
Therapeutic Targeting of DNA Damage Response in Cancer


Abstract
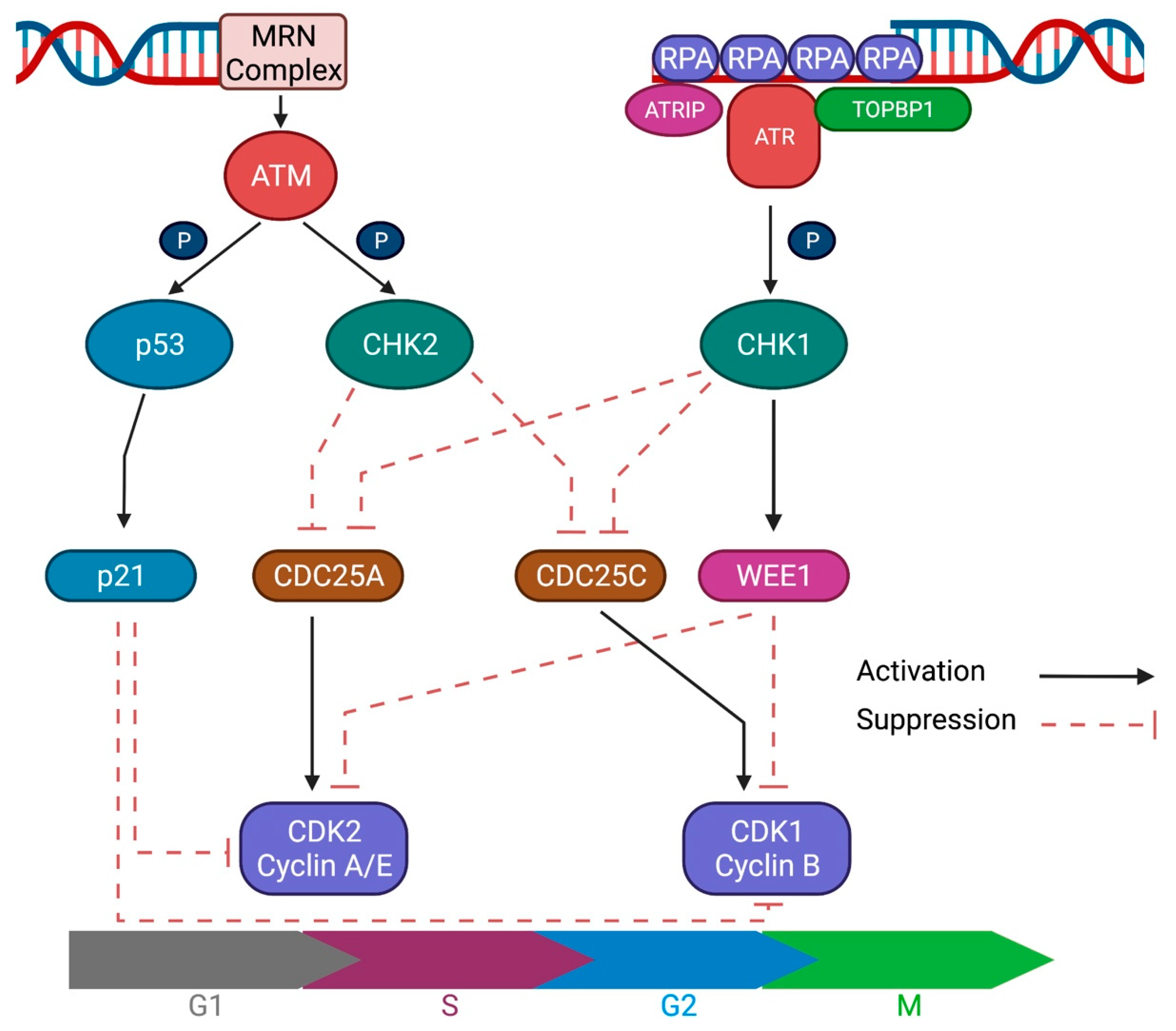
:1. Introduction
2. Mechanisms of DNA Damage Response
2.1. Single-Strand Break
2.1.1. Base Excision Repair (BER)
2.1.2. Nucleotide Excision Repair (NER)
2.2. Double-Strand Break (DSB)
2.2.1. Non-Homologous End Joining (NHEJ)
2.2.2. Homologous Recombination (HR)
2.3. DNA Damage Response Signaling
3. Therapeutic Exploitation of DNA Damage Response
3.1. PARP Inhibitors
3.2. DDR Pathway Inhibitors beyond PARP Inhibitors
3.2.1. ATR Inhibitors

{kind=link}
| Trial Phase | Disease Setting | Treatments | Most Common Grade ≥ 3 Toxicity | Efficacy | Reference |
|---|---|---|---|---|---|
| Berzosertib/M6620/VX-970 | |||||
| 1 | Solid tumors | Escalating doses of M6620 with topotecan | Anemia (19%), Leukopenia (19%), Neutropenia (19%) | PR 2/21 (10%) SD 7/21 (33%) | [51] |
| 1 | Solid tumors | Escalating doses of M6620, or combination with carboplatin | Monotherapy: None With carboplatin: Neutropenia (22%) | Monotherapy: PR 1/17 (6%) SD 5/17 (29%) With carboplatin: PR 1/21 (5%) SD 15/21 (71%) | [52] |
| 1 | Solid tumors | Escalating doses of berzosertib with cisplatin | Neutropenia (20%), Anemia (17%) | PR 4/26 (15%) SD 15/26 (58%) | [53] |
| 1 | Solid tumors | Escalating doses of berzosertib with gemcitabine +/− cisplatin | With gemcitabine Neutropenia (16%) ALT increased (16%) Fatigue (16%) With gemcitabine + cisplatin Neutropenia (63%) Thrombocytopenia (38%) | With gemcitabine PR 4/48 (8%) SD 29/48 (60%) With gemcitabine + cisplatin PR 1/7 (14%) SD 4/7 (57%) | [54] |
| 2 | Ovarian cancer (Platinum-resistant) | Randomization (1:1) Gemcitabine +/− berzosertib | Gemcitabine + berzosertib: Neutropenia (35%) Anemia (15%) Gemcitabine alone: Neutropenia (28%) Anemia (11%) | Gemcitabine + berzosertib PFS 22.9 weeks Gemcitabine alone PFS 14.7 weeks (HR 0.57, 90% CI 0.33–0.98) | [55] |
| 2 | Urothelial carcinoma | Randomization (1:1) Gemcitabine + Cisplatin +/− berzosertib | Gemcitabine + cisplatin + berzosertib: Anemia (57%) Gemcitabine + cisplatin: Anemia (25%) | Gemcitabine + cisplatin + berzosertib: PFS 8.0 months OS 14.4 months Gemcitabine + cisplatin: PFS 8.0 months OS 19.8 months HR for PFS: 1.22 (95% CI 0.72–2.08) HR for OS: 1.42 (95% CI 0.76–2.68) | [56] |
| Ceralasertib/AZD6738 | |||||
| 1 | Solid tumors | Escalating doses of berzosertib with paclitaxel | Neutropenia (30%) Anemia (23%) | CR 1/57 (2%) PR 12/57 (21%) SD 18/57 (32%) | [59] |
| 1 | Solid tumors | Escalating doses of berzosertib with carboplatin | Anemia (39%) Thrombocytopenia (36%) Neutropenia (25%) | PR 2/34 (6%) SD 18/34 (53%) | [60] |
| 2 | Melanoma | Ceralasertib + Durvalumab | Anemia (33%) Thrombocytopenia (23%) | PR 9/30 (30%) SD 10/30 (33%) Median PFS 7.1 months (95% CI 3.6–10.6) Median OS 14.2 months (95% CI 9.3–19.1) | [65] |
| BAY-1895344 | |||||
| 1 | Solid tumors | Escalating doses of BAY-1895344 | Neutropenia (55%) Leukopenia (18%) Thrombocytopenia (18%) | PR 4/21 (19%) SD 8/21 (38%) | [63] |
| M4344 | |||||
| 1 | Solid tumors | Escalating doses of M4344 or combination with carboplatin | Trial ongoing (Not reported) | Trial ongoing (Not reported) | NCT 02278250 |
3.2.2. ATM Inhibitors
3.2.3. DNA-PK Inhibitors
| Trial Phase | Disease Setting | Treatments | Most Common Grade ≥ 3 Toxicity | Efficacy | Reference |
|---|---|---|---|---|---|
| CC-115 | |||||
| 1 | Refractory/relapsed CLL/SLL | CC-115 monotherapy | Not reported | PR 4/8 (50%) 1 PR with IWCLL criteria 3 PR with lymphocytosis SD 3/8 (38%) | [76] |
| 1 | Cohort A (dose-escalation): solid tumors Cohort B (dose-expansion): selected refractory solid tumors | CC-115 monotherapy | Cohort A: Patients with at least one related Gr 3 AE (41%) Cohort B: Patients with at least one related Gr 3 AE (26%) | Cohort A: CR 1/39 (3%) PR 1/39 (3%) SD 18/39 (46%) Cohort B: -CLL/SLL PR 5/16 (31%) SD 4/16 (25%) -Overall PR 7/78 (9%) SD 29/78 (37%) | [77] |
| M3814/Nedisertib/Peposertib | |||||
| 1 | Solid tumors | Escalating doses of peposertib | Maculo-papular rash (13%) Nausea (7%) | SD 12/31 (39%) | [81] |
| AZD7648 | |||||
| 1/2 | Solid tumors | AZD7648 alone or in combination with pegylated liposomal doxorubicin | Trial ongoing (Not reported) | Trial ongoing (Not reported) | NCT 03907969 |
3.2.4. CHK1/2 Inhibitors
3.2.5. WEE1 Inhibitors
| Trial Phase | Disease Setting | Treatments | Most Common Grade ≥ 3 Toxicity | Efficacy | Reference |
|---|---|---|---|---|---|
| Adavosertib/MK-1775/AZD1775 | |||||
| 1 | Solid tumors | Escalating doses of AZD1775 | Lymphopenia (20%) Neutropenia (16%) Anemia (16%) | PR 2/25 (8%) | [104] |
| 1 | Solid tumors | AZD1775 alone or in combination with standard chemotherapy | AZD1775 + Gemcitabine: Neutropenia (33%) AZD1775 + Cisplatin: Neutropenia (12%) AZD1775 + Carboplatiin: Thrombocytopenia (31%) Neutropenia (18%) | AZD1775 + Gemcitabine: PR 4/81 (5%) AZD1775 + Cisplatin: PR 9/58 (16%) AZD1775 + Carboplatiin: PR 4/62 (6%) | [105] |
| 1 | Locally advanced pancreatic cancer | Escalating doses of AZD1775 with gemcitabine and radiation | Neutropenia (12%) Fatigue (9%) Fever (9%) Anorexia/Nausea/ Vomiting (9%) | Median OS 21.7 months (90% CI 16.7–24.8) Median PFS 9.4 months (90% CI 8.0–9.9) | [106] |
| 1 | Locally advanced head and neck cancer | Escalating doses of AZD1775 in combination with radiation | Lymphopenia (92%) | CR 8/10 (80%) PR 2/10 (20%) | [107] |
| 2 | TP53-mutated refractory ovarian cancer | AZD1775 in combination with carboplatin | Thrombocytopenia (48%) Neutropenia (39%) | CR 1/21 (5%) PR 8/21 (38%) SD 7/21 (33%) | [108] |
| 2 | TP53-mutated, platinum-sensitive ovarian cancer | Randomization (1:1) Paclitaxel and Carboplatin +/− AZD1775 | AZD1775 + Chemotherapy: Neutropenia (36%) Placebo + Chemotherapy: Neutropenia (33%) | AZD1775 + Chemotherapy: PFS 7.9 months Placebo + Chemotherapy: PFS 7.3 months HR for PFS: 0.63 (95% CI 0.38–1.06) | [109] |
| 2 | Platinum-refractory ovarian cancer | Randomization (2:1) Gemcitabine +/− Adavosertib | AZD1775 + Gemcitabine: Neutropenia (62%) Placebo + Gemcitabine Neutropenia (30%) | AZD1775 + Gemcitabine: PFS 4.6 months Placebo + Gemcitabine: PFS 3.0 months HR for PFS: 0.55 (95% CI 0.35–0.90) | [110] |
| 2 | Platinum-resistant ovarian cancer | Adavosertib in combination with - Gemcitabine - Paclitaxel - Carboplatin - Pegylated liposomal doxorubicin (PLD) | with Gemcitabine Neutropenia (78%) with Paclitaxel Neutropenia (53%) with Carboplatin Thrombocytopenia (63%) with PLD Neutropenia (17%) | with Gemcitabine PR 1/9 (11%) with Paclitaxel PR 10/38 (26%) with Carboplatin PR 13/35 (37%) with PLD PR 3/12 (25%) | [111] |
| 2 | TNBC | Adavosertib in combination with cisplatin | Diarrhea (21%) Neutropenia (18%) | CR 3/34 (9%) PR 6/34 (18%) SD 13/34 (38%) | [112] |
| 2 | Colorectal cancer with TP53 and RAS mutations | Randomization (2:1) (maintenance) Adavosertib or active monitoring | Adavosertib Diarrhea (9%) | Adavosertib PFS 3.6 months OS 14.0 months Active monitoring PFS 1.9 months OS 12.8 months HR for PFS: 0.35 (95% CI 0.18–0.68) HR for OS: 0.92 (95% CI 0.44–1.94) | [113] |
| 2 | Small cell lung cancer | Adavosertib monotherapy (biomarker-selected patients) | Diarrhea 1/31 (3%) | PR 0/31 (0%) SD 9/31 (29%) | [114] |
| 2 | Uterine serous sarcoma | Adavosertib monotherapy | Neutropenia (32%) | CR 1/34 (3%) PR 9/34 (26%) | [115] |
| 2 | Solid tumor with mutations in DNA repair genes | Adavosertib in combination with carboplatin | Anemia (39%) Thrombocytopenia (39%) Neutropenia (32%) | PR 0/24 (0%) | [116] |
3.2.6. PLK1 Inhibitors
| Trial Phase | Disease Setting | Treatments | Most Common Grade ≥ 3 Toxicity | Efficacy | Reference |
|---|---|---|---|---|---|
| Volasertib/BI 6727 | |||||
| 1 | Solid tumors | Escalating doses of Volasertib | Neutropenia (14%) Thrombocytopenia (14%) | PR 3/65 (5%) SD 26/65 (40%) | [118] |
