
Cardioprotection of Immature Heart by Simultaneous Activation of PKA and Epac: A Role for the Mitochondrial Permeability Transition Pore



Abstract
1. Introduction
2. Results
2.1. The Effects of cAMP Analogue Results in Whole Heart
2.1.1. Effects of the cAMP Analogues on CK Activity in Coronary Effluent
2.1.2. Effects of the cAMP Analogues on Infarct Size following Perfusion
2.2. Mitochondrial Permeability Transition Pore Inhibition
2.2.1. The Effect of Isoprenaline-Induced Cardiac β-Adrenoreceptor Stimulation on MPTP Opening in Adult and Immature Hearts Not Exposed to I/R Injury
2.2.2. Amelioration of the Effect of I/R injury on MPTP Opening by Isoprenaline or 8-Br-cAMP Perfusion
2.2.3. Effects of CPT and 6-Bnz
3. Discussion
3.1. Combined Stimulation of PKA and Epac Provides Maximal Protection against Injury in the Adult Heart
3.2. The Immature Perfused Heart Shows Increased Resistance to Injury
3.3. A Combined Action of PKA and Epac Is Needed to Protect an Immature Heart against Ischaemia/Reperfusion
3.4. The Immature Heart’s Mitochondria Are Less Susceptible to Ca2+-Induced Swelling via the MPTP Than the Adult Heart’s
3.5. Ischaemia and Reperfusion Injury Sensitises the MPTP to Ca2+ in Both Immature and Adult Hearts
3.6. The MPTP Is less Likely to Open in the Immature Heart Exposed to Injury Than the Adult Heart
3.7. MPTP Inhibition Is Replicated by cAMP Analogues but Requires PKA and Epac Synergy
4. Materials and Methods
4.1. Animals Used for Experimentation
4.2. Extraction of Hearts
4.3. Langendorff Perfusion
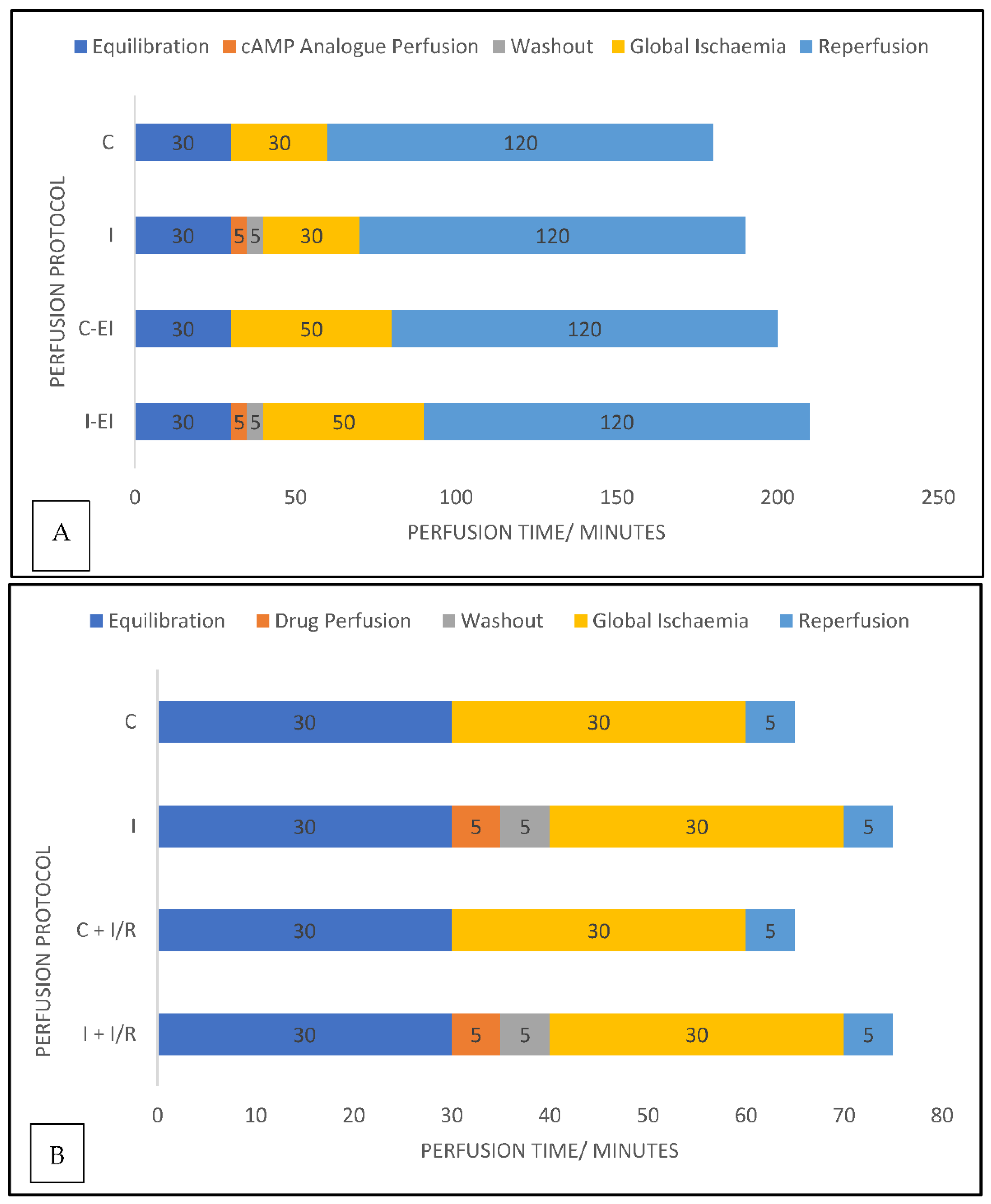
4.4. Experimental Protocols
4.5. Measurement of Creatine Kinase Activity
4.6. Measurement of Infarct Size
4.7. Perfusion Protocols for Mitochondrial Isolation Experiments
4.8. Mitochondrial Isolation
4.9. Mitochondrial Swelling Assay
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Depre, C.; Taegtmeyer, H. Metabolic aspects of programmed cell survival and cell death in the heart. Cardiovasc. Res. 2000, 45, 538–548. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion—from mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Pasdois, P. The role of the mitochondrial permeability transition pore in heart disease. Biochim. Biophys. Acta 2009, 1787, 1402–1415. [Google Scholar] [CrossRef] [PubMed]
- Honda, H.M.; Korge, P.; Weiss, J.N. Mitochondria and Ischemia/Reperfusion Injury. Ann. N. Y. Acad. Sci. 2005, 1047, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Clarke, S.J.; Javadov, S.A. Mitochondrial permeability transition pore opening during myocardial reperfusion—a target for cardioprotection. Cardiovasc. Res. 2004, 61, 372–385. [Google Scholar] [CrossRef]
- Lochner, A.; Genade, S.; Tromp, E.; Podzuweit, T.; Moolman, J.A. Ischemic preconditioning and the beta-adrenergic signal transduction pathway. Circulation 1999, 100, 958–966. [Google Scholar] [CrossRef]
