
R-Loop-Associated Genomic Instability and Implication of WRN and WRNIP1

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Replication Stress
3. Transcription as a Source of Replication Stress
4. R-Loops as Inducers of Transcription-Associated Genome Instability
5. Mechanisms of Resolution and Removal of R-Loops
6. A Role of WRN in Limiting R-Loop-Associated Genomic Instability
7. WRNIP1-Mediated Response Is Involved in Counteracting R-Loop-Associated Genomic Instability
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Branzei, D.; Foiani, M. Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell Biol. 2010, 11, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A.; García-Muse, T. R loops: From transcription byproducts to threats to genome stability. Mol. Cell 2012, 46, 115–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaillard, H.; Aguilera, A. Transcription as a Threat to Genome Integrity. Annu. Rev. Biochem. 2016, 85, 291–317. [Google Scholar] [CrossRef] [PubMed]
- Allison, D.F.; Wang, G.G. R-loops: Formation, function, and relevance to cell stress. Cell Stress 2019, 3, 38–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez-González, B.; Aguilera, A. Transcription-mediated replication hindrance: A major driver of genome instability. Genes Dev. 2019, 33, 1008–1026. [Google Scholar] [CrossRef] [Green Version]
- Crossley, M.P.; Bocek, M.; Cimprich, K.A. R-Loops as Cellular Regulators and Genomic Threats. Mol. Cell 2019, 73, 398–411. [Google Scholar] [CrossRef] [Green Version]
- García-Muse, T.; Aguilera, A. R Loops: From Physiological to Pathological Roles. Cell 2019, 179, 604–618. [Google Scholar] [CrossRef]
- Brambati, A.; Zardoni, L.; Nardini, E.; Pellicioli, A.; Liberi, G. The dark side of RNA:DNA hybrids. Mutat. Res. Rev. Mutat. Res. 2020, 784, 108300. [Google Scholar] [CrossRef]
- Lecona, E.; Fernández-Capetillo, O. Replication stress and cancer: It takes two to tango. Exp. Cell Res. 2014, 329, 26–34. [Google Scholar] [CrossRef] [Green Version]
- Técher, H.; Koundrioukoff, S.; Nicolas, A.; Debatisse, M. The impact of replication stress on replication dynamics and DNA damage in vertebrate cells. Nat. Rev. Genet. 2017, 18, 535–550. [Google Scholar] [CrossRef]
- Magdalou, I.; Lopez, B.S.; Pasero, P.; Lambert, S.a.E. The causes of replication stress and their consequences on genome stability and cell fate. Semin. Cell Dev. Biol. 2014, 30, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Ubhi, T.; Brown, G.W. Exploiting DNA replication stress for cancer treatment. Cancer Res. 2019, 79, 1730–1739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.K.; Hickson, I.D. RecQ helicases: Multifunctional genome caretakers. Nat. Rev. Cancer 2009, 9, 644–654. [Google Scholar] [CrossRef]
- Wei, X.; Samarabandu, J.; Devdhar, R.S.; Siegel, A.J.; Acharya, R.; Berezney, R. Segregation of transcription and replication sites into higher order domains. Science 1998, 281, 1502–1505. [Google Scholar] [CrossRef]
- Akamatsu, Y.; Kobayashi, T. The Human RNA Polymerase I Transcription Terminator Complex Acts as a Replication Fork Barrier That Coordinates the Progress of Replication with rRNA Transcription Activity. Mol. Cell. Biol. 2015, 35, 1871–1881. [Google Scholar] [CrossRef] [Green Version]
