
Fanconi Anemia Patients from an Indigenous Community in Mexico Carry a New Founder Pathogenic Variant in FANCG

 , ,
, ,  ,
,  , , , and
, , , and
Abstract
:1. Introduction
2. Results
2.1. Chromosomal Breakage Analysis Confirmed a FA Diagnosis
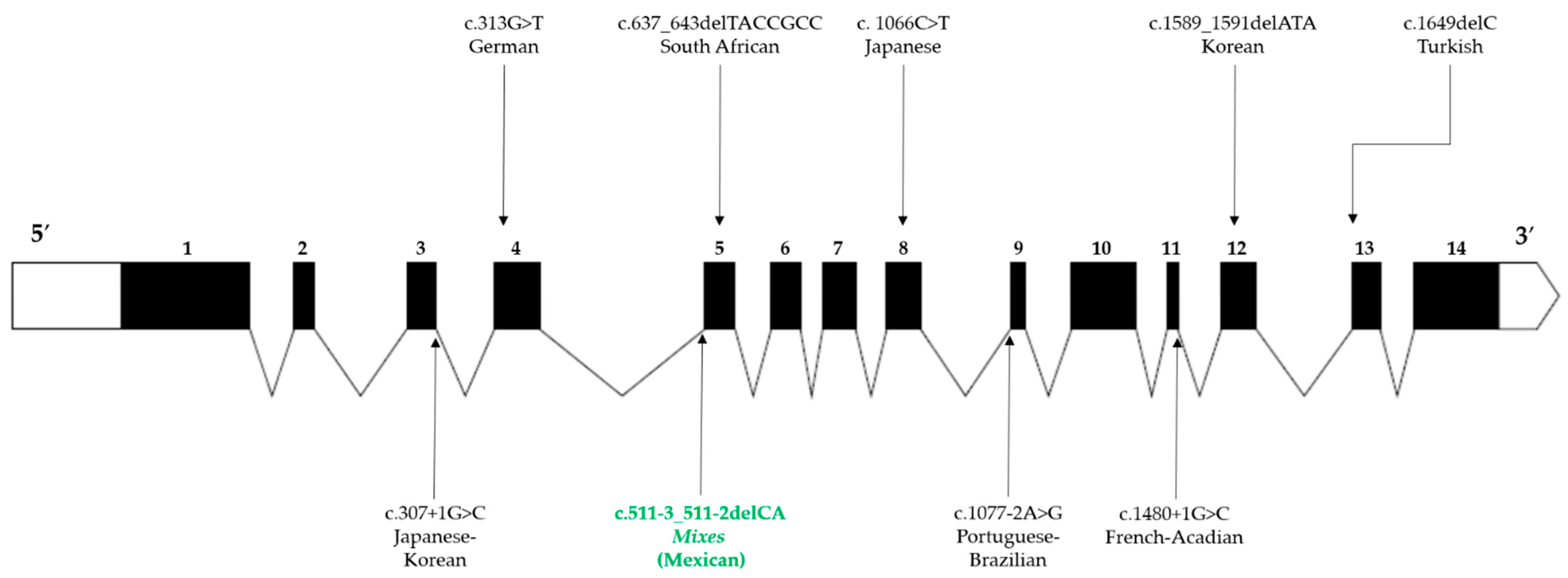
2.2. Pathogenic Variant Found in FANCG: c.511-3_511-2delCA Is Predicted to Induce Exon 5 Skipping
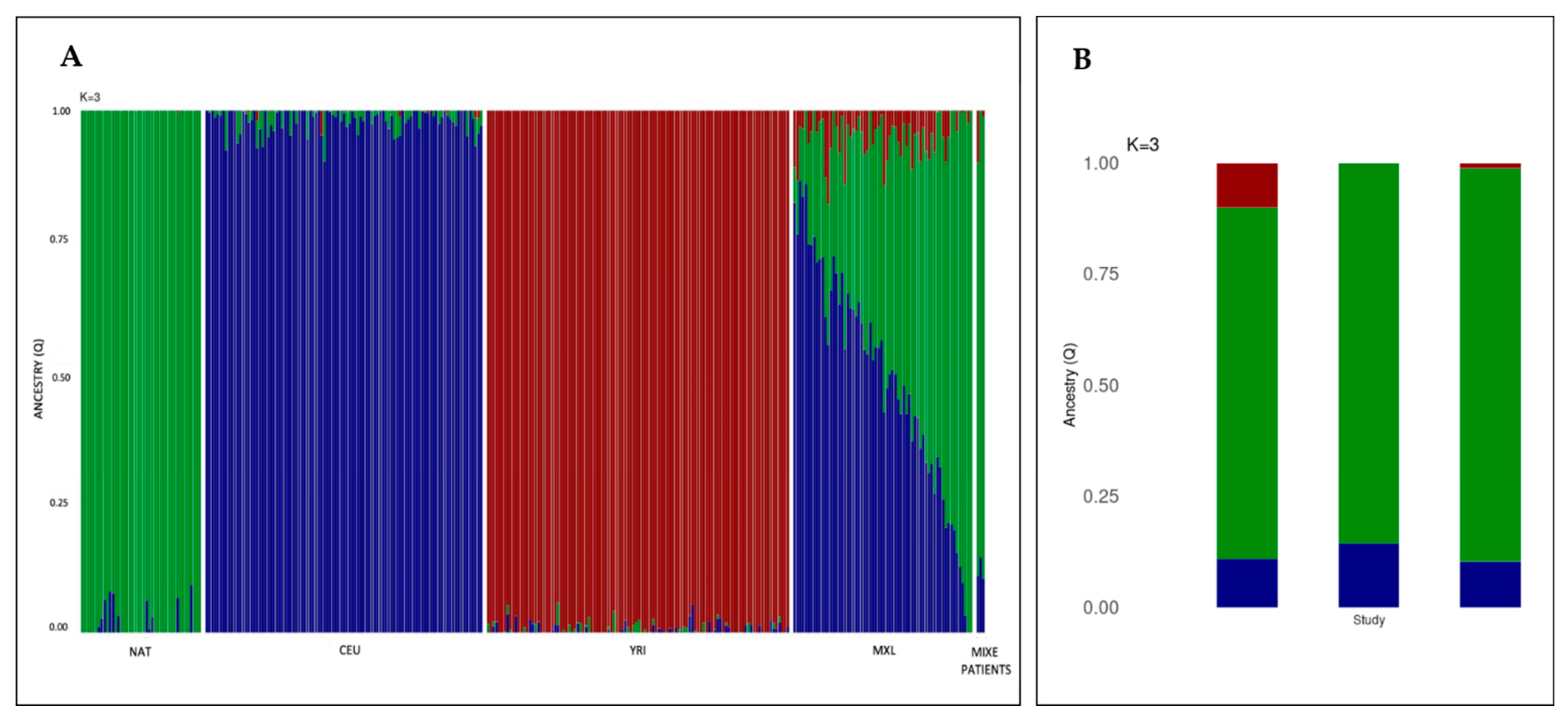
2.3. Reduced Genetic Variation in Locus g.29754068-38771831 Supporting a Founder Effect for FANCG:c.511-3_511-2delCA in the Mixe Population
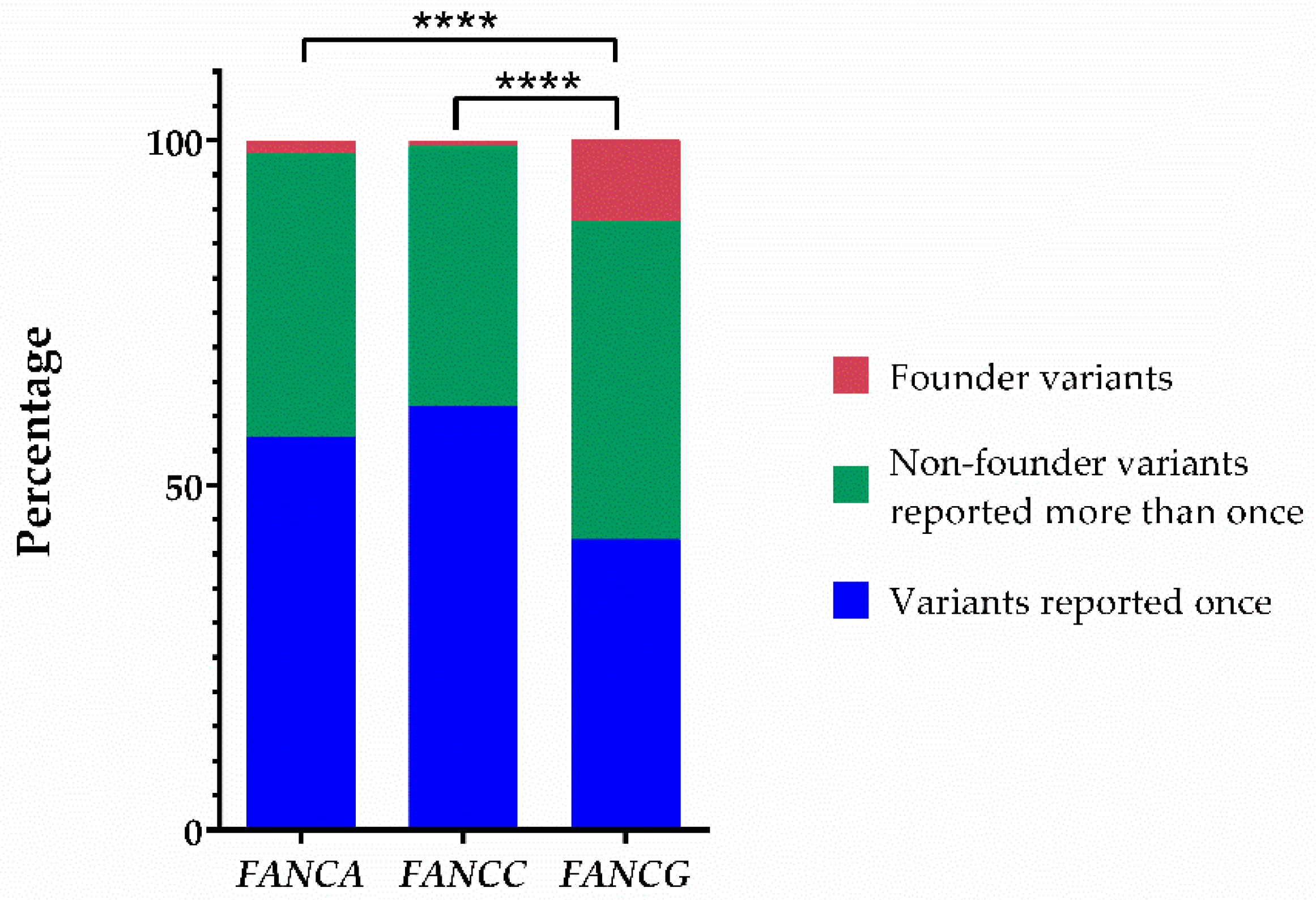
2.4. The Fraction of Pathogenic Variants Linked to a Founder Effect in FANCG Is the Highest among the Most Common FANC Genes
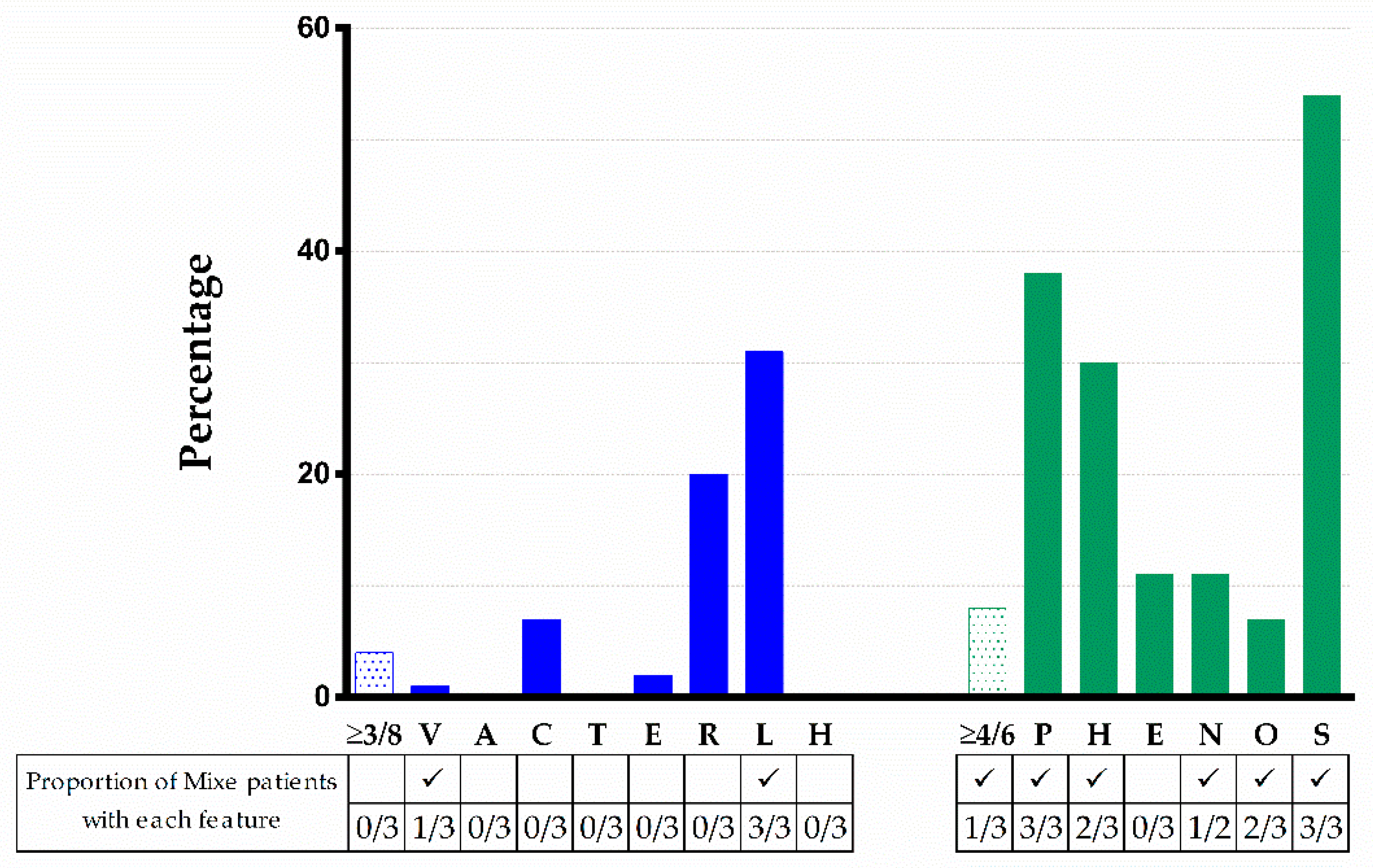
2.5. Evaluation of the Phenotype of Mixe Patients with FA in the Framework of the Reported FANCG Pathogenic Variant
3. Discussion
4. Materials and Methods
4.1. Editorial Policies and Ethical Considerations
4.2. Patients
4.3. Chromosomal Breakage Test
4.4. Genotyping
4.4.1. Genomic DNA Extraction
4.4.2. Targeted Next-Generation Sequencing
4.4.3. Sanger Sequencing
