
Human Endothelial Progenitor Cells Protect the Kidney against Ischemia-Reperfusion Injury via the NLRP3 Inflammasome in Mice

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
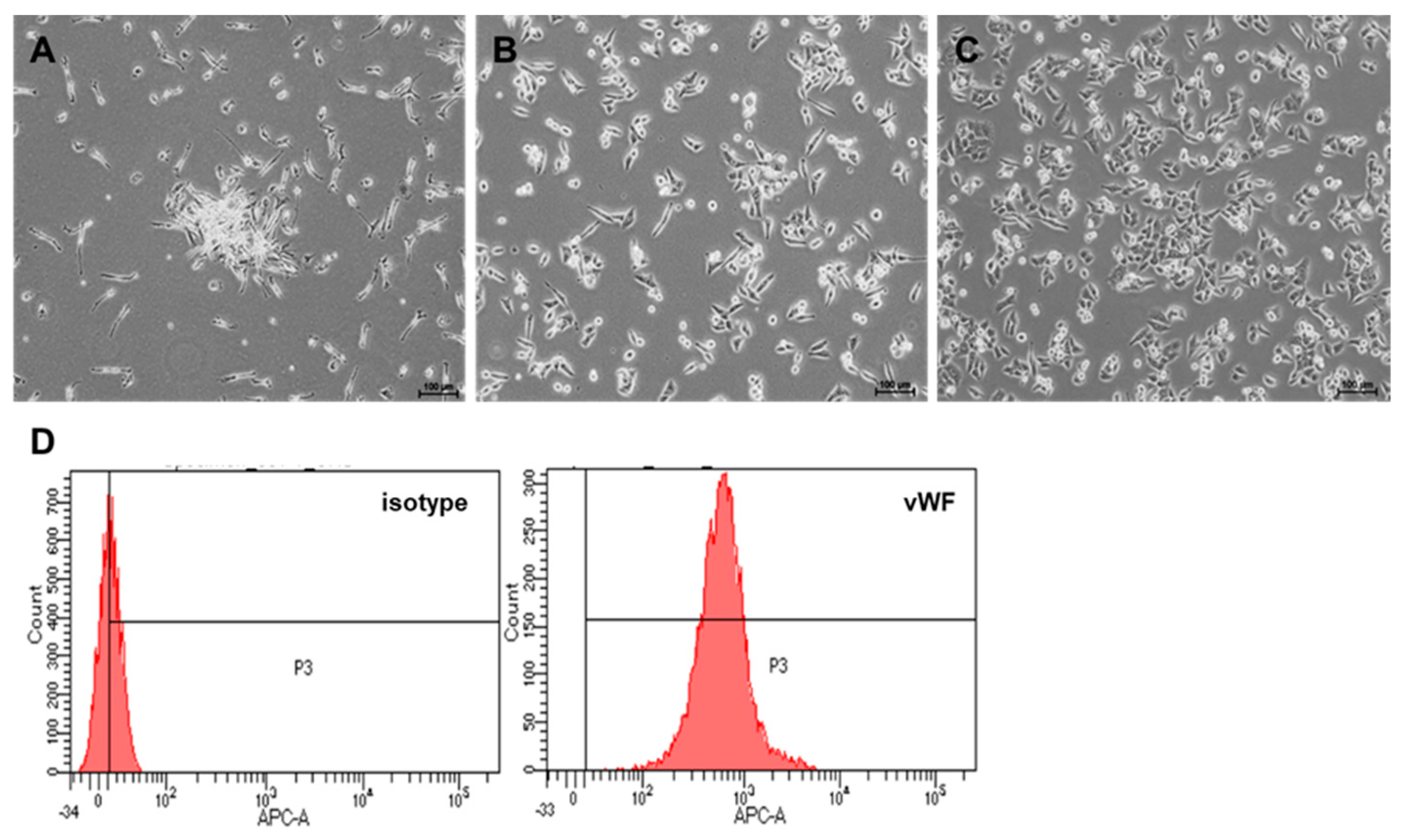
2.1. Culture and Differentiation of EPCs from PBMNCs
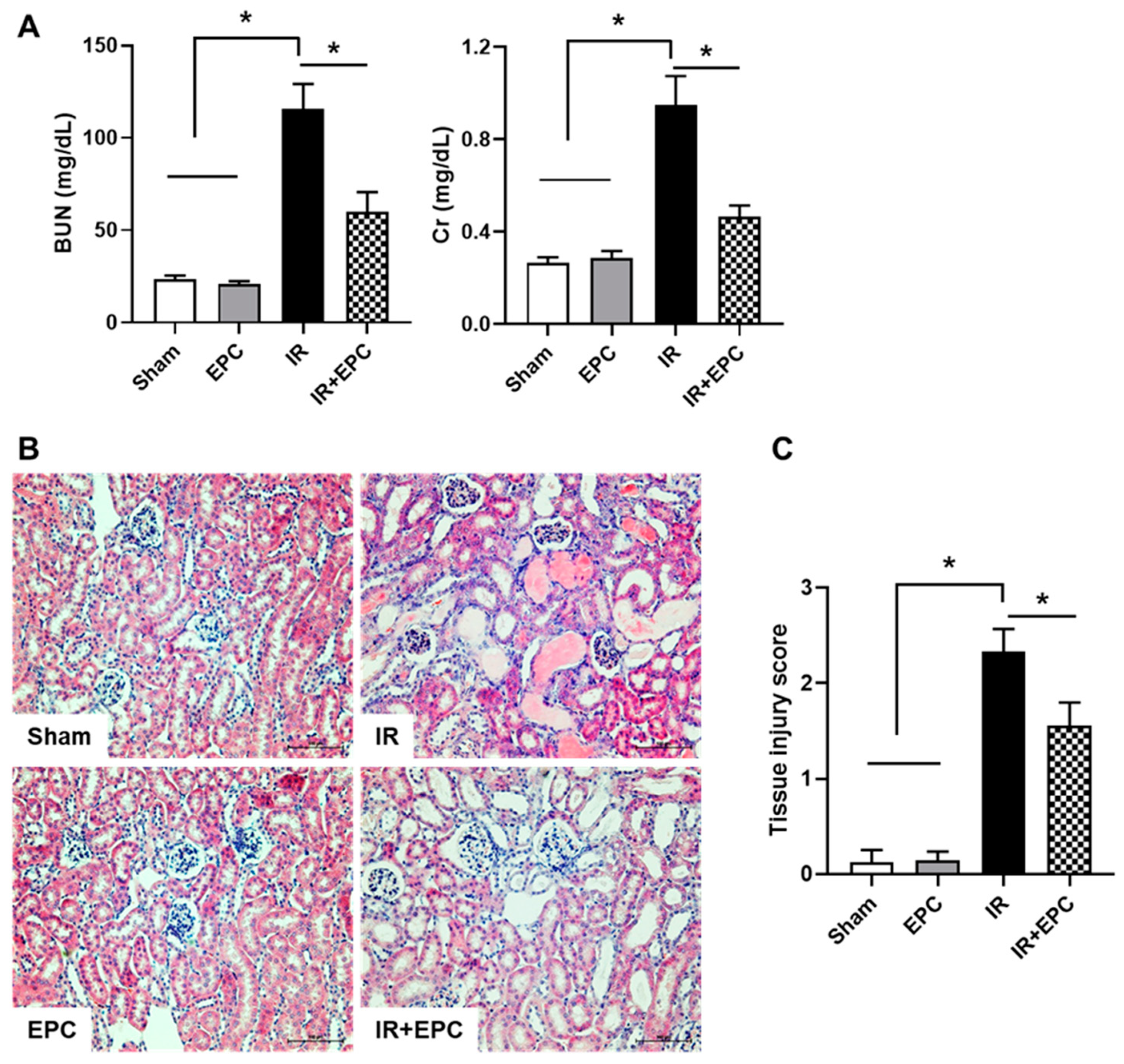
2.2. EPCs Ameliorate IRI-Induced Renal Dysfunction and Tissue Damage
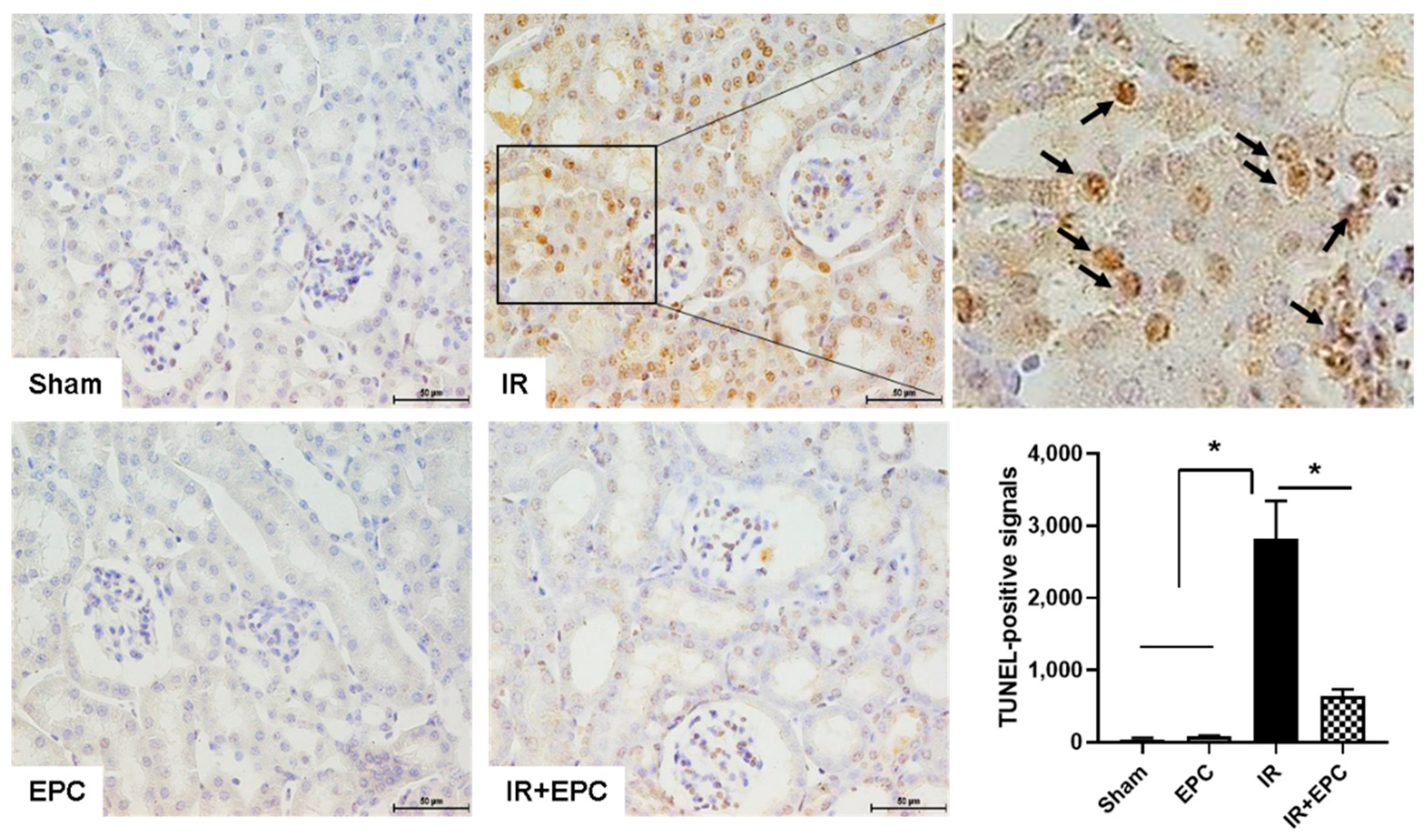
2.3. EPCs Reduce IRI-Induced Apoptosis
2.4. EPCs Attenuate IRI-Induced Oxidative Stress
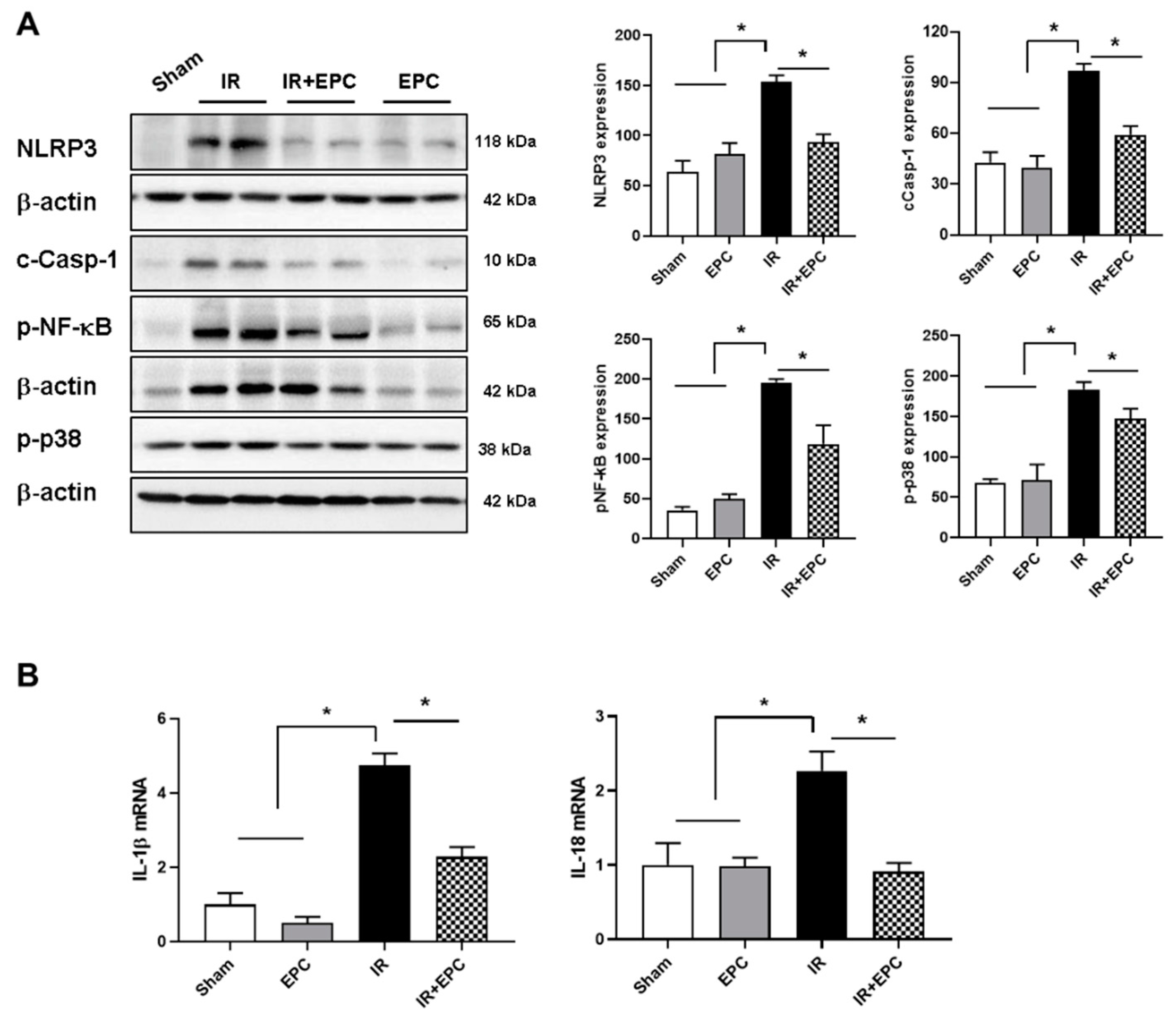
2.5. EPCs Decrease Inflammasome Activation
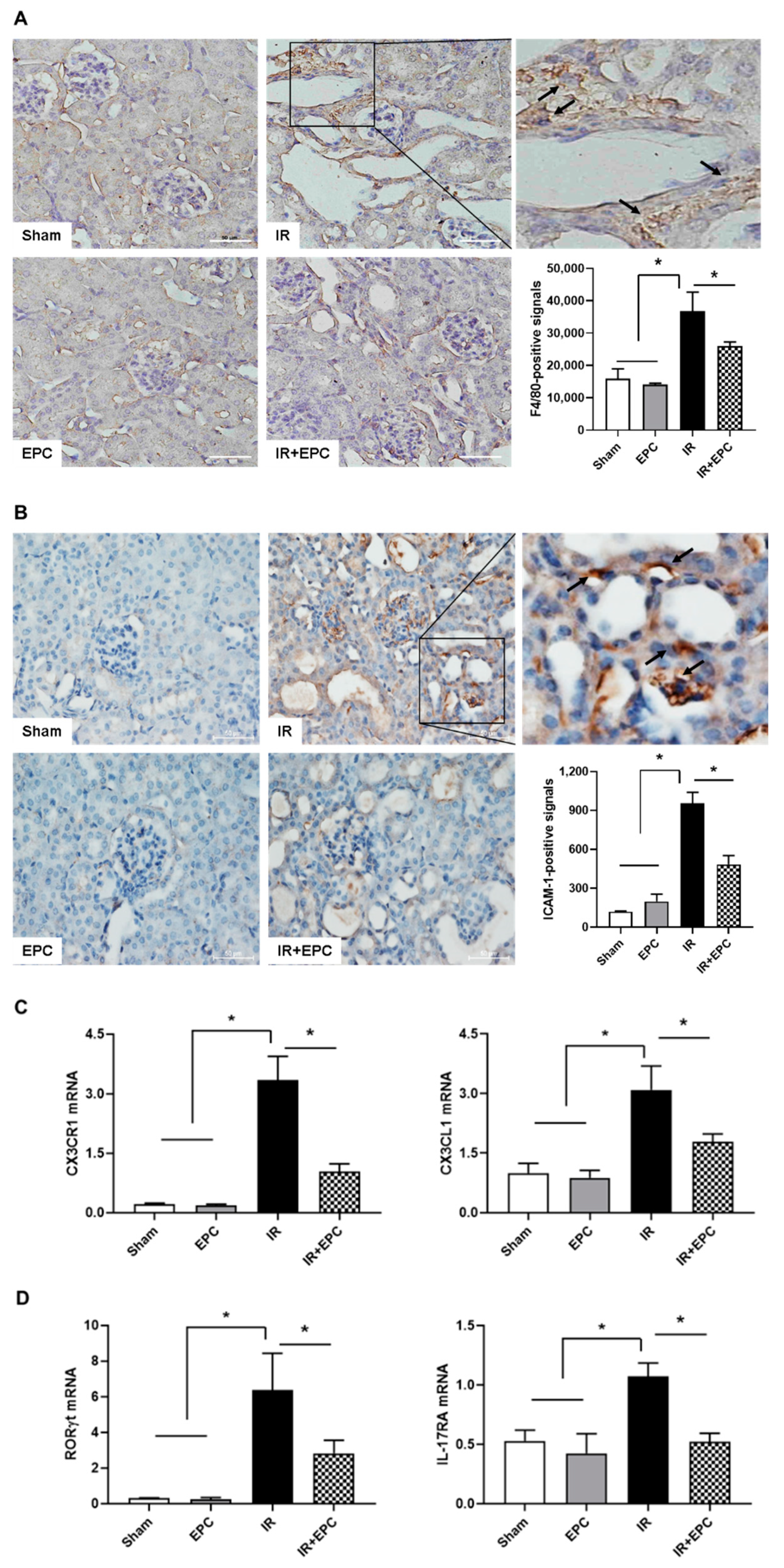
2.6. EPCs Reduce Infiltration of Inflammation-Related Immune Cells
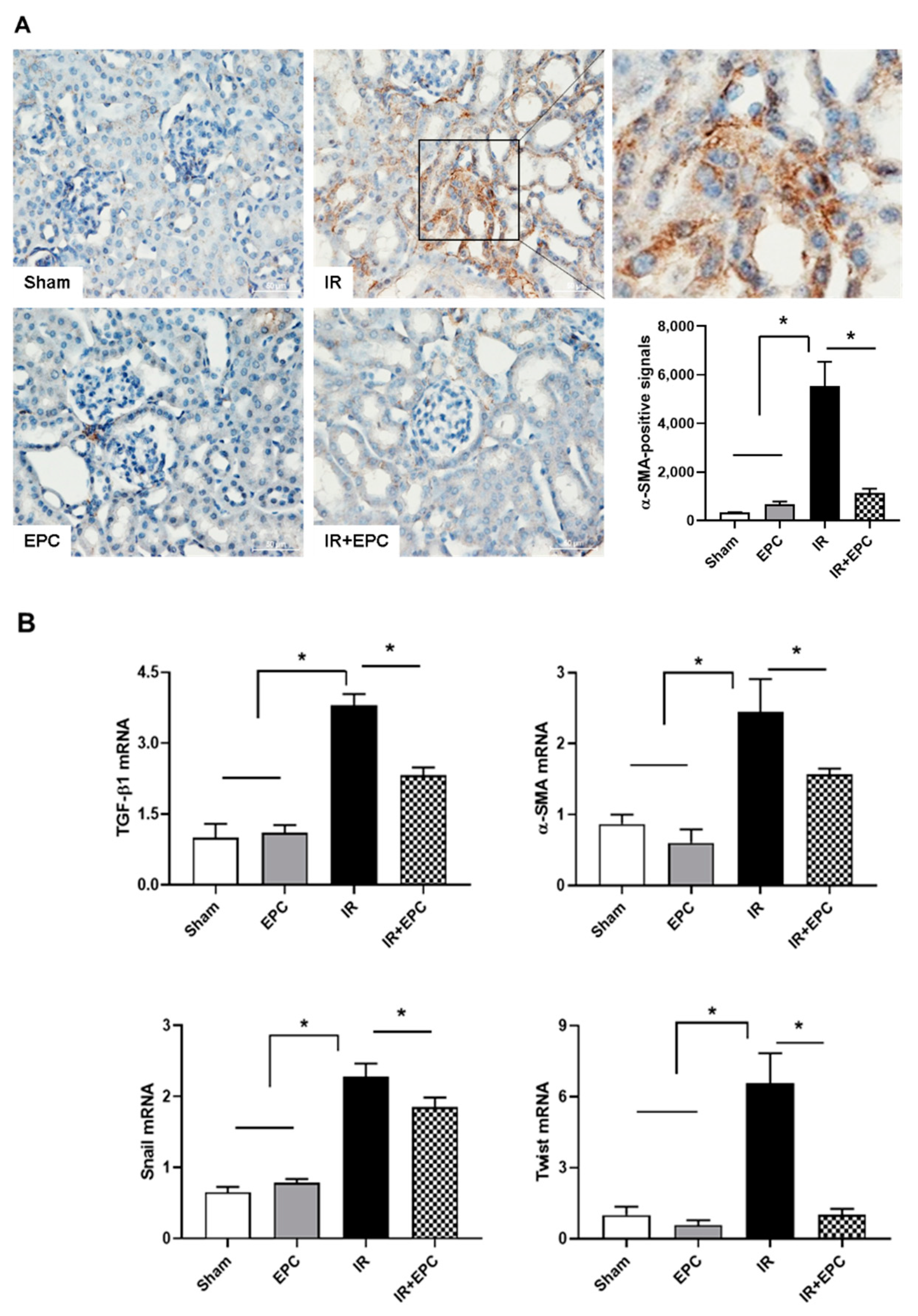
2.7. EPCs Reduce IRI-Induced Renal Fibrosis via the EMT Pathway
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Isolation and Culture of EPCs from Human PBMNCs
4.3. IRI-Induced AKI
4.4. Histopathology
4.5. TUNEL Assay
4.6. Immunoblotting
4.7. Immunohistochemistry
4.8. Quantitative Real-Time-Polymerase Chain Reaction (PCR)