| 2 | Urothelial cancer | Volasertib 300mg every 3 weeks | Neutropenia (28%) Thrombocytopenia (20%) | PR 7/50 (14%) SD 13/50 (26%) Median PFS 1.4 months (95% CI 1.3–2.6) Median OS 8.5 months (95% CI 3.9–12.1) | [119] |
| 2 | AML ineligible for induction chemotherapy | Randomization (1:1) LDAC +/− Volasertib | Volasertib + LDAC: Febrile neutropenia (55%) LDAC: Febrile Neutropenia (16%) | Volasertib + LDAC: CR + CRi 6/45 (13%) LDAC: CR + CRi 13/42 (31%) | [120] |
| 3 | AML ineligible for induction chemotherapy | Randomization (2:1) Low-dose cytarabine +/− Volasertib | Volasertib + LDAC: Febrile neutropenia (59%) Thrombocytopenia (39%) Placebo + LDAC: Thrombocytopenia (29%) Febrile Neutropenia (28%) | Volasertib + LDAC: CR + CRi 123/444 (28%) Median OS: 5.6 months (95% CI 4.5–6.8) Placebo + LDAC: CR + CRi 38/222 (17%) Median OS: 4.8 months (95% CI 3.8–6.4) HR for OS: 0.97 (95% CI 0.8–1.2) | [121] |
| 2 | NSCLC, second-line treatment | Randomization - Volasertib - Pemetrexed - Volasertib + Pemetrexed | Volasertib Neutropenia (14%) Pemetrexed Fatigue (9%) Volasertib + Pemetrexed Neutropenia (11%) | Volasertib Median PFS: 1.4 months Pemetrexed Median PFS: 5.3 months Volasertib + Pemetrexed Median PFS: 3.3 months | [122] |
| 2 | Platinum-resistant ovarian cancer | Randomization (1:1) Volasertib vs. Chemotherapy (non-platinum) | Volasertib Neutropenia (44%) Chemotherapy Neutropenia (6%) | Volasertib 24 week DCR: 30.6% Chemotherapy 24 week DCR: 43.1% | [123] |
| Onvansertib/PCM-075/NMS-1286937 | |||||
| 1 | Solid tumors | Escalating doses of NMS-1286937 | Neutropenia (16%) Thrombocytopenia (16%) | SD 5/16 (31%) | [125] |
| 1 | AML | Escalating doses of onvansertib with either LDAC or decitabnie | Anemia (35%) Thrombocytopenia (25%) Neutropenia (25%) | Onvansertib + LDAC CR + CRi 1/15 (7%) Onvansertib + Decitabine CR + CRi 5/21 (24%) | [126] |
4. Biomarkers for DDR-Targeted Therapies: Beyond BRCA1/2 Mutations
4.1. Homologous Recombination Deficiency (HRD) Scores
4.2. Sequencing-Based Mutational Signatures
5. Combination Strategies of DDR-Targeted Therapies
5.1. Combination with DDR Inhibitors
5.2. Combination with Targeted Agents
5.2.1. Antiangiogenic Agents
5.2.2. PI3K Inhibitors
5.2.3. Antiandrogen Therapies
5.2.4. MAPK Pathway Inhibitors
5.3. Combination with Immune Checkpoint Inhibitors
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martincorena, I.; Campbell, P.J. Somatic mutation in cancer and normal cells. Science 2015, 349, 1483–1489. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [Green Version]
- Pilie, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef]
- Caldecott, K.W. Mammalian DNA base excision repair: Dancing in the moonlight. DNA Repair 2020, 93, 102921. [Google Scholar] [CrossRef]
- Beard, W.A.; Horton, J.K.; Prasad, R.; Wilson, S.H. Eukaryotic Base Excision Repair: New Approaches Shine Light on Mechanism. Annu. Rev. Biochem. 2019, 88, 137–162. [Google Scholar] [CrossRef]
- Grundy, G.J.; Parsons, J.L. Base excision repair and its implications to cancer therapy. Essays Biochem. 2020, 64, 831–843. [Google Scholar] [CrossRef]
- Mu, H.; Geacintov, N.E.; Broyde, S.; Yeo, J.E.; Scharer, O.D. Molecular basis for damage recognition and verification by XPC-RAD23B and TFIIH in nucleotide excision repair. DNA Repair 2018, 71, 33–42. [Google Scholar] [CrossRef]
- Kusakabe, M.; Onishi, Y.; Tada, H.; Kurihara, F.; Kusao, K.; Furukawa, M.; Iwai, S.; Yokoi, M.; Sakai, W.; Sugasawa, K. Mechanism and regulation of DNA damage recognition in nucleotide excision repair. Genes Env. 2019, 41, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, N.; Raja, S.; Van Houten, B. The involvement of nucleotide excision repair proteins in the removal of oxidative DNA damage. Nucleic Acids Res. 2020, 48, 11227–11243. [Google Scholar] [CrossRef]
- Sugasawa, K. Mechanism and regulation of DNA damage recognition in mammalian nucleotide excision repair. Enzymes 2019, 45, 99–138. [Google Scholar] [CrossRef] [PubMed]
- Apostolou, Z.; Chatzinikolaou, G.; Stratigi, K.; Garinis, G.A. Nucleotide Excision Repair and Transcription-Associated Genome Instability. Bioessays 2019, 41, e1800201. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Rothenberg, E.; Ramsden, D.A.; Lieber, M.R. The molecular basis and disease relevance of non-homologous DNA end joining. Nat. Rev. Mol. Cell Biol. 2020, 21, 765–781. [Google Scholar] [CrossRef]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Pannunzio, N.R.; Watanabe, G.; Lieber, M.R. Nonhomologous DNA end-joining for repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10512–10523. [Google Scholar] [CrossRef] [Green Version]
- Difilippantonio, M.J.; Zhu, J.; Chen, H.T.; Meffre, E.; Nussenzweig, M.C.; Max, E.E.; Ried, T.; Nussenzweig, A. DNA repair protein Ku80 suppresses chromosomal aberrations and malignant transformation. Nature 2000, 404, 510–514. [Google Scholar] [CrossRef] [Green Version]
- Li, G.C.; Ouyang, H.; Li, X.; Nagasawa, H.; Little, J.B.; Chen, D.J.; Ling, C.C.; Fuks, Z.; Cordon-Cardo, C. Ku70: A candidate tumor suppressor gene for murine T cell lymphoma. Mol. Cell 1998, 2, 1–8. [Google Scholar] [CrossRef]
- Espejel, S.; Martin, M.; Klatt, P.; Martin-Caballero, J.; Flores, J.M.; Blasco, M.A. Shorter telomeres, accelerated ageing and increased lymphoma in DNA-PKcs-deficient mice. EMBO Rep. 2004, 5, 503–509. [Google Scholar] [CrossRef] [Green Version]
- Myung, K.; Ghosh, G.; Fattah, F.J.; Li, G.; Kim, H.; Dutia, A.; Pak, E.; Smith, S.; Hendrickson, E.A. Regulation of telomere length and suppression of genomic instability in human somatic cells by Ku86. Mol. Cell Biol. 2004, 24, 5050–5059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Ghosh, G.; Hendrickson, E.A. Ku86 represses lethal telomere deletion events in human somatic cells. Proc. Natl. Acad. Sci. USA 2009, 106, 12430–12435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Wiese, C.; Kwon, Y.; Hromas, R.; Sung, P. The BRCA Tumor Suppressor Network in Chromosome Damage Repair by Homologous Recombination. Annu. Rev. Biochem. 2019, 88, 221–245. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, W.D.; Shah, S.S.; Heyer, W.D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10524–10535. [Google Scholar] [CrossRef] [Green Version]
- Semmler, L.; Reiter-Brennan, C.; Klein, A. BRCA1 and Breast Cancer: A Review of the Underlying Mechanisms Resulting in the Tissue-Specific Tumorigenesis in Mutation Carriers. J. Breast Cancer 2019, 22, 1–14. [Google Scholar] [CrossRef]
- Singh, A.K.; Yu, X. Tissue-Specific Carcinogens as Soil to Seed BRCA1/2-Mutant Hereditary Cancers. Trends Cancer 2020, 6, 559–568. [Google Scholar] [CrossRef]
- Kantidze, O.L.; Velichko, A.K.; Luzhin, A.V.; Petrova, N.V.; Razin, S.V. Synthetically Lethal Interactions of ATM, ATR, and DNA-PKcs. Trends Cancer 2018, 4, 755–768. [Google Scholar] [CrossRef]
- Menolfi, D.; Zha, S. ATM, ATR and DNA-PKcs kinases-the lessons from the mouse models: Inhibition not equal deletion. Cell Biosci. 2020, 10, 8. [Google Scholar] [CrossRef]
- Bradbury, A.; Hall, S.; Curtin, N.; Drew, Y. Targeting ATR as Cancer Therapy: A new era for synthetic lethality and synergistic combinations? Pharm. Ther. 2020, 207, 107450. [Google Scholar] [CrossRef]
- Ma, H.T.; Poon, R.Y.C. Aurora kinases and DNA damage response. Mutat. Res. Mol. Mech. Mutagen. 2020, 821, 111716. [Google Scholar] [CrossRef] [PubMed]