4.5. In Silico Splice Site Analysis
4.6. High-Resolution Microarray Analysis
4.6.1. Analysis of Long-Contiguous Stretch of Homozygosity
4.6.2. Estimation of Coefficient of Inbreeding
4.6.3. Haplotype Inference
4.7. Ancestry Analysis
4.8. Fanconi Anemia Variant Database Analysis
4.9. Phenotype Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 1KGP | 1000 Genomes Project |
| ACMG | American College of Medical Genetics |
| AMR | Ad Mixed American (1KGP super population) |
| ANC | Absolut Neutrophils Counts |
| bp | base pair |
| BMF | Bone marrow failure |
| CNS | Central nervous system |
| CEU | Utah residents (CEPH) with Northern and Western European ancestry (1KGP population) |
| ChAS | Chromosome Analysis Suite |
| CHB | Han Chinese in Beijing, China (1KGP population) |
| DEB | Diepoxibutane |
| FA | Fanconi anemia |
| FA/BRCA | Fanconi anemia/Breast Cancer pathway |
| FPV | Founder pathogenic variant |
| Hb | Hemoglobin |
| HGVS | Human Genome Variation Society |
| HSCT | Hematopoietic stem-cell transplantation |
| HSF | Human Splicing Finder |
| Hg19 | Homo sapiens (human) genome assembly GRCh37 |
| INP | Instituto Nacional de Pediatría |