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AKI | acute kidney injury |
| α-SMA | α-smooth muscle actin |
| CX3CL1 | chemokine (C-X3-C motif) ligand 1 |
| CX3CR1 | CX3C chemokine receptor 1 |
| EPC | endothelial progenitor cell |
| ICAM-1 | intracellular adhesion molecule-1 |
| IL-1β | interleukin-1beta |
| IL-17RA | interleukin-17 receptor A |
| IL-18 | interleukin-18 |
| IRI | ischemia-reperfusion injury |
| NF-κB | nuclear factor kappa B |
| NLRP | nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) proteins |
| RORγT | RAR-related orphan receptor gamma T |
| ROS | reactive oxygen species |
| vWF | von Willebrand factor |
References
- Wang, H.E.; Muntner, P.; Chertow, G.M.; Warnock, D.G. Acute kidney injury and mortality in hospitalized patients. Am. J. Nephrol. 2012, 35, 349–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickkers, P.; Darmon, M.; Hoste, E.; Joannidis, M.; Legrand, M.; Ostermann, M.; Prowle, J.R.; Schneider, A.; Schetz, M. Acute kidney injury in the critically ill: An updated review on pathophysiology and management. Intensive Care Med. 2021, 47, 835–850. [Google Scholar] [CrossRef] [PubMed]