- Krystyniak, A.; Garcia-Echeverria, C.; Prigent, C.; Ferrari, S. Inhibition of Aurora A in response to DNA damage. Oncogene 2006, 25, 338–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macurek, L.; Lindqvist, A.; Lim, D.; Lampson, M.A.; Klompmaker, R.; Freire, R.; Clouin, C.; Taylor, S.S.; Yaffe, M.B.; Medema, R.H. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature 2008, 455, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Seki, A.; Coppinger, J.A.; Jang, C.Y.; Yates, J.R.; Fang, G. Bora and the kinase Aurora a cooperatively activate the kinase Plk1 and control mitotic entry. Science 2008, 320, 1655–1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, C.; Kurimasa, A.; Brenneman, M.A.; Chen, D.J.; Nickoloff, J.A. DNA-dependent protein kinase suppresses double-strand break-induced and spontaneous homologous recombination. Proc. Natl. Acad. Sci. USA 2002, 99, 3758–3763. [Google Scholar] [CrossRef] [Green Version]
- Stiff, T.; O’Driscoll, M.; Rief, N.; Iwabuchi, K.; Lobrich, M.; Jeggo, P.A. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004, 64, 2390–2396. [Google Scholar] [CrossRef] [Green Version]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [Green Version]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murai, J.; Huang, S.Y.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pommier, Y.; O’Connor, M.J.; de Bono, J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016, 8, 362ps317. [Google Scholar] [CrossRef] [PubMed]
- Cong, K.; Peng, M.; Kousholt, A.N.; Lee, W.T.C.; Lee, S.; Nayak, S.; Krais, J.; VanderVere-Carozza, P.S.; Pawelczak, K.S.; Calvo, J.; et al. Replication gaps are a key determinant of PARP inhibitor synthetic lethality with BRCA deficiency. Mol. Cell 2021, 81, 3128–3144. [Google Scholar] [CrossRef]
- Panzarino, N.J.; Krais, J.J.; Cong, K.; Peng, M.; Mosqueda, M.; Nayak, S.U.; Bond, S.M.; Calvo, J.A.; Doshi, M.B.; Bere, M.; et al. Replication Gaps Underlie BRCA Deficiency and Therapy Response. Cancer Res. 2021, 81, 1388–1397. [Google Scholar] [CrossRef]
- Cliby, W.A.; Roberts, C.J.; Cimprich, K.A.; Stringer, C.M.; Lamb, J.R.; Schreiber, S.L.; Friend, S.H. Overexpression of a kinase-inactive ATR protein causes sensitivity to DNA-damaging agents and defects in cell cycle checkpoints. EMBO J. 1998, 17, 159–169. [Google Scholar] [CrossRef] [Green Version]
- Rundle, S.; Bradbury, A.; Drew, Y.; Curtin, N.J. Targeting the ATR-CHK1 Axis in Cancer Therapy. Cancers 2017, 9, 41. [Google Scholar] [CrossRef]
- Charrier, J.D.; Durrant, S.J.; Golec, J.M.; Kay, D.P.; Knegtel, R.M.; MacCormick, S.; Mortimore, M.; O’Donnell, M.E.; Pinder, J.L.; Reaper, P.M.; et al. Discovery of potent and selective inhibitors of ataxia telangiectasia mutated and Rad3 related (ATR) protein kinase as potential anticancer agents. J. Med. Chem. 2011, 54, 2320–2330. [Google Scholar] [CrossRef]
- Fokas, E.; Prevo, R.; Pollard, J.R.; Reaper, P.M.; Charlton, P.A.; Cornelissen, B.; Vallis, K.A.; Hammond, E.M.; Olcina, M.M.; Gillies McKenna, W.; et al. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis. 2012, 3, e441. [Google Scholar] [CrossRef] [Green Version]
- Knegtel, R.; Charrier, J.D.; Durrant, S.; Davis, C.; O’Donnell, M.; Storck, P.; MacCormick, S.; Kay, D.; Pinder, J.; Virani, A.; et al. Rational Design of 5-(4-(Isopropylsulfonyl)phenyl)-3-(3-(4-((methylamino)methyl)phenyl)isoxazol-5-yl)pyrazin-2-amine (VX-970, M6620): Optimization of Intra- and Intermolecular Polar Interactions of a New Ataxia Telangiectasia Mutated and Rad3-Related (ATR) Kinase Inhibitor. J. Med. Chem. 2019, 62, 5547–5561. [Google Scholar] [CrossRef]
- Thomas, A.; Redon, C.E.; Sciuto, L.; Padiernos, E.; Ji, J.; Lee, M.J.; Yuno, A.; Lee, S.; Zhang, Y.; Tran, L.; et al. Phase I Study of ATR Inhibitor M6620 in Combination With Topotecan in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2018, 36, 1594–1602. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; O’Carrigan, B.; Penney, M.S.; Lim, J.S.; Brown, J.S.; de Miguel Luken, M.J.; Tunariu, N.; Perez-Lopez, R.; Rodrigues, D.N.; Riisnaes, R.; et al. Phase I Trial of First-in-Class ATR Inhibitor M6620 (VX-970) as Monotherapy or in Combination With Carboplatin in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2020, 38, 3195–3204. [Google Scholar] [CrossRef]
- Shapiro, G.I.; Wesolowski, R.; Devoe, C.; Lord, S.; Pollard, J.; Hendriks, B.S.; Falk, M.; Diaz-Padilla, I.; Plummer, R.; Yap, T.A. Phase 1 study of the ATR inhibitor berzosertib in combination with cisplatin in patients with advanced solid tumours. Br. J. Cancer 2021, 125, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Middleton, M.R.; Dean, E.; Evans, T.R.J.; Shapiro, G.I.; Pollard, J.; Hendriks, B.S.; Falk, M.; Diaz-Padilla, I.; Plummer, R. Phase 1 study of the ATR inhibitor berzosertib (formerly M6620, VX-970) combined with gemcitabine +/- cisplatin in patients with advanced solid tumours. Br. J. Cancer 2021, 125, 510–519. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Cheng, S.C.; Wahner Hendrickson, A.E.; Penson, R.T.; Schumer, S.T.; Doyle, L.A.; Lee, E.K.; Kohn, E.C.; Duska, L.R.; Crispens, M.A.; et al. Berzosertib plus gemcitabine versus gemcitabine alone in platinum-resistant high-grade serous ovarian cancer: A multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020, 21, 957–968. [Google Scholar] [CrossRef]
- Pal, S.K.; Frankel, P.H.; Mortazavi, A.; Milowsky, M.; Vaishampayan, U.; Parikh, M.; Lyou, Y.; Weng, P.; Parikh, R.; Teply, B.; et al. Effect of Cisplatin and Gemcitabine With or Without Berzosertib in Patients With Advanced Urothelial Carcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2021, 7, 1536. [Google Scholar] [CrossRef] [PubMed]
- Foote, K.M.; Blades, K.; Cronin, A.; Fillery, S.; Guichard, S.S.; Hassall, L.; Hickson, I.; Jacq, X.; Jewsbury, P.J.; McGuire, T.M.; et al. Discovery of 4-{4-[(3R)-3-Methylmorpholin-4-yl]-6-[1-(methylsulfonyl)cyclopropyl]pyrimidin-2-y l}-1H-indole (AZ20): A potent and selective inhibitor of ATR protein kinase with monotherapy in vivo antitumor activity. J. Med. Chem. 2013, 56, 2125–2138. [Google Scholar] [CrossRef]
- Foote, K.M.; Nissink, J.W.M.; McGuire, T.; Turner, P.; Guichard, S.; Yates, J.W.T.; Lau, A.; Blades, K.; Heathcote, D.; Odedra, R.; et al. Discovery and Characterization of AZD6738, a Potent Inhibitor of Ataxia Telangiectasia Mutated and Rad3 Related (ATR) Kinase with Application as an Anticancer Agent. J. Med. Chem. 2018, 61, 9889–9907. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.T.; Smith, S.A.; Mortimer, P.; Loembe, A.B.; Cho, H.; Kim, K.M.; Smith, C.; Willis, S.; Irurzun-Arana, I.; Berges, A.; et al. Phase I Study of Ceralasertib (AZD6738), a Novel DNA Damage Repair Agent, in Combination with Weekly Paclitaxel in Refractory Cancer. Clin. Cancer Res. 2021, 27, 4700–4709. [Google Scholar] [CrossRef]
- Yap, T.A.; Krebs, M.G.; Postel-Vinay, S.; El-Khouiery, A.; Soria, J.C.; Lopez, J.; Berges, A.; Cheung, S.A.; Irurzun-Arana, I.; Goldwin, A.; et al. Ceralasertib (AZD6738), an Oral ATR Kinase Inhibitor, in Combination with Carboplatin in Patients with Advanced Solid Tumors: A Phase I Study. Clin. Cancer Res. 2021, 27, 5213–5224. [Google Scholar] [CrossRef]
- Lucking, U.; Wortmann, L.; Wengner, A.M.; Lefranc, J.; Lienau, P.; Briem, H.; Siemeister, G.; Bomer, U.; Denner, K.; Schafer, M.; et al. Damage Incorporated: Discovery of the Potent, Highly Selective, Orally Available ATR Inhibitor BAY 1895344 with Favorable Pharmacokinetic Properties and Promising Efficacy in Monotherapy and in Combination Treatments in Preclinical Tumor Models. J. Med. Chem. 2020, 63, 7293–7325. [Google Scholar] [CrossRef] [PubMed]