| kb | kilobases |
| LCSH | Long-contiguous stretches of homozygosity |
| MCV | Mean Corpuscular Volume |
| MRI | Magnetic resonance imaging |
| mRNA | messenger RNA |
| MXL | Mexican Ancestry in Los Angeles CA, USA (1 KGP population) |
| MT | Mutant |
| NAT | Native Americans (1KGP population) |
| NGS | Next generation sequencing |
| NMD | Non-sense mediated decay |
| OMIM | Online Mendelian Inheritance in Man |
| PCR | Polymerase chain reaction |
| PHENOS | Skin Pigmentation, small Head, small Eyes, Nervous system, Otology, Short stature |
| PLT | Platelets |
| PVS1 | Pathogenic Very Strong 1 |
| PM2 | Pathogenic Moderate 2 |
| PP3 | Pathogenic Supporting 3 |
| PDB | Protein Data Bank |
| PV | Pathogenic variants |
| SNV | Single nucleotide variant |
| SDS | Standard deviation score |
| TPR | Tetratricopeptide repeat |
| URTI | Upper respiratory tract infection |
| VACTERL-H | Vertebral, Anal, Cardiac, Tracheo-esophageal fistula, Esophageal atresia, Renal, upper Limb, and Hydrocephalus |
| WG | Weeks of gestation |
| WT | Wild type |
| y.o. | Year old |
| YRI | Yoruba in Ibadan, Nigeria (1KGP population) |
References
- Auerbach, A.D. Fanconi anemia and its diagnosis. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2009, 668, 4–10. [Google Scholar] [CrossRef] [Green Version]
- Alter, B.P.; Giri, N.; Savage, S.; Rosenberg, P. Cancer in the National Cancer Institute inherited bone marrow failure syndrome cohort after fifteen years of follow-up. Haematologica 2017, 103, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Savage, S.A.; Walsh, M.F. Myelodysplastic Syndrome, Acute Myeloid Leukemia, and Cancer Surveillance in Fanconi Anemia. Hematol. Clin. N. Am. 2018, 32, 657–668. [Google Scholar] [CrossRef]
- Kottemann, M.C.; Smogorzewska, A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 2013, 493, 356–363. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, A.; D’Andrea, A. Fanconi anemia pathway. Curr. Biol. 2017, 27, R986–R988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sroka, I.; Frohnmayer, L.; Van Ravenhorst, S.; Wirkkula, L. Fanconi Anemia Clinical Care Guidelines, 5th ed. Available online: https://www.fanconi.org/images/uploads/other/Fanconi_Anemia_Clinical_Care_Guidelines_5thEdition_web.pdf (accessed on 24 December 2021).