- Conti, C.; Saccà, B.; Herrick, J.; Lalou, C.; Pommier, Y.; Bensimon, A. Replication fork velocities at adjacent replication origins are coordinately modified during DNA replication in human cells. Mol. Biol. Cell 2007, 18, 3059–3067. [Google Scholar] [CrossRef] [Green Version]
- Singh, J.; Padgett, R.A. Rates of in situ transcription and splicing in large human genes. Nat. Struct. Mol. Biol. 2009, 16, 1128–1133. [Google Scholar] [CrossRef] [Green Version]
- Helmrich, A.; Ballarino, M.; Tora, L. Collisions between Replication and Transcription Complexes Cause Common Fragile Site Instability at the Longest Human Genes. Mol. Cell 2011, 44, 966–977. [Google Scholar] [CrossRef] [Green Version]
- Gan, W.; Guan, Z.; Liu, J.; Gui, T.; Shen, K.; Manley, J.L.; Li, X. R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev. 2011, 25, 2041–2056. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, A.; Grosso, A.R.; Elkaoutari, A.; Coleno, E.; Presle, A.; Sridhara, S.C.; Janbon, G.; Géli, V.; de Almeida, S.F.; Palancade, B. Introns Protect Eukaryotic Genomes from Transcription-Associated Genetic Instability. Mol. Cell 2017, 67, 608–621.e6. [Google Scholar] [CrossRef] [PubMed]
- Belotserkovskii, B.P.; Tornaletti, S.; D’Souza, A.D.; Hanawalt, P.C. R-loop generation during transcription: Formation, processing and cellular outcomes. DNA Repair 2018, 71, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Zardoni, L.; Nardini, E.; Brambati, A.; Lucca, C.; Choudhary, R.; Loperfido, F.; Sabbioneda, S.; Liberi, G. Elongating RNA polymerase II and RNA:DNA hybrids hinder fork progression and gene expression at sites of head-on replication-transcription collisions. Nucleic Acids Res. 2021, 49, 12769–12784. [Google Scholar] [CrossRef] [PubMed]
- Skourti-Stathaki, K.; Kamieniarz-Gdula, K.; Proudfoot, N.J. R-loops induce repressive chromatin marks over mammalian gene terminators. Nature 2014, 516, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Santos-Pereira, J.M.; Aguilera, A. R loops: New modulators of genome dynamics and function. Nat. Rev. Genet. 2015, 16, 583–597. [Google Scholar] [CrossRef]
- Helmrich, A.; Ballarino, M.; Nudler, E.; Tora, L. Transcription-replication encounters, consequences and genomic instability. Nat. Struct. Mol. Biol. 2013, 20, 412–418. [Google Scholar] [CrossRef]
- Sollier, J.; Stork, C.T.; García-Rubio, M.L.; Paulsen, R.D.; Aguilera, A.; Cimprich, K.A. Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol. Cell 2014, 56, 777–785. [Google Scholar] [CrossRef] [Green Version]
- Stein, H.; Hausen, P. Enzyme from calf thymus degrading the RNA moiety of DNA-RNA hybrids: Effect on DNA-dependent RNA polymerase. Science 1969, 166, 393–395. [Google Scholar] [CrossRef]
- Chang, E.; Stirling, P. Replication Fork Protection Factors Controlling R-Loop Bypass and Suppression. Genes 2017, 8, 33. [Google Scholar] [CrossRef] [Green Version]
- Cristini, A.; Groh, M.; Kristiansen, M.S.; Gromak, N. RNA/DNA Hybrid Interactome Identifies DXH9 as a Molecular Player in Transcriptional Termination and R-Loop-Associated DNA Damage. Cell Rep. 2018, 23, 1891–1905. [Google Scholar] [CrossRef] [Green Version]