- Basile, D.P.; Anderson, M.D.; Sutton, T.A. Pathophysiology of acute kidney injury. Compr. Physiol. 2012, 2, 1303–1353. [Google Scholar] [PubMed] [Green Version]
- Kanagasundaram, N.S. Pathophysiology of ischaemic acute kidney injury. Ann. Clin. Biochem. 2015, 52, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Bonventre, J.V.; Yang, L. Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Investig. 2011, 121, 4210–4221. [Google Scholar] [CrossRef]
- Komada, T.; Muruve, D.A. The role of inflammasomes in kidney disease. Nat. Rev. Nephrol. 2019, 15, 501–520. [Google Scholar] [CrossRef]
- Anders, H.J.; Muruve, D.A. The inflammasomes in kidney disease. J. Am. Soc. Nephrol. 2011, 22, 1007–1018. [Google Scholar] [CrossRef]
- Urbich, C.; Dimmeler, S. Endothelial progenitor cells: Characterization and role in vascular biology. Circ. Res. 2004, 95, 343–353. [Google Scholar] [CrossRef] [Green Version]
- Ozkok, A.; Yildiz, A. Endothelial Progenitor Cells and Kidney Diseases. Kidney Blood Press Res. 2018, 43, 701–718. [Google Scholar] [CrossRef]