- Niraj, J.; Färkkilä, A.; D’Andrea, A.D. The Fanconi Anemia Pathway in Cancer. Annu. Rev. Cancer Biol. 2019, 3, 457–478. [Google Scholar] [CrossRef]
- Gille, J.J.P.; Floor, K.; Kerkhoven, L.; Ameziane, N.; Joenje, H.; De Winter, J.P. Diagnosis of Fanconi Anemia: Mutation Analysis by Multiplex Ligation-Dependent Probe Amplification and PCR-Based Sanger Sequencing. Anemia 2012, 2012, 603253. [Google Scholar] [CrossRef] [PubMed]
- Callén, E.; Casado, J.A.; Tischkowitz, M.D.; Bueren, J.A.; Creus, A.; Marcos, R.; Dasí, A.; Estella, J.M.; Muñoz, A.; Ortega, J.J.; et al. A common founder mutation in FANCA underlies the world’s highest prevalence of Fanconi anemia in Gypsy families from Spain. Blood 2005, 105, 1946–1949. [Google Scholar] [CrossRef] [Green Version]
- Whitney, M.A.; Saito, H.; Jakobs, P.M.; Gibson, R.; Moses, R.E.; Grompe, M. A common mutation in the FACC gene causes Fanconi anaemia in Ashkenazi Jews. Nat. Genet. 1993, 4, 202–205. [Google Scholar] [CrossRef] [PubMed]
- De Vries, Y.; Lwiwski, N.; Levitus, M.; Kuyt, B.; Israels, S.; Arwert, F.; Zwaan, M.; Greenberg, C.R.; Alter, B.P.; Joenje, H.; et al. A Dutch Fanconi AnemiaFANCCFounder Mutation in Canadian Manitoba Mennonites. Anemia 2012, 2012, 865170. [Google Scholar] [CrossRef] [Green Version]
- Teresa, B.G.; Frias, S.; Molina, T.V.; Villarreal, M.T.; Rodriguez, A.; Carnevale, A.; López-Hernández, G.; Vollbrechtshausen, L.; Olaya-Vargas, A.; Torres, L. FANCC Dutch founder mutation in a Mennonite family from Tamaulipas, México. Mol. Genet. Genom. Med. 2019, 7, e710. [Google Scholar] [CrossRef] [Green Version]
- Auerbach, A.D.; Greenbaum, J.; Pujara, K.; Batish, S.D.; Bitencourt, M.A.; Kokemohr, I.; Schneider, H.; Lobitzc, S.; Pasquini, R.; Giampietro, P.F.; et al. Spectrum of sequence variation in theFANCG gene: An International Fanconi Anemia Registry (IFAR) study. Hum. Mutat. 2003, 21, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Morgan, N.V.; Essop, F.; Demuth, I.; de Ravel, T.; Jansen, S.; Tischkowitz, M.; Lewis, C.M.; Wainwright, L.; Poole, J.; Joenje, H.; et al. A common Fanconi anemia mutation in black populations of sub-Saharan Africa. Blood 2005, 105, 3542–3544. [Google Scholar] [CrossRef]
- Wilson, J.B.; Blom, E.; Cunningham, R.; Xiao, Y.; Kupfer, G.M.; Jones, N.J. Several tetratricopeptide repeat (TPR) motifs of FANCG are required for assembly of the BRCA2/D1-D2-G-X3 complex, FANCD2 monoubiquitylation and phleomycin resistance. Mutat. Res. Mol. Mech. Mutagen. 2010, 689, 12–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blom, E.; van de Vrugt, H.J.; de Vries, Y.; de Winter, J.P.; Arwert, F.; Joenje, H. Multiple TPR motifs characterize the Fanconi anemia FANCG protein. DNA Repair 2003, 3, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Shakeel, S.; Rajendra, E.; Alcón, P.; O’Reilly, F.J.; Chorev, D.; Maslen, S.; Degliesposti, G.; Russo, C.J.; He, S.; Hill, C.H.; et al. Structure of the Fanconi anaemia monoubiquitin ligase complex. Nature 2019, 575, 234–237. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Sarangi, P.; D’Andrea, A.D. The Fanconi anaemia pathway: New players and new functions. Nat. Rev. Mol. Cell Biol. 2016, 17, 337–349. [Google Scholar] [CrossRef]
- García-De-Teresa, B.; Rodríguez, A.; Frias, S. Chromosome Instability in Fanconi Anemia: From Breaks to Phenotypic Consequences. Genes 2020, 11, 1528. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [Green Version]
- Li, L.-H.; Ho, S.-F.; Chen, C.-H.; Wei, C.-Y.; Wong, W.-C.; Li, L.-Y.; Hung, S.-I.; Chung, W.-H.; Pan, W.-H.; Lee, M.T.M.; et al. Long contiguous stretches of homozygosity in the human genome. Hum. Mutat. 2006, 27, 1115–1121. [Google Scholar] [CrossRef]