- Tedeschi, F.A.; Cloutier, S.C.; Tran, E.J.; Jankowsky, E. The DEAD-box protein Dbp2p is linked to noncoding RNAs, the helicase Sen1p, and R-loops. RNA 2018, 24, 1693–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, P.L.T.; Pohl, T.J.; Chen, C.-F.; Chan, A.; Pott, S.; Zakian, V.A. PIF1 family DNA helicases suppress R-loop mediated genome instability at tRNA genes. Nat. Commun. 2017, 8, 15025. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.X.; Grunseich, C.; Fox, J.; Burdick, J.; Zhu, Z.; Ravazian, N.; Hafner, M.; Cheung, V.G. Human proteins that interact with RNA/DNA hybrids. Genome Res. 2018, 28, 1405–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodroj, D.; Recolin, B.; Serhal, K.; Martinez, S.; Tsanov, N.; Abou Merhi, R.; Maiorano, D. An ATR -dependent function for the Ddx19 RNA helicase in nuclear R-loop metabolism. EMBO J. 2017, 36, 1182–1198. [Google Scholar] [CrossRef] [PubMed]
- Lafuente-Barquero, J.; Luke-Glaser, S.; Graf, M.; Silva, S.; Gómez-González, B.; Lockhart, A.; Lisby, M.; Aguilera, A.; Luke, B. The Smc5/6 complex regulates the yeast Mph1 helicase at RNA-DNA hybrid-mediated DNA damage. PLoS Genet. 2017, 13, e1007136. [Google Scholar] [CrossRef]
- Li, L.; Matsui, M.; Corey, D.R. Activating frataxin expression by repeat-targeted nucleic acids. Nat. Commun. 2016, 7, 10606. [Google Scholar] [CrossRef]
- Alzu, A.; Bermejo, R.; Begnis, M.; Lucca, C.; Piccini, D.; Carotenuto, W.; Saponaro, M.; Brambati, A.; Cocito, A.; Foiani, M.; et al. Senataxin associates with replication forks to protect fork integrity across RNA-polymerase-II-transcribed genes. Cell 2012, 151, 835–846. [Google Scholar] [CrossRef] [Green Version]
- Mischo, H.E.; Gómez-González, B.; Grzechnik, P.; Rondón, A.G.; Wei, W.; Steinmetz, L.; Aguilera, A.; Proudfoot, N.J. Yeast Sen1 helicase protects the genome from transcription-associated instability. Mol. Cell 2011, 41, 21–32. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro de Almeida, C.; Dhir, S.; Dhir, A.; Moghaddam, A.E.; Sattentau, Q.; Meinhart, A.; Proudfoot, N.J. RNA Helicase DDX1 Converts RNA G-Quadruplex Structures into R-Loops to Promote IgH Class Switch Recombination. Mol. Cell 2018, 70, 650–662.e8. [Google Scholar] [CrossRef] [Green Version]
- Sridhara, S.C.; Carvalho, S.; Grosso, A.R.; Gallego-Paez, L.M.; Carmo-Fonseca, M.; de Almeida, S.F. Transcription Dynamics Prevent RNA-Mediated Genomic Instability through SRPK2-Dependent DDX23 Phosphorylation. Cell Rep. 2017, 18, 334–343. [Google Scholar] [CrossRef] [Green Version]
- Schwab, R.A.; Nieminuszczy, J.; Shah, F.; Langton, J.; Lopez Martinez, D.; Liang, C.; Cohn, M.A.; Gibbons, R.J.; Deans, A.J.; Niedzwiedz, W. The Fanconi Anemia Pathway Maintains Genome Stability by Coordinating Replication and Transcription. Mol. Cell 2015, 60, 351–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Rubio, M.L.; Pérez-Calero, C.; Barroso, S.I.; Tumini, E.; Herrera-Moyano, E.; Rosado, I.V.; Aguilera, A. The Fanconi Anemia Pathway Protects Genome Integrity from R-loops. PLoS Genet. 2015, 11, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatia, V.; Barroso, S.I.; García-Rubio, M.L.; Tumini, E.; Herrera-Moyano, E.; Aguilera, A. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 2014, 511, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Hatchi, E.; Skourti-Stathaki, K.; Ventz, S.; Pinello, L.; Yen, A.; Kamieniarz-Gdula, K.; Dimitrov, S.; Pathania, S.; McKinney, K.M.; Eaton, M.L.; et al. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol. Cell 2015, 57, 636–647. [Google Scholar] [CrossRef] [Green Version]