- Wengner, A.M.; Siemeister, G.; Lucking, U.; Lefranc, J.; Wortmann, L.; Lienau, P.; Bader, B.; Bomer, U.; Moosmayer, D.; Eberspacher, U.; et al. The Novel ATR Inhibitor BAY 1895344 Is Efficacious as Monotherapy and Combined with DNA Damage-Inducing or Repair-Compromising Therapies in Preclinical Cancer Models. Mol. Cancer Ther. 2020, 19, 26–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yap, T.A.; Tan, D.S.P.; Terbuch, A.; Caldwell, R.; Guo, C.; Goh, B.C.; Heong, V.; Haris, N.R.M.; Bashir, S.; Drew, Y.; et al. First-in-Human Trial of the Oral Ataxia Telangiectasia and RAD3-Related (ATR) Inhibitor BAY 1895344 in Patients with Advanced Solid Tumors. Cancer Discov. 2021, 11, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Jo, U.; Senatorov, I.S.; Zimmermann, A.; Saha, L.K.; Murai, Y.; Kim, S.H.; Rajapakse, V.N.; Elloumi, F.; Takahashi, N.; Schultz, C.W.; et al. Novel and Highly Potent ATR Inhibitor M4344 Kills Cancer Cells With Replication Stress, and Enhances the Chemotherapeutic Activity of Widely Used DNA Damaging Agents. Mol. Cancer Ther. 2021, 20, 1431–1441. [Google Scholar] [CrossRef]
- Kim, R.; Kwon, M.; An, M.; Kim, S.T.; Smith, S.A.; Loembe, A.B.; Mortimer, P.G.S.; Armenia, J.; Lukashchuk, N.; Shah, N.; et al. Phase II study of ceralasertib (AZD6738) in combination with durvalumab in patients with advanced/metastatic melanoma who have failed prior anti-PD-1 therapy. Ann. Oncol. 2021, 33, 192–203. [Google Scholar] [CrossRef]
- Sarkaria, J.N.; Eshleman, J.S. ATM as a target for novel radiosensitizers. Semin. Radiat. Oncol. 2001, 11, 316–327. [Google Scholar] [CrossRef]
- Hickson, I.; Zhao, Y.; Richardson, C.J.; Green, S.J.; Martin, N.M.; Orr, A.I.; Reaper, P.M.; Jackson, S.P.; Curtin, N.J.; Smith, G.C. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004, 64, 9152–9159. [Google Scholar] [CrossRef] [Green Version]
- Golding, S.E.; Rosenberg, E.; Valerie, N.; Hussaini, I.; Frigerio, M.; Cockcroft, X.F.; Chong, W.Y.; Hummersone, M.; Rigoreau, L.; Menear, K.A.; et al. Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol. Cancer Ther. 2009, 8, 2894–2902. [Google Scholar] [CrossRef] [Green Version]
- Pike, K.G.; Barlaam, B.; Cadogan, E.; Campbell, A.; Chen, Y.; Colclough, N.; Davies, N.L.; de-Almeida, C.; Degorce, S.L.; Didelot, M.; et al. The Identification of Potent, Selective, and Orally Available Inhibitors of Ataxia Telangiectasia Mutated (ATM) Kinase: The Discovery of AZD0156 (8-{6-[3-(Dimethylamino)propoxy]pyridin-3-yl}-3-methyl-1-(tetrahydro-2 H-pyran-4-yl)-1,3-dihydro-2 H-imidazo[4,5-c]quinolin-2-one). J. Med. Chem. 2018, 61, 3823–3841. [Google Scholar] [CrossRef]
- Durant, S.T.; Zheng, L.; Wang, Y.; Chen, K.; Zhang, L.; Zhang, T.; Yang, Z.; Riches, L.; Trinidad, A.G.; Fok, J.H.L.; et al. The brain-penetrant clinical ATM inhibitor AZD1390 radiosensitizes and improves survival of preclinical brain tumor models. Sci. Adv. 2018, 4, eaat1719. [Google Scholar] [CrossRef] [Green Version]
- Fuchss, T.; Mederski, W.W.; Zenke, F.T.; Dahmen, H.; Zimmermann, A.; Blaukat, A. Abstract 329: Highly potent and selective ATM kinase inhibitor M3541: A clinical candidate drug with strong antitumor activity in combination with radiotherapy. Cancer Res. 2018, 78, 329. [Google Scholar] [CrossRef]
- Zimmermann, A.; Zenke, F.; Dahmen, H.; Sirrenberg, C.; Grombacher, T.; Vassilev, L.T.; Fuchss, T.; Blaukat, A. Abstract 338: A new investigational ATM Inhibitor, M3541, synergistically potentiates fractionated radiotherapy and chemotherapy in cancer cells and animal models. Cancer Res. 2018, 78, 338. [Google Scholar] [CrossRef]
- Medova, M.; Medo, M.; Hovhannisyan, L.; Munoz-Maldonado, C.; Aebersold, D.M.; Zimmer, Y. DNA-PK in human malignant disorders: Mechanisms and implications for pharmacological interventions. Pharm. Ther. 2020, 215, 107617. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, D.S.; Perrin-Ninkovic, S.M.; Shevlin, G.; Elsner, J.; Zhao, J.; Whitefield, B.; Tehrani, L.; Sapienza, J.; Riggs, J.R.; Parnes, J.S.; et al. Optimization of a Series of Triazole Containing Mammalian Target of Rapamycin (mTOR) Kinase Inhibitors and the Discovery of CC-115. J. Med. Chem. 2015, 58, 5599–5608. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, T.; Sapinoso, L.M.; Tran, T.; Gaffney, B.; Wong, L.; Sankar, S.; Raymon, H.K.; Mortensen, D.S.; Xu, S. CC-115, a dual inhibitor of mTOR kinase and DNA-PK, blocks DNA damage repair pathways and selectively inhibits ATM-deficient cell growth in vitro. Oncotarget 2017, 8, 74688–74702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thijssen, R.; Ter Burg, J.; Garrick, B.; van Bochove, G.G.; Brown, J.R.; Fernandes, S.M.; Rodriguez, M.S.; Michot, J.M.; Hallek, M.; Eichhorst, B.; et al. Dual TORK/DNA-PK inhibition blocks critical signaling pathways in chronic lymphocytic leukemia. Blood 2016, 128, 574–583. [Google Scholar] [CrossRef]
- Munster, P.; Mita, M.; Mahipal, A.; Nemunaitis, J.; Massard, C.; Mikkelsen, T.; Cruz, C.; Paz-Ares, L.; Hidalgo, M.; Rathkopf, D.; et al. First-In-Human Phase I Study Of A Dual mTOR Kinase And DNA-PK Inhibitor (CC-115) In Advanced Malignancy. Cancer Manag. Res. 2019, 11, 10463–10476. [Google Scholar] [CrossRef] [Green Version]
- Klein, C.; Dokic, I.; Mairani, A.; Mein, S.; Brons, S.; Haring, P.; Haberer, T.; Jakel, O.; Zimmermann, A.; Zenke, F.; et al. Overcoming hypoxia-induced tumor radioresistance in non-small cell lung cancer by targeting DNA-dependent protein kinase in combination with carbon ion irradiation. Radiat. Oncol. 2017, 12, 208. [Google Scholar] [CrossRef] [Green Version]
- Wise, H.C.; Iyer, G.V.; Moore, K.; Temkin, S.M.; Gordon, S.; Aghajanian, C.; Grisham, R.N. Activity of M3814, an Oral DNA-PK Inhibitor, In Combination with Topoisomerase II Inhibitors in Ovarian Cancer Models. Sci. Rep. 2019, 9, 18882. [Google Scholar] [CrossRef] [Green Version]
- Zenke, F.T.; Zimmermann, A.; Sirrenberg, C.; Dahmen, H.; Kirkin, V.; Pehl, U.; Grombacher, T.; Wilm, C.; Fuchss, T.; Amendt, C.; et al. Pharmacologic Inhibitor of DNA-PK, M3814, Potentiates Radiotherapy and Regresses Human Tumors in Mouse Models. Mol. Cancer Ther. 2020, 19, 1091–1101. [Google Scholar] [CrossRef] [Green Version]
- van Bussel, M.T.J.; Awada, A.; de Jonge, M.J.A.; Mau-Sorensen, M.; Nielsen, D.; Schoffski, P.; Verheul, H.M.W.; Sarholz, B.; Berghoff, K.; El Bawab, S.; et al. A first-in-man phase 1 study of the DNA-dependent protein kinase inhibitor peposertib (formerly M3814) in patients with advanced solid tumours. Br. J. Cancer 2021, 124, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Fok, J.H.L.; Ramos-Montoya, A.; Vazquez-Chantada, M.; Wijnhoven, P.W.G.; Follia, V.; James, N.; Farrington, P.M.; Karmokar, A.; Willis, S.E.; Cairns, J.; et al. AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat. Commun. 2019, 10, 5065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, F.W.; Finlay, M.R.V.; Ting, A.K.T.; Beattie, D.; Lamont, G.M.; Fallan, C.; Wrigley, G.L.; Schimpl, M.; Howard, M.R.; Williamson, B.; et al. The Discovery of 7-Methyl-2-[(7-methyl[1,2,4]triazolo[1,5-a]pyridin-6-yl)amino]-9-(tetrahydro-2H-p yran-4-yl)-7,9-dihydro-8H-purin-8-one (AZD7648), a Potent and Selective DNA-Dependent Protein Kinase (DNA-PK) Inhibitor. J. Med. Chem. 2020, 63, 3461–3471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welch, S.; Hirte, H.W.; Carey, M.S.; Hotte, S.J.; Tsao, M.S.; Brown, S.; Pond, G.R.; Dancey, J.E.; Oza, A.M. UCN-01 in combination with topotecan in patients with advanced recurrent ovarian cancer: A study of the Princess Margaret Hospital Phase II consortium. Gynecol. Oncol. 2007, 106, 305–310. [Google Scholar] [CrossRef]
- Ma, C.X.; Ellis, M.J.; Petroni, G.R.; Guo, Z.; Cai, S.R.; Ryan, C.E.; Craig Lockhart, A.; Naughton, M.J.; Pluard, T.J.; Brenin, C.M.; et al. A phase II study of UCN-01 in combination with irinotecan in patients with metastatic triple negative breast cancer. Breast Cancer Res. Treat. 2013, 137, 483–492. [Google Scholar] [CrossRef] [Green Version]
- Seto, T.; Esaki, T.; Hirai, F.; Arita, S.; Nosaki, K.; Makiyama, A.; Kometani, T.; Fujimoto, C.; Hamatake, M.; Takeoka, H.; et al. Phase I, dose-escalation study of AZD7762 alone and in combination with gemcitabine in Japanese patients with advanced solid tumours. Cancer Chemother. Pharmacol. 2013, 72, 619–627. [Google Scholar] [CrossRef]