- Pajusalu, S.; Žilina, O.; Yakoreva, M.; Tammur, P.; Kuuse, K.; Mölter-Väär, T.; Nõukas, M.; Reimand, T.; Õunap, K. The Diagnostic Utility of Single Long Contiguous Stretches of Homozygosity in Patients without Parental Consanguinity. Mol. Syndr. 2015, 6, 135–140. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.-C.; Ross, L.; Mahon, L.W.; Owen, R.; Hemmat, M.; Wang, B.T.; El Naggar, M.; A Kopita, K.; Randolph, L.M.; Chase, J.M.; et al. Regions of homozygosity identified by oligonucleotide SNP arrays: Evaluating the incidence and clinical utility. Eur. J. Hum. Genet. 2014, 23, 663–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaves, T.F.; Oliveira, L.F.; Ocampos, M.; Barbato, I.T.; De Luca, G.R.; Filho, J.H.B.; Pinto, L.L.D.C.; Bernardi, P.; Maris, A.F. Long contiguous stretches of homozygosity detected by chromosomal microarrays (CMA) in patients with neurodevelopmental disorders in the South of Brazil. BMC Med. Genom. 2019, 12, 50. [Google Scholar] [CrossRef]
- Silva-Zolezzi, I.; Hidalgo-Miranda, A.; Estrada-Gil, J.; Fernandez-Lopez, J.C.; Uribe-Figueroa, L.; Contreras, A.; Balam-Ortiz, E.; del Bosque-Plata, L.; Velazquez-Fernandez, D.; Lara, C.; et al. Analysis of genomic diversity in Mexican Mestizo populations to develop genomic medicine in Mexico. Proc. Natl. Acad. Sci. USA 2009, 106, 8611–8616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spear, M.L.; Diaz-Papkovich, A.; Ziv, E.; Yracheta, J.M.; Gravel, S.; Torgerson, D.G.; Hernandez, R.D. Recent shifts in the genomic ancestry of Mexican Americans may alter the genetic architecture of biomedical traits. eLife 2020, 9, e56029. [Google Scholar] [CrossRef]
- Yagasaki, H.; Hamanoue, S.; Oda, T.; Nakahata, T.; Asano, S.; Yamashita, T. Identification and characterization of novel mutations of the major Fanconi anemia gene FANCA in the Japanese population. Hum. Mutat. 2004, 24, 481–490. [Google Scholar] [CrossRef]
- Demuth, I.; Wlodarski, M.; Tipping, A.J.; Morgan, N.; De Winter, J.P.; Thiel, M.; Gräsl, S.; Schindler, D.; D’Andrea, A.D.; Altay, C.; et al. Spectrum of mutations in the Fanconi anaemia group G gene, FANCG/XRCC9. Eur. J. Hum. Genet. 2000, 8, 861–868. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Kim, M.; Jang, W.; Chae, H.; Kim, Y.; Chung, N.-G.; Lee, J.-W.; Cho, B.; Jeong, D.-C.; Park, I.Y.; et al. Founder Haplotype Analysis of Fanconi Anemia in the Korean Population Finds Common Ancestral Haplotypes for a FANCG Variant: Ancestral Haplotypes for AFANCGVariant. Ann. Hum. Genet. 2015, 79, 153–161. [Google Scholar] [CrossRef]
- Alter, B.P.; Giri, N. Thinking of VACTERL-H? Rule out Fanconi Anemia According to PHENOS: VACTERL-H, PHENOS, and Fanconi Anemia. Am. J. Med. Genet. Part A 2016, 170, 1520–1524. [Google Scholar] [CrossRef]
- Aguilar-Ordoñez, I.; Pérez-Villatoro, F.; García-Ortiz, H.; Barajas-Olmos, F.; Ballesteros-Villascán, J.; González-Buenfil, R.; Fresno, C.; Garcíarrubio, A.; Fernández-López, J.C.; Tovar, H.; et al. Whole genome variation in 27 Mexican indigenous populations, demographic and biomedical insights. PLoS ONE 2021, 16, e0249773. [Google Scholar] [CrossRef] [PubMed]
- Torres-Cisneros, G. Mixes. In Pueblos Indigenas del Mexico Contemporaneo; Comisión Nacional para el Desarrollo de los Pueblos Indígenas, PNUD: Mexico City, Mexico, 2004; pp. 1–43. [Google Scholar]
- Quinto-Cortés, C.D.; Arriola, L.A.; García-Hughes, G.; García-López, R.; Molina, D.P.; Flores, M.; Palacios, R.; Piñero, D. Genetic Characterization of Indigenous Peoples from Oaxaca, Mexico, and Its Relation to Linguistic and Geographic Isolation. Hum. Biol. 2010, 82, 409–432. [Google Scholar] [CrossRef]
- Cuentame INEGI. Available online: http://www.cuentame.inegi.org.mx/monografias/informacion/oax/territorio/div_municipal.aspx?tema=me&e=20 (accessed on 8 December 2021).