- Khaliulin, I.; Halestrap, A.P.; Bryant, S.M.; Dudley, D.J.; James, A.F.; Suleiman, M.-S. Clinically-relevant consecutive treatment with isoproterenol and adenosine protects the failing heart against ischaemia and reperfusion. J. Transl. Med. 2014, 12, 139. [Google Scholar] [CrossRef]
- Khaliulin, I.; Parker, J.E.; Halestrap, A.P. Consecutive pharmacological activation of PKA and PKC mimics the potent cardioprotection of temperature preconditioning. Cardiovasc. Res. 2010, 88, 324–333. [Google Scholar] [CrossRef]
- Cannavo, A.; Liccardo, D.; Koch, W.J. Targeting cardiac β-adrenergic signaling via GRK2 inhibition for heart failure therapy. Front. Physiol. 2013, 4, 264. [Google Scholar] [CrossRef]
- Lymperopoulos, A.; Rengo, G.; Koch, W.J. Adrenergic nervous system in heart failure: Pathophysiology and therapy. Circ. Res. 2013, 113, 739–753. [Google Scholar] [CrossRef]
- Bers, D.M. Going to cAMP just got more complicated. J. Physiol. 2007, 583 Pt 2, 415–416. [Google Scholar] [CrossRef] [PubMed]
- De Rooij, J.; Zwartkruis, F.J.; Verheijen, M.H.; Cool, R.H.; Nijman, S.M.; Wittinghofer, A.; Bos, J.L. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar] [CrossRef] [PubMed]
- Okumura, S.; Fujita, T.; Cai, W.; Jin, M.; Namekata, I.; Mototani, Y.; Jin, H.; Ohnuki, Y.; Tsuneoka, Y.; Kurotani, R.; et al. Epac1-dependent phospholamban phosphorylation mediates the cardiac response to stresses. J. Clin. Investig. 2014, 124, 2785–2801. [Google Scholar] [CrossRef]
- Pereira, L.; Ruiz-Hurtado, G.; Morel, E.; Laurent, A.-C.; Métrich, M.; Domínguez-Rodríguez, A.; Lauton-Santos, S.; Lucas, A.; Benitah, J.-P.; Bers, D.M.; et al. Epac enhances excitation–transcription coupling in cardiac myocytes. J. Mol. Cell. Cardiol. 2012, 52, 283–291. [Google Scholar] [CrossRef]
- Birnbaum, Y.; Ye, Y.; Rosanio, S.; Tavackoli, S.; Hu, Z.-Y.; Schwarz, E.R.; Uretsky, B.F. Prostaglandins mediate the cardioprotective effects of atorvastatin against ischemia?reperfusion injury. Cardiovasc. Res. 2005, 65, 345–355. [Google Scholar] [CrossRef]
- Xiao, C.-Y.; Yuhki, K.-I.; Hara, A.; Fujino, T.; Kuriyama, S.; Yamada, T.; Takayama, K.; Takahata, O.; Karibe, H.; Taniguchi, T.; et al. Prostaglandin E 2 Protects the Heart From Ischemia-Reperfusion Injury via Its Receptor Subtype EP 4. Circulation 2004, 109, 2462–2468. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Harding, P.; LaPointe, M.C. PKA, Rap1, ERK1/2, and p90RSK mediate PGE2 and EP4 signaling in neonatal ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H136–H143. [Google Scholar] [CrossRef]
- Chang, G.; Liu, J.; Qin, S.; Jiang, Y.; Zhang, P.; Yu, H.; Lu, K.; Zhang, N.; Cao, L.; Wang, Y.; et al. Cardioprotection by exenatide: A novel mechanism via improving mitochondrial function involving the GLP-1 receptor/cAMP/PKA pathway. Int. J. Mol. Med. 2017, 41, 1693–1703. [Google Scholar] [CrossRef]