- Liang, Z.; Liang, F.; Teng, Y.; Chen, X.; Liu, J.; Longerich, S.; Rao, T.; Green, A.M.; Collins, N.B.; Xiong, Y.; et al. Binding of FANCI-FANCD2 Complex to RNA and R-Loops Stimulates Robust FANCD2 Monoubiquitination. Cell Rep. 2019, 26, 564–572.e5. [Google Scholar] [CrossRef] [Green Version]
- Madireddy, A.; Kosiyatrakul, S.T.; Boisvert, R.A.; Herrera-Moyano, E.; Garcia-Rubio, M.L.; Gerhardt, J.; Vuono, E.A.; Owen, N.; Yan, Z.; Olson, S.; et al. FANCD2 Facilitates Replication through Common Fragile Sites. Mol. Cell 2016, 64, 388–404. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, Y.; Abe, M.; Itaya, A.; Tomida, J.; Ishiai, M.; Takaori-Kondo, A.; Taoka, M.; Isobe, T.; Takata, M. FANCD2 protects genome stability by recruiting RNA processing enzymes to resolve R-loops during mild replication stress. FEBS J. 2019, 286, 139–150. [Google Scholar] [CrossRef] [Green Version]
- Tresini, M.; Warmerdam, D.O.; Kolovos, P.; Snijder, L.; Vrouwe, M.G.; Demmers, J.A.A.; van IJcken, W.F.J.; Grosveld, F.G.; Medema, R.H.; Hoeijmakers, J.H.J.; et al. The core spliceosome as target and effector of non-canonical ATM signalling. Nature 2015, 523, 53–58. [Google Scholar] [CrossRef] [Green Version]
- Barroso, S.; Herrera-Moyano, E.; Muñoz, S.; García-Rubio, M.; Gómez-González, B.; Aguilera, A. The DNA damage response acts as a safeguard against harmful DNA–RNA hybrids of different origins. EMBO Rep. 2019, 20, e47250. [Google Scholar] [CrossRef]
- Marabitti, V.; Lillo, G.; Malacaria, E.; Palermo, V.; Sanchez, M.; Pichierri, P.; Franchitto, A. ATM pathway activation limits R-loop-associated genomic instability in Werner syndrome cells. Nucleic Acids Res. 2019, 47, 3485–3502. [Google Scholar] [CrossRef]
- Marabitti, V.; Lillo, G.; Malacaria, E.; Palermo, V.; Pichierri, P.; Franchitto, A. Checkpoint Defects Elicit a WRNIP1-Mediated Response to Counteract R-Loop-Associated Genomic Instability. Cancers 2020, 12, 389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, L.S.; Mason, P.A. The Role of WRN Helicase/Exonuclease in DNA Replication. In The Mechanisms of DNA Replication; BoD—Books on Demand: Norderstedt, Germany, 2013. [Google Scholar]
- Shen, J.C.; Gray, M.D.; Oshima, J.; Kamath-Loeb, A.S.; Fry, M.; Loeb, L.A. Werner syndrome protein. I. DNA helicase and dna exonuclease reside on the same polypeptide. J. Biol. Chem. 1998, 273, 34139–34144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Li, B.; Gray, M.D.; Oshima, J.; Mian, I.S.; Campisi, J. The premature ageing syndrome protein, WRN, is a 3′→5′ exonuclease. Nat. Genet. 1998, 20, 114–116. [Google Scholar] [CrossRef] [PubMed]
- David, A.; Vincent, M.; Arrigoni, P.P.; Barbarot, S.; Pistorius, M.A.; Isidor, B.; Frampas, E. Werner syndrome: Clinical features, pathogenesis and potential therapeutic interventions. Ageing Res. Rev. 2017, 33, 105–114. [Google Scholar]
- Baynton, K.; Otterlei, M.; Bjørås, M.; Von Kobbe, C.; Bohr, V.a.; Seeberg, E. WRN interacts physically and functionally with the recombination mediator protein RAD52. J. Biol. Chem. 2003, 278, 36476–36486. [Google Scholar] [CrossRef] [Green Version]
- Otterlei, M.; Bruheim, P.; Ahn, B.; Bussen, W.; Karmakar, P.; Baynton, K.; Bohr, V.A. Werner syndrome protein participates in a complex with RAD51, RAD54, RAD54B and ATR in response to ICL-induced replication arrest. J. Cell Sci. 2006, 119, 5137–5146. [Google Scholar] [CrossRef] [Green Version]
