dictates tumorigenesis and multipotency. Oncogene 2017, 36, 570–584. [Google Scholar] [CrossRef] [Green Version]
- Novak, M.; Koprivnikar Krajnc, M.; Hrastar, B.; Breznik, B.; Majc, B.; Mlinar, M.; Rotter, A.; Porčnik, A.; Mlakar, J.; Stare, K.; et al. CCR5-Mediated Signaling Is Involved in Invasion of Glioblastoma Cells in Its Microenvironment. Int. J. Mol. Sci. 2020, 21, 4199. [Google Scholar] [CrossRef]
- Yan, T.; Skaftnesmo, K.O.; Leiss, L.; Sleire, L.; Wang, J.; Li, X.; Enger, P.T. Neuronal markers are expressed in human gliomas and NSE knockdown sensitizes glioblastoma cells to radiotherapy and temozolomide. BMC Cancer 2011, 11, 524. [Google Scholar] [CrossRef] [Green Version]
- Expression of ENO2 in Cancer. The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000111674-ENO2/pathology (accessed on 5 January 2022).
- Sehnal, D.; Bittrich, S.; Deshpande, M.; Svobodová, R.; Berka, K.; Bazgier, V.; Velankar, S.; Burley, S.K.; Koča, J.; Rose, A.S. Mol* Viewer: Modern web app for 3D visualization and analysis of large biomolecular structures. Nucleic Acids Res. 2021, 49, W431–W437. [Google Scholar] [CrossRef]
- Chai, G.; Brewer, J.M.; Lovelace, L.L.; Aoki, T.; Minor, W.; Lebioda, L. Expression, purification and the 1.8 angstroms resolution crystal structure of human neuron specific enolase. J. Mol. Biol. 2004, 341, 1015–1021. [Google Scholar] [CrossRef]
- Hafner, A.; Obermajer, N.; Kos, J. Gamma-1-syntrophin mediates trafficking of gamma-enolase towards the plasma membrane and enhances its neurotrophic activity. NeuroSignals 2011, 18, 246–258. [Google Scholar] [CrossRef]
- Nägler, D.K.; Krüger, S.; Kellner, A.; Ziomek, E.; Menard, R.; Buhtz, P.; Krams, M.; Roessner, A.; Kellner, U. Up-regulation of cathepsin X in prostate cancer and prostatic intraepithelial neoplasia. Prostate 2004, 60, 109–119. [Google Scholar] [CrossRef]
- Krueger, S.; Kalinski, T.; Hundertmark, T.; Wex, T.; Küster, D.; Peitz, U.; Ebert, M.; Nägler, D.K.; Kellner, U.; Malfertheiner, P.; et al. Up-regulation of cathepsin X in Helicobacter pylori gastritis and gastric cancer. J. Pathol. 2005, 207, 32–42. [Google Scholar] [CrossRef]
- Vizin, T.; Christensen, I.; Nielsen, H.; Kos, J. Cathepsin X in serum from patients with colorectal cancer: Relation to prognosis. Radiol. Oncol. 2012, 46, 207–212. [Google Scholar] [CrossRef] [Green Version]
- Akkari, L.; Gocheva, V.; Kester, J.C.; Hunter, K.E.; Quick, M.L.; Sevenich, L.; Wang, H.W.; Peters, C.; Tang, L.H.; Klimstra, D.S.; et al. Distinct functions of macrophage-derived and cancer cell-derived cathepsin Z combine to promote tumor malignancy via interactions with the extracellular matrix. Genes Dev. 2014, 28, 2134–2150. [Google Scholar] [CrossRef] [Green Version]
- Turk, B. Targeting proteases: Successes, failures and future prospects. Nat. Rev. Drug Discov. 2006, 5, 785–799. [Google Scholar] [CrossRef]