- Dudley, D.J.; Suleiman, M.S.; Bond, M.; James, A.F.; Khaliulin, I. 10 Cell-Permeable Cyclic AMP as a Novel Cardioprotective Agent. Heart 2014, 100, A4. [Google Scholar] [CrossRef]
- Khaliulin, I.; Ascione, R.; Maslov, L.N.; Amal, H.; Suleiman, M.S. Preconditioning or Postconditioning with 8-Br-cAMP-AM Protects the Heart against Regional Ischemia and Reperfusion: A Role for Mitochondrial Permeability Transition. Cells 2021, 10, 1223. [Google Scholar] [CrossRef]
- Khaliulin, I.; Bond, M.; James, A.F.; Dyar, Z.; Amini, R.; Johnson, J.L.; Suleiman, M. Functional and cardioprotective effects of simultaneous and individual activation of protein kinase A and Epac. J. Cereb. Blood Flow Metab. 2017, 174, 438–453. [Google Scholar] [CrossRef] [PubMed]
- Ostadal, B.; Ostadalova, I.; Kolar, F.; Sedmera, D. Developmental determinants of cardiac sensitivity to hypoxia. Can. J. Physiol. Pharmacol. 2014, 92, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Ostadalova, I.; Ostadal, B.; Kolar, F.; Parratt, J.; Wilson, S. Tolerance to Ischaemia and Ischaemic Preconditioning in Neonatal Rat Heart. J. Mol. Cell. Cardiol. 1998, 30, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Vetter, R.; Studer, R.; Reinecke, H.; Kolar, F.; Otadalova, I.; Drexler, H.; Ostádalová, I. Reciprocal changes in the postnatal expression of the sarcolemmal Na+-Ca2+-exchanger and SERCA2 in rat heart. J. Mol. Cell. Cardiol. 1995, 27, 1689–1701. [Google Scholar] [CrossRef]
- Studer, R.; Reinecke, H.; Vetter, R.; Holtz, J.; Drexler, H. Expression and function of the cardiac Na+/Ca2+ exchanger in postnatal development of the rat, in experimental-induced cardiac hypertrophy and in the failing human heart. Basic Res. Cardiol. 1997, 92 (Suppl. 1), 53–58. [Google Scholar] [CrossRef]
- Balaska, D.; Halestrap, A.; Suleiman, S.; Griffths, E. Increased susceptibility to pore-opening in heart mitochondria from neonatal compared with adult rats. J. Physiol. 2005, 567, PC31. [Google Scholar] [CrossRef]
- Lewis, M.; Szobi, A.; Balaska, D.; Khaliulin, I.; Adameova, A.; Griffiths, E.; Orchard, C.H.; Suleiman, M.-S. Consecutive Isoproterenol and Adenosine Treatment Confers Marked Protection against Reperfusion Injury in Adult but Not in Immature Heart: A Role for Glycogen. Int. J. Mol. Sci. 2018, 19, 494. [Google Scholar] [CrossRef]
- Dorn, G.W., 2nd; Tepe, N.M.; Wu, G.; Yatani, A.; Liggett, S.B. Mechanisms of impaired beta-adrenergic receptor signaling in G(alphaq)-mediated cardiac hypertrophy and ventricular dysfunction. Mol. Pharmacol. 2000, 57, 278–287. [Google Scholar]
- Appukuttan, A.; Kasseckert, S.A.; Micoogullari, M.; Flacke, J.-P.; Kumar, S.; Woste, A.; Abdallah, Y.; Pott, L.; Reusch, H.P.; Ladilov, Y. Type 10 adenylyl cyclase mediates mitochondrial Bax translocation and apoptosis of adult rat cardiomyocytes under simulated ischaemia/reperfusion. Cardiovasc. Res. 2011, 93, 340–349. [Google Scholar] [CrossRef]