- Pichierri, P.; Rosselli, F.; Franchitto, A. Werner’s syndrome protein is phosphorylated in an ATR/ATM-dependent manner following replication arrest and DNA damage induced during the S phase of the cell cycle. Oncogene 2003, 22, 1491–1500. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, S.; Nishikawa, K.; Heo, S.J.; Goto, M.; Furuichi, Y.; Shimamoto, A. Werner helicase relocates into nuclear foci in response to DNA damaging agents and co-localizes with RPA and Rad51. Genes Cells 2001, 6, 421–430. [Google Scholar] [CrossRef]
- Poot, M.; Gollahon, K.a.; Rabinovitch, P.S. Werner syndrome lymphoblastoid cells are sensitive to camptothecin-induced apoptosis in S-phase. Hum. Genet. 1999, 104, 10–14. [Google Scholar] [CrossRef]
- Pichierri, P.; Franchitto, A.; Mosesso, P.; Palitti, F. Werner’s syndrome cell lines are hypersensitive to camptothecin-induced chromosomal damage. Mutat. Res.—Fundam. Mol. Mech. Mutagen. 2000, 456, 45–57. [Google Scholar] [CrossRef]
- Pichierri, P.; Franchitto, A.; Mosesso, P.; Palitti, F. Werner’s syndrome protein is required for correct recovery after replication arrest and DNA damage induced in S-phase of cell cycle. Mol. Biol. Cell 2001, 12, 2412–2421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flynn, R.L.; Zou, L. ATR: A master conductor of cellular responses to DNA replication stress. Trends Biochem. Sci. 2011, 36, 133–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirzio, L.M.; Pichierri, P.; Bignami, M.; Franchitto, A. Werner syndrome helicase activity is essential in maintaining fragile site stability. J. Cell Biol. 2008, 180, 305–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ammazzalorso, F.; Pirzio, L.M.; Bignami, M.; Franchitto, A.; Pichierri, P. ATR and ATM differently regulate WRN to prevent DSBs at stalled replication forks and promote replication fork recovery. EMBO J. 2010, 29, 3156–3169. [Google Scholar] [CrossRef]
- Patro, B.S.; Frohlich, R.; Bohr, V.A.; Stevnsner, T. WRN helicase regulates the ATR-CHK1-induced S-phase checkpoint pathway in response to topoisomerase-I-DNA covalent complexes. J. Cell Sci. 2011, 124, 3967–3979. [Google Scholar] [CrossRef] [Green Version]
- Basile, G.; Leuzzi, G.; Pichierri, P.; Franchitto, A. Checkpoint-dependent and independent roles of the Werner syndrome protein in preserving genome integrity in response to mild replication stress. Nucleic Acids Res. 2014, 42, 12628–12639. [Google Scholar] [CrossRef] [Green Version]
- Casper, A.M.; Nghiem, P.; Arlt, M.F.; Glover, T.W. ATR regulates fragile site stability. Cell 2002, 111, 779–789. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Lopez, A.M.; Jackson, D.a; Iborra, F.J.; Cox, L.S. Asymmetry of DNA replication fork progression in Werner’s syndrome. Aging Cell 2002, 1, 30–39. [Google Scholar] [CrossRef]
- Kamath-Loeb, A.S.; Loeb, L.A.; Johansson, E.; Burgers, P.M.; Fry, M. Interactions between the Werner syndrome helicase and DNA polymerase delta specifically facilitate copying of tetraplex and hairpin structures of the d(CGG)n trinucleotide repeat sequence. J. Biol. Chem. 2001, 276, 16439–16446. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, V.; Herrera-Moyano, E.; Aguilera, A.; Gómez-González, B. The Role of Replication-Associated Repair Factors on R-Loops. Genes 2017, 8, 171. [Google Scholar] [CrossRef] [Green Version]