- Becherucci, F.; Mazzinghi, B.; Ronconi, E.; Peired, A.; Lazzeri, E.; Sagrinati, C.; Romagnani, P.; Lasagni, L. The role of endothelial progenitor cells in acute kidney injury. Blood Purif. 2009, 27, 261–270. [Google Scholar] [CrossRef]
- Patschan, D.; Schwarze, K.; Henze, E.; Patschan, S.; Muller, G.A. The endothelial-to-mesenchymal transition and endothelial cilia in EPC-mediated postischemic kidney protection. Am. J. Physiol. Ren. Physiol. 2016, 310, F679–F687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devarajan, P. Update on mechanisms of ischemic acute kidney injury. J. Am. Soc. Nephrol. 2006, 17, 1503–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silver, S.A.; Cardinal, H.; Colwell, K.; Burger, D.; Dickhout, J.G. Acute kidney injury: Preclinical innovations, challenges, and opportunities for translation. Can. J. Kidney Health Dis. 2015, 2, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Zhao, D.; Na, N.; Li, H.; Miao, B.; Hong, L.; Huang, Z. Renoprotective effect of erythropoietin via modulation of the STAT6/MAPK/NF-kappaB pathway in ischemia/reperfusion injury after renal transplantation. Int. J. Mol. Med. 2018, 41, 25–32. [Google Scholar] [PubMed] [Green Version]
- Lee, D.W.; Faubel, S.; Edelstein, C.L. Cytokines in acute kidney injury (AKI). Clin. Nephrol. 2011, 76, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.M.; Luo, S.F.; Hsieh, H.L.; Chi, P.L.; Lin, C.C.; Wu, C.C.; Hsiao, L.D. Interleukin-1beta induces ICAM-1 expression enhancing leukocyte adhesion in human rheumatoid arthritis synovial fibroblasts: Involvement of ERK, JNK, AP-1, and NF-kappaB. J. Cell Physiol. 2010, 224, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.J.; Williams, W.W., Jr.; Colvin, R.B.; Bonventre, J.V. Antibody to intercellular adhesion molecule 1 protects the kidney against ischemic injury. Proc. Natl. Acad. Sci. USA 1994, 91, 812–816. [Google Scholar] [CrossRef] [Green Version]