- Rau, C.D.; Wang, J.; Avetisyan, R.; Romay, M.C.; Martin, L.; Ren, S.; Wang, Y.; Lusis, A.J. Mapping genetic contributions to cardiac pathology induced by Beta-adrenergic stimulation in mice. Circ. Cardiovasc. Genet. 2015, 8, 40–49. [Google Scholar] [CrossRef]
- Ošťádal, B.; Ošťádalová, I.; Kolar, F.; Charvátová, Z.; Netuka, I. Ontogenetic development of cardiac tolerance to oxygen deprivation—possible mechanisms. Physiol. Res. 2009, 58 (Suppl. 2), S1–S12. [Google Scholar] [CrossRef] [PubMed]
- Modi, P.; Suleiman, M.-S. Myocardial taurine, development and vulnerability to ischemia. Amino Acids 2003, 26, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Imura, H.; Caputo, M.; Parry, A.; Pawade, A.; Angelini, G.D.; Suleiman, M.-S. Age-Dependent and Hypoxia-Related Differences in Myocardial Protection during Pediatric Open Heart Surgery. Circulation 2001, 103, 1551–1556. [Google Scholar] [CrossRef] [PubMed]
- Juhaszova, M.; Zorov, D.B.; Kim, S.H.; Pepe, S.; Fu, Q.; Fishbein, K.W.; Ziman, B.D.; Wang, S.; Ytrehus, K.; Antos, C.L.; et al. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J. Clin. Investig. 2004, 113, 1535–1549. [Google Scholar] [CrossRef]
- Gomez, L.; Paillard, M.; Thibault, H.; Derumeaux, G.; Ovize, M. Inhibition of GSK3beta by postconditioning is required to prevent opening of the mitochondrial permeability transition pore during reperfusion. Circulation 2008, 117, 2761–2768. [Google Scholar] [CrossRef] [PubMed]
- Omori, K.; Kotera, J. Overview of PDEs and Their Regulation. Circ. Res. 2007, 100, 309–327. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Nweze, I.; Lakshmanan, J.; Harbrecht, B.G. Activation of a Cyclic Amp-Guanine Exchange Factor in Hepatocytes Decreases Nitric Oxide Synthase Expression. Shock 2013, 39, 70–76. [Google Scholar] [CrossRef]
- Li, L.; Cai, H.; Liu, H.; Guo, T. β-adrenergic stimulation activates protein kinase Cε and induces extracellular signal-regulated kinase phosphorylation and cardiomyocyte hypertrophy. Mol. Med. Rep. 2015, 11, 4373–4380. [Google Scholar] [CrossRef][Green Version]
- Duquesnes, N.; Derangeon, M.; Métrich, M.; Lucas, A.; Mateo, P.; Li, L.; Morel, E.; Lezoualc’H, F.; Crozatier, B. Epac stimulation induces rapid increases in connexin43 phosphorylation and function without preconditioning effect. Pflügers Arch.-Eur. J. Physiol. 2010, 460, 731–741. [Google Scholar] [CrossRef]
- Khaliulin, I.; Clarke, S.J.; Lin, H.; Parker, J.; Suleiman, M.-S.; Halestrap, A.P. Temperature preconditioning of isolated rat hearts—A potent cardioprotective mechanism involving a reduction in oxidative stress and inhibition of the mitochondrial permeability transition pore. J. Physiol. 2007, 581, 1147–1161. [Google Scholar] [CrossRef]
- Cazorla, O.; Lucas, A.; Poirier, F.; Lacampagne, A.; Lezoualc’H, F. The cAMP binding protein Epac regulates cardiac myofilament function. Proc. Natl. Acad. Sci. USA 2009, 106, 14144–14149. [Google Scholar] [CrossRef] [PubMed]