- Sausville, E.; Lorusso, P.; Carducci, M.; Carter, J.; Quinn, M.F.; Malburg, L.; Azad, N.; Cosgrove, D.; Knight, R.; Barker, P.; et al. Phase I dose-escalation study of AZD7762, a checkpoint kinase inhibitor, in combination with gemcitabine in US patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2014, 73, 539–549. [Google Scholar] [CrossRef] [Green Version]
- Laquente, B.; Lopez-Martin, J.; Richards, D.; Illerhaus, G.; Chang, D.Z.; Kim, G.; Stella, P.; Richel, D.; Szcylik, C.; Cascinu, S.; et al. A phase II study to evaluate LY2603618 in combination with gemcitabine in pancreatic cancer patients. BMC Cancer 2017, 17, 137. [Google Scholar] [CrossRef] [Green Version]
- Wehler, T.; Thomas, M.; Schumann, C.; Bosch-Barrera, J.; Vinolas Segarra, N.; Dickgreber, N.J.; Dalhoff, K.; Sebastian, M.; Corral Jaime, J.; Alonso, M.; et al. A randomized, phase 2 evaluation of the CHK1 inhibitor, LY2603618, administered in combination with pemetrexed and cisplatin in patients with advanced nonsquamous non-small cell lung cancer. Lung Cancer 2017, 108, 212–216. [Google Scholar] [CrossRef]
- Daud, A.I.; Ashworth, M.T.; Strosberg, J.; Goldman, J.W.; Mendelson, D.; Springett, G.; Venook, A.P.; Loechner, S.; Rosen, L.S.; Shanahan, F.; et al. Phase I dose-escalation trial of checkpoint kinase 1 inhibitor MK-8776 as monotherapy and in combination with gemcitabine in patients with advanced solid tumors. J. Clin. Oncol. 2015, 33, 1060–1066. [Google Scholar] [CrossRef]
- Italiano, A.; Infante, J.R.; Shapiro, G.I.; Moore, K.N.; LoRusso, P.M.; Hamilton, E.; Cousin, S.; Toulmonde, M.; Postel-Vinay, S.; Tolaney, S.; et al. Phase I study of the checkpoint kinase 1 inhibitor GDC-0575 in combination with gemcitabine in patients with refractory solid tumors. Ann. Oncol. 2018, 29, 1304–1311. [Google Scholar] [CrossRef] [PubMed]
- King, C.; Diaz, H.B.; McNeely, S.; Barnard, D.; Dempsey, J.; Blosser, W.; Beckmann, R.; Barda, D.; Marshall, M.S. LY2606368 Causes Replication Catastrophe and Antitumor Effects through CHK1-Dependent Mechanisms. Mol. Cancer 2015, 14, 2004–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, D.; Infante, J.; Janku, F.; Jones, S.; Nguyen, L.M.; Burris, H.; Naing, A.; Bauer, T.M.; Piha-Paul, S.; Johnson, F.M.; et al. Phase I Study of LY2606368, a Checkpoint Kinase 1 Inhibitor, in Patients With Advanced Cancer. J. Clin. Oncol. 2016, 34, 1764–1771. [Google Scholar] [CrossRef] [PubMed]
- Iwasa, S.; Yamamoto, N.; Shitara, K.; Tamura, K.; Matsubara, N.; Tajimi, M.; Lin, A.B.; Asou, H.; Cai, Z.; Inoue, K.; et al. Dose-finding study of the checkpoint kinase 1 inhibitor, prexasertib, in Japanese patients with advanced solid tumors. Cancer Sci. 2018, 109, 3216–3223. [Google Scholar] [CrossRef]
- Lee, J.M.; Nair, J.; Zimmer, A.; Lipkowitz, S.; Annunziata, C.M.; Merino, M.J.; Swisher, E.M.; Harrell, M.I.; Trepel, J.B.; Lee, M.J.; et al. Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: A first-in-class proof-of-concept phase 2 study. Lancet Oncol. 2018, 19, 207–215. [Google Scholar] [CrossRef]
- Gatti-Mays, M.E.; Karzai, F.H.; Soltani, S.N.; Zimmer, A.; Green, J.E.; Lee, M.J.; Trepel, J.B.; Yuno, A.; Lipkowitz, S.; Nair, J.; et al. A Phase II Single Arm Pilot Study of the CHK1 Inhibitor Prexasertib (LY2606368) in BRCA Wild-Type, Advanced Triple-Negative Breast Cancer. Oncologist 2020, 25, 1013–e1824. [Google Scholar] [CrossRef]
- Byers, L.A.; Navarro, A.; Schaefer, E.; Johnson, M.; Ozguroglu, M.; Han, J.Y.; Bondarenko, I.; Cicin, I.; Dragnev, K.H.; Abel, A.; et al. A Phase II Trial of Prexasertib (LY2606368) in Patients With Extensive-Stage Small-Cell Lung Cancer. Clin. Lung Cancer 2021, 22, 531–540. [Google Scholar] [CrossRef]
- Moore, K.N.; Hong, D.S.; Patel, M.R.; Pant, S.; Ulahannan, S.V.; Jones, S.; Meric-Bernstam, F.; Wang, J.S.; Aljumaily, R.; Hamilton, E.P.; et al. A Phase 1b Trial of Prexasertib in Combination with Standard-of-Care Agents in Advanced or Metastatic Cancer. Target Oncol. 2021, 16, 569–589. [Google Scholar] [CrossRef]
- Do, K.T.; Kochupurakkal, B.; Kelland, S.; de Jonge, A.; Hedglin, J.; Powers, A.; Quinn, N.; Gannon, C.; Vuong, L.; Parmar, K.; et al. Phase 1 Combination Study of the CHK1 Inhibitor Prexasertib and the PARP Inhibitor Olaparib in High-grade Serous Ovarian Cancer and Other Solid Tumors. Clin. Cancer Res. 2021, 27, 4710–4716. [Google Scholar] [CrossRef]
- Do, K.T.; Manuszak, C.; Thrash, E.; Giobbie-Hurder, A.; Hu, J.; Kelland, S.; Powers, A.; de Jonge, A.; Shapiro, G.I.; Severgnini, M. Immune modulating activity of the CHK1 inhibitor prexasertib and anti-PD-L1 antibody LY3300054 in patients with high-grade serous ovarian cancer and other solid tumors. Cancer Immunol. Immunother. 2021, 70, 2991–3000. [Google Scholar] [CrossRef]
- Esposito, F.; Giuffrida, R.; Raciti, G.; Puglisi, C.; Forte, S. Wee1 Kinase: A Potential Target to Overcome Tumor Resistance to Therapy. Int. J. Mol. Sci. 2021, 22, 10689. [Google Scholar] [CrossRef] [PubMed]
- Aarts, M.; Sharpe, R.; Garcia-Murillas, I.; Gevensleben, H.; Hurd, M.S.; Shumway, S.D.; Toniatti, C.; Ashworth, A.; Turner, N.C. Forced mitotic entry of S-phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer Discov. 2012, 2, 524–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirai, H.; Iwasawa, Y.; Okada, M.; Arai, T.; Nishibata, T.; Kobayashi, M.; Kimura, T.; Kaneko, N.; Ohtani, J.; Yamanaka, K.; et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol. Cancer Ther. 2009, 8, 2992–3000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do, K.; Wilsker, D.; Ji, J.; Zlott, J.; Freshwater, T.; Kinders, R.J.; Collins, J.; Chen, A.P.; Doroshow, J.H.; Kummar, S. Phase I Study of Single-Agent AZD1775 (MK-1775), a Wee1 Kinase Inhibitor, in Patients With Refractory Solid Tumors. J. Clin. Oncol. 2015, 33, 3409–3415. [Google Scholar] [CrossRef] [Green Version]
- Leijen, S.; van Geel, R.M.; Pavlick, A.C.; Tibes, R.; Rosen, L.; Razak, A.R.; Lam, R.; Demuth, T.; Rose, S.; Lee, M.A.; et al. Phase I Study Evaluating WEE1 Inhibitor AZD1775 As Monotherapy and in Combination With Gemcitabine, Cisplatin, or Carboplatin in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2016, 34, 4371–4380. [Google Scholar] [CrossRef]
- Cuneo, K.C.; Morgan, M.A.; Sahai, V.; Schipper, M.J.; Parsels, L.A.; Parsels, J.D.; Devasia, T.; Al-Hawaray, M.; Cho, C.S.; Nathan, H.; et al. Dose Escalation Trial of the Wee1 Inhibitor Adavosertib (AZD1775) in Combination With Gemcitabine and Radiation for Patients With Locally Advanced Pancreatic Cancer. J. Clin. Oncol. 2019, 37, 2643–2650. [Google Scholar] [CrossRef]
- Chera, B.S.; Sheth, S.H.; Patel, S.A.; Goldin, D.; Douglas, K.E.; Green, R.L.; Shen, C.J.; Gupta, G.P.; Moore, D.T.; Grilley Olson, J.E.; et al. Phase 1 trial of adavosertib (AZD1775) in combination with concurrent radiation and cisplatin for intermediate-risk and high-risk head and neck squamous cell carcinoma. Cancer 2021, 127, 4447–4454. [Google Scholar] [CrossRef]
- Leijen, S.; van Geel, R.M.; Sonke, G.S.; de Jong, D.; Rosenberg, E.H.; Marchetti, S.; Pluim, D.; van Werkhoven, E.; Rose, S.; Lee, M.A.; et al. Phase II Study of WEE1 Inhibitor AZD1775 Plus Carboplatin in Patients With TP53-Mutated Ovarian Cancer Refractory or Resistant to First-Line Therapy Within 3 Months. J. Clin. Oncol. 2016, 34, 4354–4361. [Google Scholar] [CrossRef] [Green Version]
- Oza, A.M.; Estevez-Diz, M.; Grischke, E.M.; Hall, M.; Marme, F.; Provencher, D.; Uyar, D.; Weberpals, J.I.; Wenham, R.M.; Laing, N.; et al. A Biomarker-enriched, Randomized Phase II Trial of Adavosertib (AZD1775) Plus Paclitaxel and Carboplatin for Women with Platinum-sensitive TP53-mutant Ovarian Cancer. Clin. Cancer Res. 2020, 26, 4767–4776. [Google Scholar] [CrossRef]