- Kramer, L.; Turk, D.; Turk, B. The Future of Cysteine Cathepsins in Disease Management. Trends Pharmacol. Sci. 2017, 38, 873–898. [Google Scholar] [CrossRef]
- Pečar Fonović, U.; Mitrović, A.; Knez, D.; Jakoš, T.; Pišlar, A.; Brus, B.; Doljak, B.; Stojan, J.; Žakelj, S.; Trontelj, J.; et al. Identification and characterization of the novel reversible and selective cathepsin X inhibitors. Sci. Rep. 2017, 7, 11459. [Google Scholar] [CrossRef] [Green Version]
- Sadaghiani, A.M.; Verhelst, S.H.L.; Gocheva, V.; Hill, K.; Majerova, E.; Stinson, S.; Joyce, J.A.; Bogyo, M. Design, synthesis, and evaluation of in vivo potency and selectivity of epoxysuccinyl-based inhibitors of papain-family cysteine proteases. Chem. Biol. 2007, 14, 499–511. [Google Scholar] [CrossRef]
- Hafner Pišlar, A.; Zidar, N.; Kikelj, D.; Kos, J. Cathepsin X promotes 6-hydroxydopamine-induced apoptosis of PC12 and SH-SY5Y cells. Neuropharmacology 2014, 82, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Cuddapah, V.A.; Robel, S.; Watkins, S.; Sontheimer, H. A neurocentric perspective on glioma invasion. Nat. Rev. Neurosci. 2014, 15, 455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obermajer, N.; Jevnikar, Z.; Doljak, B.; Sadaghiani, A.M.; Bogyo, M.; Kos, J. Cathepsin X-mediated β2 integrin activation results in nanotube outgrowth. Cell. Mol. Life Sci. 2009, 66, 1126–1134. [Google Scholar] [CrossRef] [PubMed]
- Pečar Fonović, U.; Jevnikar, Z.; Rojnik, M.; Doljak, B.; Fonović, M.; Jamnik, P.; Kos, J. Profilin 1 as a Target for Cathepsin X Activity in Tumor Cells. PLoS ONE 2013, 8, e53918. [Google Scholar] [CrossRef]
- Hattori, T.; Ohsawa, K.; Mizuno, Y.; Kato, K.; Kohsaka, S. Synthetic peptide corresponding to 30 amino acids of the C-terminal of neuron-specific enolase promotes survival of neocortical neurons in culture. Biochem. Biophys. Res. Commun. 1994, 202, 25–30. [Google Scholar] [CrossRef]
- Hattori, T.; Takei, N.; Mizuno, Y.; Kato, K.; Kohsaka, S. Neurotrophic and neuroprotective effects of neuron-specific enolase on cultured neurons from embryonic rat brain. Neurosci. Res. 1995, 21, 191–198. [Google Scholar] [CrossRef]
- Tapia, F.J.; Barbosa, A.J.A.; Marangos, P.J.; Polak, J.M.; Bloom, S.R.; Dermody, C.; Pearse, A.G.E. Neuron-specific enolase is produced by neuroendocrine tumours. Lancet 1981, 1, 808–811. [Google Scholar] [CrossRef]
- Baebler, Š.; Svalina, M.; Petek, M.; Stare, K.; Rotter, A.; Pompe-Novak, M.; Gruden, K. quantGenius: Implementation of a decision support system for qPCR-based gene quantification. BMC Bioinform. 2017, 18, 276. [Google Scholar] [CrossRef]
- Kassambara, A.; Mundt, F. factoextra: Extract and Visualize the Results of Multivariate Data Analyses. R Package Version 1.0.7. 2020. Available online: https://CRAN.R-project.org/package=factoextra (accessed on 1 February 2022).