- Novalija, E.; Kevin, L.G.; Camara, A.K.; Bosnjak, Z.J.; Kampine, J.P.; Stowe, D.F. Reactive oxygen species precede the epsilon isoform of protein kinase C in the anesthetic preconditioning signaling cascade. Anesthesiology 2003, 99, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Dekker, L.R.; Coronel, R.; VanBavel, E.; Spaan, J.A.; Opthof, T. Intracellular Ca2+ and delay of ischemia-induced electrical uncoupling in preconditioned rabbit ventricular myocardium. Cardiovasc. Res. 1999, 44, 101–112. [Google Scholar] [CrossRef]
- Suleiman, M.-S.; Halestrap, A.; Griffiths, E. Mitochondria: A target for myocardial protection. Pharmacol. Ther. 2001, 89, 29–46. [Google Scholar] [CrossRef]
- Riva, E.; Hearse, D.J. Age-dependent changes in myocardial susceptibility to ischemic injury. Cardioscience 1993, 4, 85–92. [Google Scholar]
- Ong, S.-B.; Samangouei, P.; Kalkhoran, S.B.; Hausenloy, D.J. The mitochondrial permeability transition pore and its role in myocardial ischemia reperfusion injury. J. Mol. Cell. Cardiol. 2015, 78, 23–34. [Google Scholar] [CrossRef]
- Ong, S.-B.; Dongworth, R.K.; Cabrera-Fuentes, H.A.; Hausenloy, D.J. Role of the MPTP in conditioning the heart—translatability and mechanism. J. Cereb. Blood Flow Metab. 2015, 172, 2074–2084. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| cAMP Analogue | 6-Bnz-cAMP-AM (6-Bnz) | 8-CPT-2′-O-Me-cAMP-AM (CPT) | 8-Br-cAMP-AM (8-Br) |
|---|---|---|---|
| Function | PKA activator | Epac Activator | Activator of both |
| Perfusion Concentration | 5 µM | 10 µM | 5 µM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lewis, M.J.; Khaliulin, I.; Hall, K.; Suleiman, M.S. Cardioprotection of Immature Heart by Simultaneous Activation of PKA and Epac: A Role for the Mitochondrial Permeability Transition Pore. Int. J. Mol. Sci. 2022, 23, 1720. https://doi.org/10.3390/ijms23031720
Lewis MJ, Khaliulin I, Hall K, Suleiman MS. Cardioprotection of Immature Heart by Simultaneous Activation of PKA and Epac: A Role for the Mitochondrial Permeability Transition Pore. International Journal of Molecular Sciences. 2022; 23(3):1720. https://doi.org/10.3390/ijms23031720
Chicago/Turabian StyleLewis, Martin John, Igor Khaliulin, Katie Hall, and M. Saadeh Suleiman. 2022. "Cardioprotection of Immature Heart by Simultaneous Activation of PKA and Epac: A Role for the Mitochondrial Permeability Transition Pore" International Journal of Molecular Sciences 23, no. 3: 1720. https://doi.org/10.3390/ijms23031720
APA StyleLewis, M. J., Khaliulin, I., Hall, K., & Suleiman, M. S. (2022). Cardioprotection of Immature Heart by Simultaneous Activation of PKA and Epac: A Role for the Mitochondrial Permeability Transition Pore. International Journal of Molecular Sciences, 23(3), 1720. https://doi.org/10.3390/ijms23031720