- Orphanet: Anemia de Fanconi. Available online: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=ES&Expert=84 (accessed on 19 December 2021).
- Rogers, K.J.; Fu, W.; Akey, J.M.; Monnat, J.R., Jr. Global and disease-associated genetic variation in the human Fanconi anemia gene family. Hum. Mol. Genet. 2014, 23, 6815–6825. [Google Scholar] [CrossRef] [Green Version]
- Shimamura, A.; Alter, B.P. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev. 2010, 24, 101–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiesco-Roa, M.O.; Giri, N.; McReynolds, L.J.; Best, A.F.; Alter, B.P. Genotype-phenotype associations in Fanconi anemia: A literature review. Blood Rev. 2019, 37, 100589. [Google Scholar] [CrossRef] [PubMed]
- Graf, C.M.; Nichele, S.; Siviero, R.B.; Loth, G.; Trennepohl, J.P.; Zinher, M.T.; Grandinetti, A.; Pilonetto, D.V.; Pasquini, R.; Moreira, A.T.R.; et al. Ocular Manifestations in Patients with Fanconi Anemia: A Single Center Experience Including 106 Patients. J. Pediatr. 2021. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Mehta, P.A.; Jiang, C.S.; Rosti, R.O.; Usleaman, G.; da Rosa, J.M.C.; Lach, F.P.; Goodridge, E.; Auerbach, A.D.; Davies, S.M.; et al. Comparison of the clinical phenotype and haematological course of siblings with Fanconi anaemia. Br. J. Haematol. 2020, 193, 971–975. [Google Scholar] [CrossRef] [PubMed]
- Faivre, L.; Guardiola, P.; Lewis, C.; Dokal, I.; Ebell, W.; Zatterale, A.; Altay, C.; Poole, J.; Stones, D.; Kwee, M.L.; et al. Association of complementation group and mutation type with clinical outcome in fanconi anemia. European Fanconi Anemia Research Group. Blood 2000, 96, 4064–4070. [Google Scholar]
- Feben, C.; Kromberg, J.; Wainwright, R.; Stones, D.; Sutton, C.; Poole, J.; Haw, T.; Krause, A. Phenotypic consequences in black South African Fanconi anemia patients homozygous for a founder mutation. Genet. Med. 2014, 16, 400–406. [Google Scholar] [CrossRef] [Green Version]
- Dillon, B.; Feben, C.; Segal, D.; Du Plessis, J.; Reynders, D.; Wainwright, R.; Poole, J.; Krause, A. Endocrine profiling in patients with Fanconi anemia, homozygous for a FANCG founder mutation. Mol. Genet. Genom. Med. 2020, 8, e1351. [Google Scholar] [CrossRef]
- Köhler, S.; Gargano, M.; Matentzoglu, N.; Carmody, L.C.; Lewis-Smith, D.; Vasilevsky, N.A.; Danis, D.; Balagura, G.; Baynam, G.; Brower, A.M.; et al. The Human Phenotype Ontology in 2021. Nucleic Acids Res. 2021, 49, D1207–D1217. [Google Scholar] [CrossRef]
- Esmer, C.; Sánchez, S.; Ramos, S.; Molina, B.; Frias, S.; Carnevale, A. DEB test for Fanconi anemia detection in patients with atypical phenotypes. Am. J. Med. Genet. Part A 2005, 124A, 35–39. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; Del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Van Der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- den Dunnen, J.T.; Dalgleish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; McGowan-Jordan, J.; Roux, A.-F.; Smith, T.; Antonarakis, S.E.; Taschner, P.E.; et al. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum. Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef] [Green Version]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Aguilera, M.A.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Sambrook, J. The Basic Polymerase Chain Reaction (PCR). Cold Spring Harb. Protoc. 2018, 5, pdb-prot095117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearney, H.M.; Kearney, J.B.; Conlin, L.K. Diagnostic Implications of Excessive Homozygosity Detected by SNP-Based Microarrays: Consanguinity, Uniparental Disomy, and Recessive Single-Gene Mutations. Clin. Lab. Med. 2011, 31, 595–613. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference Sequence | DNA Change (Genomic, hg19) | ACMG Criteria Variant Classification | HGVS Nomenclature |