- Stirling, P.C.; Hieter, P. Canonical DNA Repair Pathways Influence R-Loop-Driven Genome Instability. J. Mol. Biol. 2016, 429, 3132–3138. [Google Scholar] [CrossRef] [PubMed]
- García-Muse, T.; Aguilera, A. Transcription–replication conflicts: How they occur and how they are resolved. Nat. Rev. Mol. Cell Biol. 2016, 17, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, M.; Sommers, J.a; Shoemaker, R.H.; Brosh, R.M. Inhibition of helicase activity by a small molecule impairs Werner syndrome helicase (WRN) function in the cellular response to DNA damage or replication stress. Proc. Natl. Acad. Sci. USA 2011, 108, 1525–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, P.; Grosse, F. Human DHX9 helicase preferentially unwinds RNA-containing displacement loops (R-loops) and G-quadruplexes. DNA Repair 2011, 10, 654–665. [Google Scholar] [CrossRef]
- Li, M.; Pokharel, S.; Wang, J.-T.; Xu, X.; Liu, Y. RECQ5-dependent SUMOylation of DNA topoisomerase I prevents transcription-associated genome instability. Nat. Commun. 2015, 6, 6720. [Google Scholar] [CrossRef] [Green Version]
- Urban, V.; Dobrovolna, J.; Hühn, D.; Fryzelkova, J.; Bartek, J.; Janscak, P. RECQ5 helicase promotes resolution of conflicts between replication and transcription in human cells. J. Cell Biol. 2016, 214, 401–415. [Google Scholar] [CrossRef] [Green Version]
- Sordet, O.; Nakamura, A.J.; Redon, C.E.; Pommier, Y. DNA double-strand breaks and ATM activation by transcription-blocking DNA lesions. Cell Cycle 2010, 9, 274–278. [Google Scholar] [CrossRef] [Green Version]
- Sordet, O.; Redon, C.E.; Guirouilh-Barbat, J.; Smith, S.; Solier, S.; Douarre, C.; Conti, C.; Nakamura, A.J.; Das, B.B.; Nicolas, E.; et al. Ataxia telangiectasia mutated activation by transcription- and topoisomerase I-induced DNA double-strand breaks. EMBO Rep. 2009, 10, 887–893. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Gonzalez, B.; Felipe-Abrio, I.; Aguilera, A. The S-Phase Checkpoint Is Required To Respond to R-Loops Accumulated in THO Mutants. Mol. Cell. Biol. 2009, 29, 5203–5213. [Google Scholar] [CrossRef] [Green Version]
- Singh, T.R.; Ali, A.M.; Paramasivam, M.; Pradhan, A.; Wahengbam, K.; Seidman, M.M.; Meetei, A.R. ATR-Dependent Phosphorylation of FANCM at Serine 1045 Is Essential for FANCM Functions. Cancer Res. 2013, 73, 4300–4310. [Google Scholar] [CrossRef] [Green Version]
- Davies, S.L.; North, P.S.; Dart, A.; Lakin, N.D.; Hickson, I.D. Phosphorylation of the Bloom’s Syndrome Helicase and Its Role in Recovery from S-Phase Arrest. Mol. Cell. Biol. 2004, 24, 1279–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, E.Y.-C.; Novoa, C.A.; Aristizabal, M.J.; Coulombe, Y.; Segovia, R.; Chaturvedi, R.; Shen, Y.; Keong, C.; Tam, A.S.; Jones, S.J.M.; et al. RECQ-like helicases Sgs1 and BLM regulate R-loop–associated genome instability. J. Cell Biol. 2017, 216, 3991–4005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Paull, T.T. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene 2007, 26, 7741–7748. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, L.; Wiedner, M.; Velimezi, G.; Prochazkova, J.; Owusu, M.; Bauer, S.; Loizou, J.I. ATMIN is required for the ATM-mediated signaling and recruitment of 53BP1 to DNA damage sites upon replication stress. DNA Repair 2014, 24, 122–130. [Google Scholar] [CrossRef] [Green Version]