- Lheureux, S.; Cristea, M.C.; Bruce, J.P.; Garg, S.; Cabanero, M.; Mantia-Smaldone, G.; Olawaiye, A.B.; Ellard, S.L.; Weberpals, J.I.; Wahner Hendrickson, A.E.; et al. Adavosertib plus gemcitabine for platinum-resistant or platinum-refractory recurrent ovarian cancer: A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 2021, 397, 281–292. [Google Scholar] [CrossRef]
- Moore, K.N.; Chambers, S.K.; Hamilton, E.P.; Chen, L.M.; Oza, A.M.; Ghamande, S.A.; Konecny, G.E.; Plaxe, S.C.; Spitz, D.L.; Geenen, J.J.J.; et al. Adavosertib with Chemotherapy in Patients with Primary Platinum-Resistant Ovarian, Fallopian Tube, or Peritoneal Cancer: An Open-Label, Four-Arm, Phase II Study. Clin. Cancer Res. 2021, 28, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Keenan, T.E.; Li, T.; Vallius, T.; Guerriero, J.L.; Tayob, N.; Kochupurakkal, B.; Davis, J.; Pastorello, R.; Tahara, R.K.; Anderson, L.; et al. Clinical Efficacy and Molecular Response Correlates of the WEE1 Inhibitor Adavosertib Combined with Cisplatin in Patients with Metastatic Triple-Negative Breast Cancer. Clin. Cancer Res. 2021, 27, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Seligmann, J.F.; Fisher, D.J.; Brown, L.C.; Adams, R.A.; Graham, J.; Quirke, P.; Richman, S.D.; Butler, R.; Domingo, E.; Blake, A.; et al. Inhibition of WEE1 Is Effective in TP53- and RAS-Mutant Metastatic Colorectal Cancer: A Randomized Trial (FOCUS4-C) Comparing Adavosertib (AZD1775) With Active Monitoring. J. Clin. Oncol. 2021, 39, 3705–3715. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Shim, J.; Mortimer, P.G.S.; Smith, S.A.; Godin, R.E.; Hollingsworth, S.J.; Kim, H.J.; Jung, H.A.; Sun, J.M.; Park, W.Y.; et al. Biomarker-driven phase 2 umbrella trial study for patients with recurrent small cell lung cancer failing platinum-based chemotherapy. Cancer 2020, 126, 4002–4012. [Google Scholar] [CrossRef]
- Liu, J.F.; Xiong, N.; Campos, S.M.; Wright, A.A.; Krasner, C.; Schumer, S.; Horowitz, N.; Veneris, J.; Tayob, N.; Morrissey, S.; et al. Phase II Study of the WEE1 Inhibitor Adavosertib in Recurrent Uterine Serous Carcinoma. J. Clin. Oncol. 2021, 39, 1531–1539. [Google Scholar] [CrossRef]
- Chen, A.P.; Kummar, S.; Moore, N.; Rubinstein, L.V.; Zhao, Y.; Williams, P.M.; Palmisano, A.; Sims, D.; O’Sullivan Coyne, G.; Rosenberger, C.L.; et al. Molecular Profiling-Based Assignment of Cancer Therapy (NCI-MPACT): A Randomized Multicenter Phase II Trial. JCO Precis. Oncol. 2021, 5, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, D.; Steegmaier, M.; Hoffmann, M.; Grauert, M.; Baum, A.; Quant, J.; Haslinger, C.; Garin-Chesa, P.; Adolf, G.R. BI 6727, a Polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin. Cancer Res. 2009, 15, 3094–3102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoffski, P.; Awada, A.; Dumez, H.; Gil, T.; Bartholomeus, S.; Wolter, P.; Taton, M.; Fritsch, H.; Glomb, P.; Munzert, G. A phase I, dose-escalation study of the novel Polo-like kinase inhibitor volasertib (BI 6727) in patients with advanced solid tumours. Eur. J. Cancer 2012, 48, 179–186. [Google Scholar] [CrossRef]
- Stadler, W.M.; Vaughn, D.J.; Sonpavde, G.; Vogelzang, N.J.; Tagawa, S.T.; Petrylak, D.P.; Rosen, P.; Lin, C.C.; Mahoney, J.; Modi, S.; et al. An open-label, single-arm, phase 2 trial of the Polo-like kinase inhibitor volasertib (BI 6727) in patients with locally advanced or metastatic urothelial cancer. Cancer 2014, 120, 976–982. [Google Scholar] [CrossRef] [Green Version]
- Dohner, H.; Lubbert, M.; Fiedler, W.; Fouillard, L.; Haaland, A.; Brandwein, J.M.; Lepretre, S.; Reman, O.; Turlure, P.; Ottmann, O.G.; et al. Randomized, phase 2 trial of low-dose cytarabine with or without volasertib in AML patients not suitable for induction therapy. Blood 2014, 124, 1426–1433. [Google Scholar] [CrossRef]
- Dohner, H.; Symeonidis, A.; Deeren, D.; Demeter, J.; Sanz, M.A.; Anagnostopoulos, A.; Esteve, J.; Fiedler, W.; Porkka, K.; Kim, H.J.; et al. Adjunctive Volasertib in Patients With Acute Myeloid Leukemia not Eligible for Standard Induction Therapy: A Randomized, Phase 3 Trial. Hemasphere 2021, 5, e617. [Google Scholar] [CrossRef] [PubMed]
- Ellis, P.M.; Leighl, N.B.; Hirsh, V.; Reaume, M.N.; Blais, N.; Wierzbicki, R.; Sadrolhefazi, B.; Gu, Y.; Liu, D.; Pilz, K.; et al. A Randomized, Open-Label Phase II Trial of Volasertib as Monotherapy and in Combination With Standard-Dose Pemetrexed Compared With Pemetrexed Monotherapy in Second-Line Treatment for Non-Small-Cell Lung Cancer. Clin. Lung Cancer 2015, 16, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Pujade-Lauraine, E.; Selle, F.; Weber, B.; Ray-Coquard, I.L.; Vergote, I.; Sufliarsky, J.; Del Campo, J.M.; Lortholary, A.; Lesoin, A.; Follana, P.; et al. Volasertib Versus Chemotherapy in Platinum-Resistant or -Refractory Ovarian Cancer: A Randomized Phase II Groupe des Investigateurs Nationaux pour l’Etude des Cancers de l’Ovaire Study. J. Clin. Oncol. 2016, 34, 706–713. [Google Scholar] [CrossRef] [PubMed]
- Valsasina, B.; Beria, I.; Alli, C.; Alzani, R.; Avanzi, N.; Ballinari, D.; Cappella, P.; Caruso, M.; Casolaro, A.; Ciavolella, A.; et al. NMS-P937, an orally available, specific small-molecule polo-like kinase 1 inhibitor with antitumor activity in solid and hematologic malignancies. Mol. Cancer Ther. 2012, 11, 1006–1016. [Google Scholar] [CrossRef] [Green Version]
- Weiss, G.J.; Jameson, G.; Von Hoff, D.D.; Valsasina, B.; Davite, C.; Di Giulio, C.; Fiorentini, F.; Alzani, R.; Carpinelli, P.; Di Sanzo, A.; et al. Phase I dose escalation study of NMS-1286937, an orally available Polo-Like Kinase 1 inhibitor, in patients with advanced or metastatic solid tumors. Invest. New Drugs 2018, 36, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Zeidan, A.M.; Ridinger, M.; Lin, T.L.; Becker, P.S.; Schiller, G.J.; Patel, P.A.; Spira, A.I.; Tsai, M.L.; Samuelsz, E.; Silberman, S.L.; et al. A Phase Ib Study of Onvansertib, a Novel Oral PLK1 Inhibitor, in Combination Therapy for Patients with Relapsed or Refractory Acute Myeloid Leukemia. Clin. Cancer Res. 2020, 26, 6132–6140. [Google Scholar] [CrossRef]
- Byrum, A.K.; Vindigni, A.; Mosammaparast, N. Defining and Modulating ‘BRCAness’. Trends Cell Biol. 2019, 29, 740–751. [Google Scholar] [CrossRef]
- Abkevich, V.; Timms, K.M.; Hennessy, B.T.; Potter, J.; Carey, M.S.; Meyer, L.A.; Smith-McCune, K.; Broaddus, R.; Lu, K.H.; Chen, J.; et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br. J. Cancer 2012, 107, 1776–1782. [Google Scholar] [CrossRef] [Green Version]
- Popova, T.; Manie, E.; Rieunier, G.; Caux-Moncoutier, V.; Tirapo, C.; Dubois, T.; Delattre, O.; Sigal-Zafrani, B.; Bollet, M.; Longy, M.; et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012, 72, 5454–5462. [Google Scholar] [CrossRef] [Green Version]
- Birkbak, N.J.; Wang, Z.C.; Kim, J.Y.; Eklund, A.C.; Li, Q.; Tian, R.; Bowman-Colin, C.; Li, Y.; Greene-Colozzi, A.; Iglehart, J.D.; et al. Telomeric allelic imbalance indicates defective DNA repair and sensitivity to DNA-damaging agents. Cancer Discov. 2012, 2, 366–375. [Google Scholar] [CrossRef] [Green Version]
- Timms, K.M.; Abkevich, V.; Hughes, E.; Neff, C.; Reid, J.; Morris, B.; Kalva, S.; Potter, J.; Tran, T.V.; Chen, J.; et al. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res. 2014, 16, 475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Telli, M.L.; Timms, K.M.; Reid, J.; Hennessy, B.; Mills, G.B.; Jensen, K.C.; Szallasi, Z.; Barry, W.T.; Winer, E.P.; Tung, N.M.; et al. Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin. Cancer Res. 2016, 22, 3764–3773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frampton, G.M.; Fichtenholtz, A.; Otto, G.A.; Wang, K.; Downing, S.R.; He, J.; Schnall-Levin, M.; White, J.; Sanford, E.M.; An, P.; et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat. Biotechnol. 2013, 31, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.E.; Leary, A.; Scott, C.L.; Serra, V.; Lord, C.J.; Bowtell, D.; Chang, D.K.; Garsed, D.W.; Jonkers, J.; Ledermann, J.A.; et al. ESMO recommendations on predictive biomarker testing for homologous recombination deficiency and PARP inhibitor benefit in ovarian cancer. Ann. Oncol. 2020, 31, 1606–1622. [Google Scholar] [CrossRef] [PubMed]