- Maechler, M.; Rousseeuw, P.; Struyf, A.; Hubert, M.; Hornik, K. cluster: Cluster Analysis Basics and Extensions. R package version 2019. [Google Scholar]
- Lah, T.T.; Novak, M.; Pena, A.M.A.; Marinelli, O.; Žvar, B.B.; Majc, B.; Mlinar, M.; Bošnjak, R.; Breznik, B.; Zomer, R.; et al. Cannabigerol Is a Potential Therapeutic Agent in a Novel Combined Therapy for Glioblastoma. Cells 2021, 10, 340. [Google Scholar] [CrossRef]
- Mitrović, A.; Sosič, I.; Kos, Š.; Tratar, U.L.; Breznik, B.; Kranjc, S.; Mirković, B.; Gobec, S.; Lah, T.T.; Serša, G.; et al. Addition of 2-(ethylamino)acetonitrile group to nitroxoline results in significantly improved anti-tumor activity in vitro and in vivo. Oncotarget 2017, 8, 59136–59147. [Google Scholar] [CrossRef] [Green Version]
- Breznik, B.; Motaln, H.; Vittori, M.; Rotter, A.; Lah, T.T. Mesenchymal stem cells differentially affect the invasion of distinct glioblastoma cell lines. Oncotarget 2017, 8, 25482–25499. [Google Scholar] [CrossRef] [Green Version]
- Moriconi, C.; Palmieri, V.; Di Santo, R.; Tornillo, G.; Papi, M.; Pilkington, G.; De Spirito, M.; Gumbleton, M. INSIDIA: A FIJI Macro Delivering High-Throughput and High-Content Spheroid Invasion Analysis. Biotechnol. J. 2017, 10. [Google Scholar] [CrossRef]

| Primary Antibodies | Source | Dilution |
|---|---|---|
| Goat polyclonal anti-cathepsin X | R&D System (AG934) | 1:200 |
| Mouse monoclonal anti-Iba1 | Abcam (ab15690) | 1:200 |
| Rabbit polyclonal anti-CD68 | Atlas antibodies (HPA048982) | 1:2500 |
| Mouse monoclonal anti-SOX2 | Abcam (ab171380) | 1:50 |
| Rabbit polyclonal anti-CD133 | Abcam (ab19898) | 1:100 |
| Rabbit polyclonal anti-GFAP | Abcam (ab211271) | 1:1000 |
| Mouse monoclonal anti-γ-enolase (NSE-P1) | Santa Cruz Biotechnology (sc-21738) | 1:250 |
| Mouse monoclonal anti-γ-enolase (NSE-P2) | Santa Cruz Biotechnology (sc-21737) | 1:250 |
| Mouse monoclonal anti-γ-enolase (D-7) | Santa Cruz Biotechnology (sc-376375) | 1:250 |
| Secondary Antibodies | Source | Dilution |
|---|---|---|
| Donkey anti-goat IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 | Thermo Fisher Scientific (A32814) | 1:200 |
| Donkey anti-mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 647 | Thermo Fisher Scientific (A32787) | 1:200 |
| Donkey anti-rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 546 | Thermo Fisher Scientific (A10040) | 1:200 |
| Donkey anti-mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 546 | Thermo Fisher Scientific (A10036) | 1:200 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Majc, B.; Habič, A.; Novak, M.; Rotter, A.; Porčnik, A.; Mlakar, J.; Župunski, V.; Pečar Fonović, U.; Knez, D.; Zidar, N.; et al. Upregulation of Cathepsin X in Glioblastoma: Interplay with γ-Enolase and the Effects of Selective Cathepsin X Inhibitors. Int. J. Mol. Sci. 2022, 23, 1784. https://doi.org/10.3390/ijms23031784
Majc B, Habič A, Novak M, Rotter A, Porčnik A, Mlakar J, Župunski V, Pečar Fonović U, Knez D, Zidar N, et al. Upregulation of Cathepsin X in Glioblastoma: Interplay with γ-Enolase and the Effects of Selective Cathepsin X Inhibitors. International Journal of Molecular Sciences. 2022; 23(3):1784. https://doi.org/10.3390/ijms23031784
Chicago/Turabian StyleMajc, Bernarda, Anamarija Habič, Metka Novak, Ana Rotter, Andrej Porčnik, Jernej Mlakar, Vera Župunski, Urša Pečar Fonović, Damijan Knez, Nace Zidar, and et al. 2022. "Upregulation of Cathepsin X in Glioblastoma: Interplay with γ-Enolase and the Effects of Selective Cathepsin X Inhibitors" International Journal of Molecular Sciences 23, no. 3: 1784. https://doi.org/10.3390/ijms23031784
APA StyleMajc, B., Habič, A., Novak, M., Rotter, A., Porčnik, A., Mlakar, J., Župunski, V., Pečar Fonović, U., Knez, D., Zidar, N., Gobec, S., Kos, J., Lah Turnšek, T., Pišlar, A., & Breznik, B. (2022). Upregulation of Cathepsin X in Glioblastoma: Interplay with γ-Enolase and the Effects of Selective Cathepsin X Inhibitors. International Journal of Molecular Sciences, 23(3), 1784. https://doi.org/10.3390/ijms23031784