- Kanu, N.; Zhang, T.; Burrell, R.A.; Chakraborty, A.; Cronshaw, J.; Costa, C.D.; Grönroos, E.; Pemberton, H.N.; Anderton, E.; Gonzalez, L.; et al. RAD18, WRNIP1 and ATMIN promote ATM signalling in response to replication stress. Oncogene 2015, 35, 4009–4019. [Google Scholar] [CrossRef] [Green Version]
- Harding, S.M.; Boiarsky, J.A.; Greenberg, R.A. ATM Dependent Silencing Links Nucleolar Chromatin Reorganization to DNA Damage Recognition. Cell Rep. 2015, 13, 251–259. [Google Scholar] [CrossRef] [Green Version]
- Abraham, R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001, 15, 2177–2196. [Google Scholar] [CrossRef] [Green Version]
- Maréchal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, 1–17. [Google Scholar] [CrossRef]
- Durkin, S.G.; Arlt, M.F.; Howlett, N.G.; Glover, T.W. Depletion of CHK1, but not CHK2, induces chromosomal instability and breaks at common fragile sites. Oncogene 2006, 25, 4381–4388. [Google Scholar] [CrossRef] [Green Version]
- Kawabe, Y.I.; Branzei, D.; Hayashi, T.; Suzuki, H.; Masuko, T.; Onoda, F.; Heo, S.J.; Ikeda, H.; Shimamoto, A.; Furuichi, Y.; et al. A Novel Protein Interacts with the Werner’s Syndrome Gene Product Physically and Functionally. J. Biol. Chem. 2001, 276, 20364–20369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimura, A.; Seki, M.; Enomoto, T. The role of WRNIP1 in genome maintenance. Cell Cycle 2017, 16, 515–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branzei, D.; Seki, M.; Onoda, F.; Yagi, H.; Kawabe, Y.; Enomoto, T. Characterization of the slow-growth phenotype of S. cerevisiae whip/mgs1 sgs1 double deletion mutants. DNA Repair 2002, 1, 671–682. [Google Scholar] [CrossRef]
- Hishida, T.; Ohno, T. Saccharomyces cerevisiae MGS1 is essential in strains deficient in the RAD6-dependent DNA damage tolerance pathway. EMBO J. 2002, 21, 2019–2029. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Seki, M.; Inoue, E.; Yoshimura, A. Vertebrate WRNIP1 and BLM are required for efficient maintenance of genome stability. Genes Genet. Syst. 2008, 1, 95–100. [Google Scholar] [CrossRef] [Green Version]
- Vijeh Motlagh, N.D.; Seki, M.; Branzei, D.; Enomoto, T. Mgs1 and Rad18/Rad5/Mms2 are required for survival of Saccharomyces cerevisiae mutants with novel temperature/cold sensitive alleles of the DNA polymerase delta subunit, Pol31. DNA Repair 2006, 5, 1459–1474. [Google Scholar] [CrossRef]
- Tsurimoto, T.; Shinozaki, A.; Yano, M.; Seki, M.; Enomoto, T. Human Werner helicase interacting protein 1 (WRNIP1) functions as novel modulator for DNA polymerase δ. Genes Cells 2005, 10, 13–22. [Google Scholar] [CrossRef]
- Yoshimura, A.; Seki, M.; Kanamori, M.; Tateishi, S.; Tsurimoto, T.; Tada, S.; Enomoto, T. Physical and functional interaction between WRNIP1 and RAD18. Genes Genet. Syst. 2009, 84, 171–178. [Google Scholar] [CrossRef] [Green Version]
- Leuzzi, G.; Marabitti, V.; Pichierri, P.; Franchitto, A. WRNIP1 protects stalled forks from degradation and promotes fork restart after replication stress. EMBO J. 2016, 35, 1437–1451. [Google Scholar] [CrossRef] [Green Version]
- Matos, D.A.; Zhang, J.-M.; Ouyang, J.; Nguyen, H.D.; Genois, M.-M.; Zou, L. ATR Protects the Genome against R Loops through a MUS81-Triggered Feedback Loop. Mol. Cell 2019, 77, 514–527.e4. [Google Scholar] [CrossRef]