- Ngoi, N.Y.L.; Tan, D.S.P. The role of homologous recombination deficiency testing in ovarian cancer and its clinical implications: Do we need it? ESMO Open 2021, 6, 100144. [Google Scholar] [CrossRef] [PubMed]
- Polak, P.; Kim, J.; Braunstein, L.Z.; Karlic, R.; Haradhavala, N.J.; Tiao, G.; Rosebrock, D.; Livitz, D.; Kubler, K.; Mouw, K.W.; et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat. Genet. 2017, 49, 1476–1486. [Google Scholar] [CrossRef]
- Gulhan, D.C.; Lee, J.J.; Melloni, G.E.M.; Cortes-Ciriano, I.; Park, P.J. Detecting the mutational signature of homologous recombination deficiency in clinical samples. Nat. Genet. 2019, 51, 912–919. [Google Scholar] [CrossRef]
- Farkkila, A.; Gulhan, D.C.; Casado, J.; Jacobson, C.A.; Nguyen, H.; Kochupurakkal, B.; Maliga, Z.; Yapp, C.; Chen, Y.A.; Schapiro, D.; et al. Immunogenomic profiling determines responses to combined PARP and PD-1 inhibition in ovarian cancer. Nat. Commun. 2020, 11, 1459. [Google Scholar] [CrossRef] [Green Version]
- Cleary, J.M.; Aguirre, A.J.; Shapiro, G.I.; D’Andrea, A.D. Biomarker-Guided Development of DNA Repair Inhibitors. Mol. Cell 2020, 78, 1070–1085. [Google Scholar] [CrossRef]
- Yazinski, S.A.; Comaills, V.; Buisson, R.; Genois, M.M.; Nguyen, H.D.; Ho, C.K.; Todorova Kwan, T.; Morris, R.; Lauffer, S.; Nussenzweig, A.; et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 2017, 31, 318–332. [Google Scholar] [CrossRef]
- Kim, H.; George, E.; Ragland, R.; Rafail, S.; Zhang, R.; Krepler, C.; Morgan, M.; Herlyn, M.; Brown, E.; Simpkins, F. Targeting the ATR/CHK1 Axis with PARP Inhibition Results in Tumor Regression in BRCA-Mutant Ovarian Cancer Models. Clin. Cancer Res. 2017, 23, 3097–3108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parmar, K.; Kochupurakkal, B.S.; Lazaro, J.B.; Wang, Z.C.; Palakurthi, S.; Kirschmeier, P.T.; Yang, C.; Sambel, L.A.; Farkkila, A.; Reznichenko, E.; et al. The CHK1 Inhibitor Prexasertib Exhibits Monotherapy Activity in High-Grade Serous Ovarian Cancer Models and Sensitizes to PARP Inhibition. Clin. Cancer Res. 2019, 25, 6127–6140. [Google Scholar] [CrossRef] [PubMed]
- Tentori, L.; Lacal, P.M.; Muzi, A.; Dorio, A.S.; Leonetti, C.; Scarsella, M.; Ruffini, F.; Xu, W.; Min, W.; Stoppacciaro, A.; et al. Poly(ADP-ribose) polymerase (PARP) inhibition or PARP-1 gene deletion reduces angiogenesis. Eur. J. Cancer 2007, 43, 2124–2133. [Google Scholar] [CrossRef]
- Chan, N.; Bristow, R.G. “Contextual” synthetic lethality and/or loss of heterozygosity: Tumor hypoxia and modification of DNA repair. Clin. Cancer Res. 2010, 16, 4553–4560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.F.; Barry, W.T.; Birrer, M.; Lee, J.M.; Buckanovich, R.J.; Fleming, G.F.; Rimel, B.J.; Buss, M.K.; Nattam, S.R.; Hurteau, J.; et al. Overall survival and updated progression-free survival outcomes in a randomized phase II study of combination cediranib and olaparib versus olaparib in relapsed platinum-sensitive ovarian cancer. Ann. Oncol. 2019, 30, 551–557. [Google Scholar] [CrossRef]
- Mirza, M.R.; Avall Lundqvist, E.; Birrer, M.J.; dePont Christensen, R.; Nyvang, G.B.; Malander, S.; Anttila, M.; Werner, T.L.; Lund, B.; Lindahl, G.; et al. Niraparib plus bevacizumab versus niraparib alone for platinum-sensitive recurrent ovarian cancer (NSGO-AVANOVA2/ENGOT-ov24): A randomised, phase 2, superiority trial. Lancet Oncol. 2019, 20, 1409–1419. [Google Scholar] [CrossRef]
- Elyashiv, O.; Ledermann, J.; Parmar, G.; Farrelly, L.; Counsell, N.; Feeney, A.; El-Khouly, F.; Macdonald, I.; Neto, A.; Arthur-Darkwa, E.; et al. ICON 9-an international phase III randomized study to evaluate the efficacy of maintenance therapy with olaparib and cediranib or olaparib alone in patients with relapsed platinum-sensitive ovarian cancer following a response to platinum-based chemotherapy. Int. J. Gynecol. Cancer 2021, 31, 134–138. [Google Scholar] [CrossRef]
- Kim, J.W.; Cardin, D.B.; Vaishampayan, U.N.; Kato, S.; Grossman, S.R.; Glazer, P.M.; Shyr, Y.; Ivy, S.P.; LoRusso, P.M. Clinical Activity and Safety of Cediranib and Olaparib Combination in Patients with Metastatic Pancreatic Ductal Adenocarcinoma without BRCA Mutation. Oncologist 2021, 26, e1104–e1109. [Google Scholar] [CrossRef]
- Kumar, A.; Fernandez-Capetillo, O.; Carrera, A.C. Nuclear phosphoinositide 3-kinase beta controls double-strand break DNA repair. Proc. Natl. Acad. Sci. USA 2010, 107, 7491–7496. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, Y.H.; Garcia-Garcia, C.; Serra, V.; He, L.; Torres-Lockhart, K.; Prat, A.; Anton, P.; Cozar, P.; Guzman, M.; Grueso, J.; et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. 2012, 2, 1036–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juvekar, A.; Burga, L.N.; Hu, H.; Lunsford, E.P.; Ibrahim, Y.H.; Balmana, J.; Rajendran, A.; Papa, A.; Spencer, K.; Lyssiotis, C.A.; et al. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov. 2012, 2, 1048–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matulonis, U.A.; Wulf, G.M.; Barry, W.T.; Birrer, M.; Westin, S.N.; Farooq, S.; Bell-McGuinn, K.M.; Obermayer, E.; Whalen, C.; Spagnoletti, T.; et al. Phase I dose escalation study of the PI3kinase pathway inhibitor BKM120 and the oral poly (ADP ribose) polymerase (PARP) inhibitor olaparib for the treatment of high-grade serous ovarian and breast cancer. Ann. Oncol. 2017, 28, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Barry, W.T.; Birrer, M.; Westin, S.N.; Cadoo, K.A.; Shapiro, G.I.; Mayer, E.L.; O’Cearbhaill, R.E.; Coleman, R.L.; Kochupurakkal, B.; et al. Olaparib and alpha-specific PI3K inhibitor alpelisib for patients with epithelial ovarian cancer: A dose-escalation and dose-expansion phase 1b trial. Lancet Oncol. 2019, 20, 570–580. [Google Scholar] [CrossRef]
- Yap, T.A.; Kristeleit, R.; Michalarea, V.; Pettitt, S.J.; Lim, J.S.J.; Carreira, S.; Roda, D.; Miller, R.; Riisnaes, R.; Miranda, S.; et al. Phase I Trial of the PARP Inhibitor Olaparib and AKT Inhibitor Capivasertib in Patients with BRCA1/2- and Non-BRCA1/2-Mutant Cancers. Cancer Discov. 2020, 10, 1528–1543. [Google Scholar] [CrossRef]
- Westin, S.N.; Labrie, M.; Litton, J.K.; Blucher, A.; Fang, Y.; Vellano, C.P.; Marszalek, J.R.; Feng, N.; Ma, X.; Creason, A.; et al. Phase Ib Dose Expansion and Translational Analyses of Olaparib in Combination with Capivasertib in Recurrent Endometrial, Triple-Negative Breast, and Ovarian Cancer. Clin. Cancer Res. 2021, 27, 6354–6365. [Google Scholar] [CrossRef]
- Goodwin, J.F.; Schiewer, M.J.; Dean, J.L.; Schrecengost, R.S.; de Leeuw, R.; Han, S.; Ma, T.; Den, R.B.; Dicker, A.P.; Feng, F.Y.; et al. A hormone-DNA repair circuit governs the response to genotoxic insult. Cancer Discov. 2013, 3, 1254–1271. [Google Scholar] [CrossRef] [Green Version]
- Polkinghorn, W.R.; Parker, J.S.; Lee, M.X.; Kass, E.M.; Spratt, D.E.; Iaquinta, P.J.; Arora, V.K.; Yen, W.F.; Cai, L.; Zheng, D.; et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov. 2013, 3, 1245–1253. [Google Scholar] [CrossRef] [Green Version]
- Asim, M.; Tarish, F.; Zecchini, H.I.; Sanjiv, K.; Gelali, E.; Massie, C.E.; Baridi, A.; Warren, A.Y.; Zhao, W.; Ogris, C.; et al. Synthetic lethality between androgen receptor signalling and the PARP pathway in prostate cancer. Nat. Commun. 2017, 8, 374. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Karanika, S.; Yang, G.; Wang, J.; Park, S.; Broom, B.M.; Manyam, G.C.; Wu, W.; Luo, Y.; Basourakos, S.; et al. Androgen receptor inhibitor-induced “BRCAness” and PARP inhibition are synthetically lethal for castration-resistant prostate cancer. Sci. Signal 2017, 10, eaam7479. [Google Scholar] [CrossRef] [Green Version]