|---|---|---|---|
| NG_007312.1 | g.35077398_35077399del | PVS1, PM2, PP3 | FANCG(NM_004629.2): c.511-3_511-2delCA |
| Patient ID | Total Autosomal LCSH > 3 Mb (kb) | Percentage of Native Component | Autosomal Homozygosity (%) | F | Probable Parental Relationship |
|---|---|---|---|---|---|
| FANC32 | 140214361 | 79.17% | 4.9% | 0.0487 | Fourth-degree |
| FANC143 | 203913825 | 85.64% | 7.1% | 0.0708 | Third-degree |
| FANC155 | 249262167 | 88.53% | 8.7% | 0.0865 | Third-degree |
| Pathogenic Variant | Location | Effect | Geographic/ Ethnic Background | Reference |
|---|---|---|---|---|
| c.307+1G > C | Intron 3 | Aberrant splicing | Japanese-Korean | [27] |
| c.313G > T (p.Glu105Ter) | Exon 4 | Truncated protein, null variant | German | [28] |
| c.637_643delTACCGCC (p.Tyr213LysfsTer6) | Exon 5 | Truncated protein, null variant | South African | [14] |
| c.1066C > T (p.Gln356Ter) | Exon 8 | Truncated protein, null variant | Japanese | [29] |
| c.1077-2A > C | Intron 8 | Aberrant splicing | Portuguese-Brazilian | [13] |
| c.1480+1G > C | Intron 11 | Aberrant splicing? | French-Acadian | [13] |
| c.1589_1591delATA (p.Asp530_Thr531delinsAla) | Exon 12 | Reduce protein activity, Hypomorphic variant | Korean | [29] |
| c.1649delC (p.Thr550IlefsTer9) | Exon 13 | Truncated protein, null variant | Turkish | [28] |
| c.511-3_511-2delCA | Intron 4 | Aberrant splicing and truncated protein | Mixe (Mexican) | Present study |
| Patient ID | Sex | Family History of FA | Age at Diagnosis | Hematologic Phenotype at Initial Evaluation | VACTERL-H Features | PHENOS Features | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Hb (g/dL) | MCV (fL) | ANC (cells/µL) | PLT (cells/µL) | BMF Status | ||||||
| FANC32 | M | Sister | 6.45 | 11.4 | 100.0 | 700 | 26,000 | Moderate BMF | L | PHS |
| FANC143 † | F | No | 3.6 | 8.9 | 94.1 | 900 | 15,000 | Moderate BMF | VL | PHNOS |
| FANC155 † | F | No | 9.3 | 11.1 | 99.5 | 220 | 19,000 | Severe BMF | L | POS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reyes, P.; García-de Teresa, B.; Juárez, U.; Pérez-Villatoro, F.; Fiesco-Roa, M.O.; Rodríguez, A.; Molina, B.; Villarreal-Molina, M.T.; Meléndez-Zajgla, J.; Carnevale, A.; et al. Fanconi Anemia Patients from an Indigenous Community in Mexico Carry a New Founder Pathogenic Variant in FANCG. Int. J. Mol. Sci. 2022, 23, 2334. https://doi.org/10.3390/ijms23042334
Reyes P, García-de Teresa B, Juárez U, Pérez-Villatoro F, Fiesco-Roa MO, Rodríguez A, Molina B, Villarreal-Molina MT, Meléndez-Zajgla J, Carnevale A, et al. Fanconi Anemia Patients from an Indigenous Community in Mexico Carry a New Founder Pathogenic Variant in FANCG. International Journal of Molecular Sciences. 2022; 23(4):2334. https://doi.org/10.3390/ijms23042334
Chicago/Turabian StyleReyes, Pedro, Benilde García-de Teresa, Ulises Juárez, Fernando Pérez-Villatoro, Moisés O. Fiesco-Roa, Alfredo Rodríguez, Bertha Molina, María Teresa Villarreal-Molina, Jorge Meléndez-Zajgla, Alessandra Carnevale, and et al. 2022. "Fanconi Anemia Patients from an Indigenous Community in Mexico Carry a New Founder Pathogenic Variant in FANCG" International Journal of Molecular Sciences 23, no. 4: 2334. https://doi.org/10.3390/ijms23042334
APA StyleReyes, P., García-de Teresa, B., Juárez, U., Pérez-Villatoro, F., Fiesco-Roa, M. O., Rodríguez, A., Molina, B., Villarreal-Molina, M. T., Meléndez-Zajgla, J., Carnevale, A., Torres, L., & Frias, S. (2022). Fanconi Anemia Patients from an Indigenous Community in Mexico Carry a New Founder Pathogenic Variant in FANCG. International Journal of Molecular Sciences, 23(4), 2334. https://doi.org/10.3390/ijms23042334