- Hamperl, S.; Bocek, M.J.; Saldivar, J.C.; Swigut, T.; Cimprich, K.A. Transcription-Replication Conflict Orientation Modulates R-Loop Levels and Activates Distinct DNA Damage Responses. Cell 2017, 170, 774–786.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wellinger, R.E.; Prado, F.; Aguilera, A. Replication Fork Progression Is Impaired by Transcription in Hyperrecombinant Yeast Cells Lacking a Functional THO Complex. Mol. Cell. Biol. 2006, 26, 3327–3334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuduri, S.; Crabbe, L.; Conti, C.; Tourrière, H.; Holtgreve-Grez, H.; Jauch, A.; Pantesco, V.; De Vos, J.; Thomas, A.; Theillet, C.; et al. Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat. Cell Biol. 2009, 11, 1315–1324. [Google Scholar] [CrossRef]
- Jensen, R.B.; Carreira, A.; Kowalczykowski, S.C. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 2010, 467, 678–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moynahan, M.E.; Jasin, M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol. 2010, 11, 196–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Doty, T.; Gibson, B.; Heyer, W.-D. Human BRCA2 protein promotes RAD51 filament formation on RPA-covered single-stranded DNA. Nat. Struct. Mol. Biol. 2010, 17, 1260–1262. [Google Scholar] [CrossRef]
- Wells, J.P.; White, J.; Stirling, P.C. R Loops and Their Composite Cancer Connections. Trends Cancer 2019, 5, 619–631. [Google Scholar] [CrossRef]
- Pugliese, G.M.; Salaris, F.; Palermo, V.; Marabitti, V.; Morina, N.; Rosa, A.; Franchitto, A.; Pichierri, P. Inducible SMARCAL1 knockdown in iPSC reveals a link between replication stress and altered expression of master differentiation genes. Dis. Model. Mech. 2019, 12, dmm039487. [Google Scholar] [CrossRef] [Green Version]
- Chatzidoukaki, O.; Stratigi, K.; Goulielmaki, E.; Niotis, G.; Akalestou-Clocher, A.; Gkirtzimanaki, K.; Zafeiropoulos, A.; Altmüller, J.; Topalis, P.; Garinis, G.A. R-loops trigger the release of cytoplasmic ssDNAs leading to chronic inflammation upon DNA damage. Sci. Adv. 2021, 7, 5769. [Google Scholar] [CrossRef]
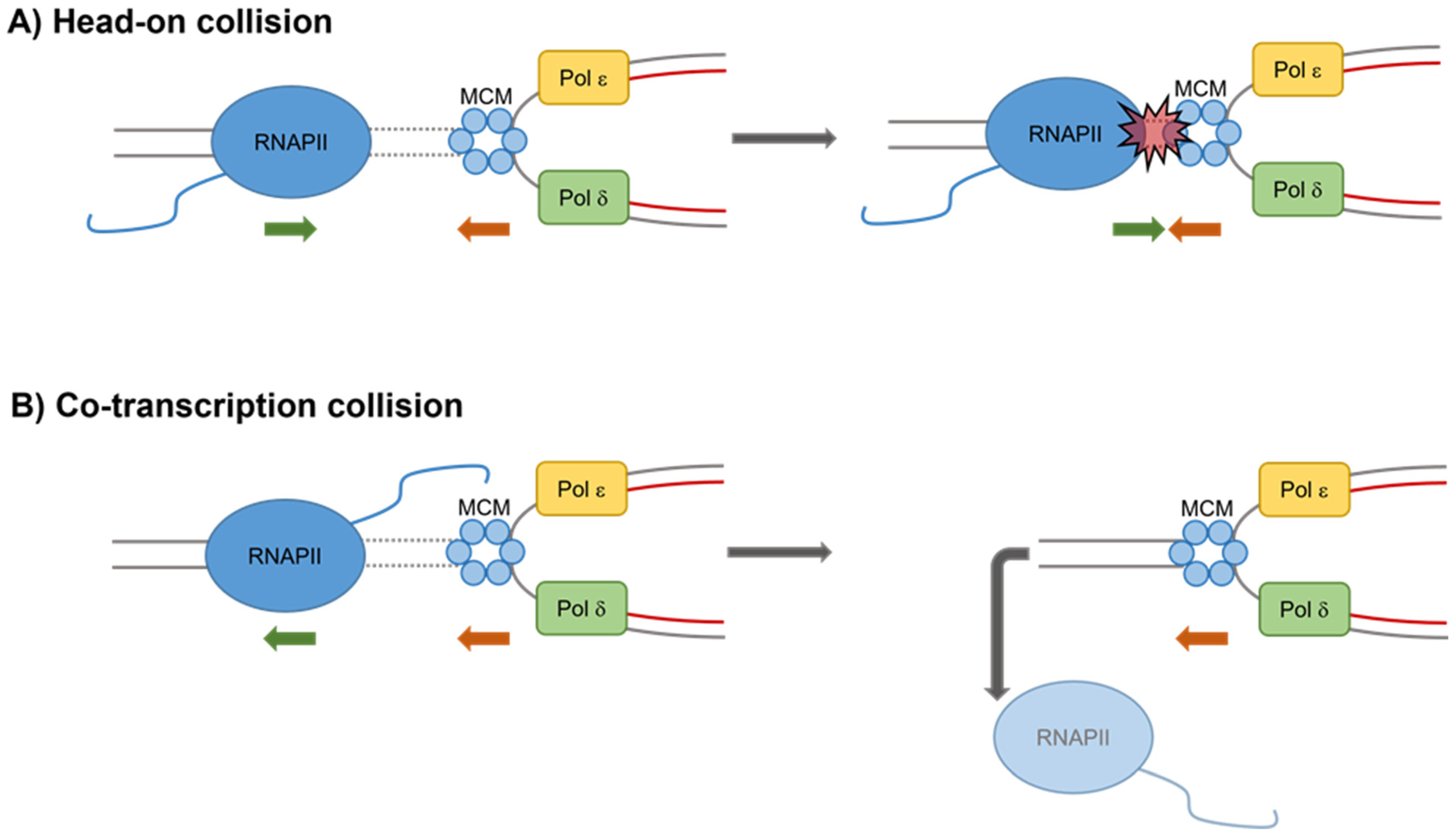

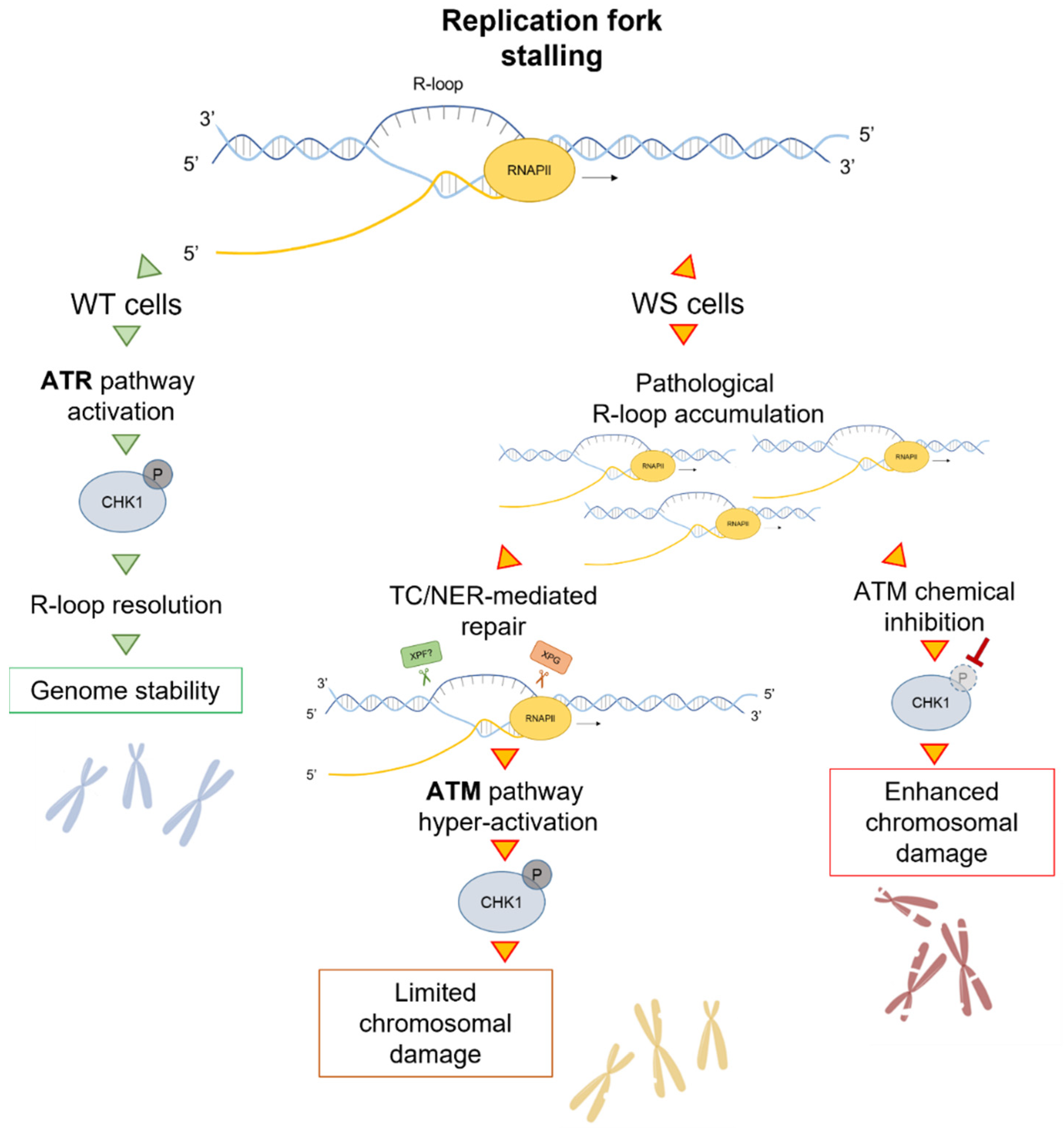

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marabitti, V.; Valenzisi, P.; Lillo, G.; Malacaria, E.; Palermo, V.; Pichierri, P.; Franchitto, A. R-Loop-Associated Genomic Instability and Implication of WRN and WRNIP1. Int. J. Mol. Sci. 2022, 23, 1547. https://doi.org/10.3390/ijms23031547
Marabitti V, Valenzisi P, Lillo G, Malacaria E, Palermo V, Pichierri P, Franchitto A. R-Loop-Associated Genomic Instability and Implication of WRN and WRNIP1. International Journal of Molecular Sciences. 2022; 23(3):1547. https://doi.org/10.3390/ijms23031547
Chicago/Turabian StyleMarabitti, Veronica, Pasquale Valenzisi, Giorgia Lillo, Eva Malacaria, Valentina Palermo, Pietro Pichierri, and Annapaola Franchitto. 2022. "R-Loop-Associated Genomic Instability and Implication of WRN and WRNIP1" International Journal of Molecular Sciences 23, no. 3: 1547. https://doi.org/10.3390/ijms23031547
APA StyleMarabitti, V., Valenzisi, P., Lillo, G., Malacaria, E., Palermo, V., Pichierri, P., & Franchitto, A. (2022). R-Loop-Associated Genomic Instability and Implication of WRN and WRNIP1. International Journal of Molecular Sciences, 23(3), 1547. https://doi.org/10.3390/ijms23031547