- Karanika, S.; Karantanos, T.; Li, L.; Wang, J.; Park, S.; Yang, G.; Zuo, X.; Song, J.H.; Maity, S.N.; Manyam, G.C.; et al. Targeting DNA Damage Response in Prostate Cancer by Inhibiting Androgen Receptor-CDC6-ATR-Chk1 Signaling. Cell Rep. 2017, 18, 1970–1981. [Google Scholar] [CrossRef] [PubMed]
- Clarke, N.; Wiechno, P.; Alekseev, B.; Sala, N.; Jones, R.; Kocak, I.; Chiuri, V.E.; Jassem, J.; Flechon, A.; Redfern, C.; et al. Olaparib combined with abiraterone in patients with metastatic castration-resistant prostate cancer: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2018, 19, 975–986. [Google Scholar] [CrossRef]
- Sun, C.; Fang, Y.; Yin, J.; Chen, J.; Ju, Z.; Zhang, D.; Chen, X.; Vellano, C.P.; Jeong, K.J.; Ng, P.K.; et al. Rational combination therapy with PARP and MEK inhibitors capitalizes on therapeutic liabilities in RAS mutant cancers. Sci. Transl. Med. 2017, 9, eaal5148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H., Jr.; et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: Prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef]
- Teo, M.Y.; Seier, K.; Ostrovnaya, I.; Regazzi, A.M.; Kania, B.E.; Moran, M.M.; Cipolla, C.K.; Bluth, M.J.; Chaim, J.; Al-Ahmadie, H.; et al. Alterations in DNA Damage Response and Repair Genes as Potential Marker of Clinical Benefit From PD-1/PD-L1 Blockade in Advanced Urothelial Cancers. J. Clin. Oncol. 2018, 36, 1685–1694. [Google Scholar] [CrossRef]
- Li, A.; Yi, M.; Qin, S.; Chu, Q.; Luo, S.; Wu, K. Prospects for combining immune checkpoint blockade with PARP inhibition. J. Hematol. Oncol. 2019, 12, 98. [Google Scholar] [CrossRef]
- Pantelidou, C.; Sonzogni, O.; De Oliveria Taveira, M.; Mehta, A.K.; Kothari, A.; Wang, D.; Visal, T.; Li, M.K.; Pinto, J.; Castrillon, J.A.; et al. PARP Inhibitor Efficacy Depends on CD8(+) T-cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov. 2019, 9, 722–737. [Google Scholar] [CrossRef] [Green Version]
- Sen, T.; Rodriguez, B.L.; Chen, L.; Corte, C.M.D.; Morikawa, N.; Fujimoto, J.; Cristea, S.; Nguyen, T.; Diao, L.; Li, L.; et al. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discov. 2019, 9, 646–661. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Zhao, W.; Ju, Z.; Wang, L.; Peng, Y.; Labrie, M.; Yap, T.A.; Mills, G.B.; Peng, G. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res. 2019, 79, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Kwon, J.; Bakhoum, S.F. The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer Discov. 2020, 10, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.K.; Hsu, J.M.; Hsu, J.L.; Yu, W.H.; Du, Y.; Lee, H.H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat. Commun. 2017, 8, 1751. [Google Scholar] [CrossRef]
- Domchek, S.M.; Postel-Vinay, S.; Im, S.A.; Park, Y.H.; Delord, J.P.; Italiano, A.; Alexandre, J.; You, B.; Bastian, S.; Krebs, M.G.; et al. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): An open-label, multicentre, phase 1/2, basket study. Lancet Oncol. 2020, 21, 1155–1164. [Google Scholar] [CrossRef]
- Drew, Y.; Kaufman, B.; Banerjee, S.; Lortholary, A.; Hong, S.H.; Park, Y.H.; Zimmermann, S.; Roxburgh, P.; Ferguson, M.; Alvarez, R.H.; et al. Phase II study of olaparib + durvalumab (MEDIOLA): Updated results in germline BRCA-mutated platinum-sensitive relapsed (PSR) ovarian cancer (OC). Ann. Oncol. 2019, 30, v485–v486. [Google Scholar] [CrossRef]
- Bang, Y.-J.; Kaufman, B.; Geva, R.; Stemmer, S.M.; Hong, S.-H.; Lee, J.-S.; Domchek, S.M.; Lanasa, M.C.; Tang, M.; Gresty, C.; et al. An open-label, phase II basket study of olaparib and durvalumab (MEDIOLA): Results in patients with relapsed gastric cancer. J. Clin. Oncol. 2019, 37, 140. [Google Scholar] [CrossRef]
- Thomas, A.; Vilimas, R.; Trindade, C.; Erwin-Cohen, R.; Roper, N.; Xi, L.; Krishnasamy, V.; Levy, E.; Mammen, A.; Nichols, S.; et al. Durvalumab in Combination with Olaparib in Patients with Relapsed SCLC: Results from a Phase II Study. J. Thorac. Oncol. 2019, 14, 1447–1457. [Google Scholar] [CrossRef] [PubMed]
- Karzai, F.; VanderWeele, D.; Madan, R.A.; Owens, H.; Cordes, L.M.; Hankin, A.; Couvillon, A.; Nichols, E.; Bilusic, M.; Beshiri, M.L.; et al. Activity of durvalumab plus olaparib in metastatic castration-resistant prostate cancer in men with and without DNA damage repair mutations. J. Immunother. Cancer 2018, 6, 141. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Waggoner, S.; Vidal, G.A.; Mita, M.; Moroney, J.W.; Holloway, R.; Van Le, L.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination With Pembrolizumab in Patients With Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol. 2019, 5, 1141–1149. [Google Scholar] [CrossRef] [Green Version]
- Vinayak, S.; Tolaney, S.M.; Schwartzberg, L.S.; Mita, M.M.; McCann, G.A.-L.; Tan, A.R.; Hendrickson, A.E.W.; Forero-Torres, A.; Anders, C.K.; Wulf, G.M.; et al. TOPACIO/Keynote-162: Niraparib + pembrolizumab in patients (pts) with metastatic triple-negative breast cancer (TNBC), a phase 2 trial. J. Clin. Oncol. 2018, 36, 1011. [Google Scholar] [CrossRef]
- Goff, P.H.; Bhakuni, R.; Pulliam, T.; Lee, J.H.; Hall, E.T.; Nghiem, P. Intersection of Two Checkpoints: Could Inhibiting the DNA Damage Response Checkpoint Rescue Immune Checkpoint-Refractory Cancer? Cancers 2021, 13, 3415. [Google Scholar] [CrossRef] [PubMed]
| Trial Phase | Disease Setting | Treatments | Most Common Grade ≥ 3 Toxicity | Efficacy | Reference |
|---|---|---|---|---|---|
| Prexasertib/LY2606368 | |||||
| 1 | Solid tumors | Escalating doses of LY2606368 | Neutropenia (89%) Leukopenia (71%) Anemia (69%) | PR 2/45 (4%) SD 15/45 (33%) | [93] |
| 1 | Solid tumors | Prexasertib monotherapy | Neutropenia (83%) Leukopenia (75%) Thrombocytopenia (33%) | SD 8/11 (73%) | [94] |
| 1 | Solid tumors | Prexasertib in combination with standard chemotherapy | Prexasertib + Cisplatin Neutropenia (67%) Prexasertib + Cetuximab Neutropenia (54%) Prexasertib + 5-FU Neutropenia (100%) | Prexasertib + Cisplatin PR 8/63 (13%) Prexasertib + Cetuximab PR 7/31 (5%) Prexasertib + 5-FU PR 1/8 (13%) | [98] |
| 1 | Solid tumors | Prexasertib in combination with olaparib | Neutropenia (79%) | BRCA-mutant HGSOC PR 4/18 (22%) SD 6/18 (33%) | [99] |
| 1 | Solid tumors | Prexasertib in combination with LY3300054 (anti-PDL1 antibody) | Neutropenia (82%) Leukopenia (76%) | PR 3/17 (18%) SD 8/17 (47%) | [100] |
| 2 | HGSOC (BRCA-wild type) | Prexasertib monotherapy | Neutropenia (93%) Leukopenia (82%) | PR 8/28 (29%) Median PFS 7.4 months (95% CI 2.1–9.4) | [95] |
| 2 | TNBC (BRCA-wild type) | Prexasertib monotherapy | Neutropenia (89%) Anemia (33%) | PR 1/9 (11%) SD 4/9 (44%) | [96] |
| 2 | Small-cell lung cancer | Prexasertib monotherapy | Neutropenia (65%) | Platinum-sensitive: PR 3/58 (5%) SD 15/58 (26%) Platinum-refractory: PR 0/60 (0%) SD 12/60 (20%) | [97] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, W.; Lee, E.S. Therapeutic Targeting of DNA Damage Response in Cancer. Int. J. Mol. Sci. 2022, 23, 1701. https://doi.org/10.3390/ijms23031701
Choi W, Lee ES. Therapeutic Targeting of DNA Damage Response in Cancer. International Journal of Molecular Sciences. 2022; 23(3):1701. https://doi.org/10.3390/ijms23031701
Chicago/Turabian StyleChoi, Wonyoung, and Eun Sook Lee. 2022. "Therapeutic Targeting of DNA Damage Response in Cancer" International Journal of Molecular Sciences 23, no. 3: 1701. https://doi.org/10.3390/ijms23031701
APA StyleChoi, W., & Lee, E. S. (2022). Therapeutic Targeting of DNA Damage Response in Cancer. International Journal of Molecular Sciences, 23(3), 1701. https://doi.org/10.3390/ijms23031701