- Collett, J.A.; Mehrotra, P.; Crone, A.; Shelley, W.C.; Yoder, M.C.; Basile, D.P. Endothelial colony-forming cells ameliorate endothelial dysfunction via secreted factors following ischemia-reperfusion injury. Am. J. Physiol. Ren. Physiol. 2017, 312, F897–F907. [Google Scholar] [CrossRef]
- Von Vietinghoff, S.; Kurts, C. Regulation and function of CX3CR1 and its ligand CX3CL1 in kidney disease. Cell Tissue Res. 2021, 385, 335–344. [Google Scholar] [CrossRef]
- Yu, Y.W.; Li, M.X.; Zhang, Z.Y.; Yu, H. The deficiency of CX3CL1/CX3CR1 system ameliorates high fructose diet-induced kidney injury by regulating NF-kappaB pathways in CX3CR1-knock out mice. Int. J. Mol. Med. 2018, 41, 3577–3585. [Google Scholar]
- Basile, D.P.; Ullah, M.M.; Collet, J.A.; Mehrotra, P. T helper 17 cells in the pathophysiology of acute and chronic kidney disease. Kidney Res. Clin. Pract. 2021, 40, 12–28. [Google Scholar] [CrossRef] [PubMed]
- Mehrotra, P.; Ullah, M.M.; Collett, J.A.; Myers, S.L.; Dwinell, M.R.; Geurts, A.M.; Basile, D.P. Mutation of RORgammaT reveals a role for Th17 cells in both injury and recovery from renal ischemia-reperfusion injury. Am. J. Physiol. Ren. Physiol. 2020, 319, F796–F808. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Lee, D.W.; Ravichandran, K.; Keys, D.O.; Akcay, A.; Nguyen, Q.; He, Z.; Jani, A.; Ljubanovic, D.; Edelstein, C.L. NLRP3 inflammasome knockout mice are protected against ischemic but not cisplatin-induced acute kidney injury. J. Pharmacol. Exp. Ther. 2013, 346, 465–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shigeoka, A.A.; Mueller, J.L.; Kambo, A.; Mathison, J.C.; King, A.J.; Hall, W.F.; Correia Jda, S.; Ulevitch, R.J.; Hoffman, H.M.; McKay, D.B. An inflammasome-independent role for epithelial-expressed Nlrp3 in renal ischemia-reperfusion injury. J. Immunol. 2010, 185, 6277–6285. [Google Scholar] [CrossRef] [Green Version]
- Tang, T.T.; Lv, L.L.; Pan, M.M.; Wen, Y.; Wang, B.; Li, Z.L.; Wu, M.; Wang, F.M.; Crowley, S.D.; Liu, B.C. Hydroxychloroquine attenuates renal ischemia/reperfusion injury by inhibiting cathepsin mediated NLRP3 inflammasome activation. Cell Death Dis. 2018, 9, 351. [Google Scholar] [CrossRef]
- Faubel, S.; Edelstein, C.L. Caspases as drug targets in ischemic organ injury. Curr. Drug Targets Immune Endocr. Metabol. Disord. 2005, 5, 269–287. [Google Scholar] [CrossRef]
- Zhang, X.; Agborbesong, E.; Li, X. The Role of Mitochondria in Acute Kidney Injury and Chronic Kidney Disease and Its Therapeutic Potential. Int. J. Mol. Sci. 2021, 22, 11253. [Google Scholar] [CrossRef]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. Inflammatory processes in renal fibrosis. Nat. Rev. Nephrol. 2014, 10, 493–503. [Google Scholar] [CrossRef]
- Burns, W.C.; Kantharidis, P.; Thomas, M.C. The role of tubular epithelial-mesenchymal transition in progressive kidney disease. Cells Tissues Organs 2007, 185, 222–231. [Google Scholar] [CrossRef]
- Grande, M.T.; Sanchez-Laorden, B.; Lopez-Blau, C.; De Frutos, C.A.; Boutet, A.; Arevalo, M.; Rowe, R.G.; Weiss, S.J.; Lopez-Novoa, J.M.; Nieto, M.A. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21, 989–997. [Google Scholar] [CrossRef] [Green Version]
- Qi, R.; Wang, J.; Jiang, Y.; Qiu, Y.; Xu, M.; Rong, R.; Zhu, T. Snai1-induced partial epithelial-mesenchymal transition orchestrates p53-p21-mediated G2/M arrest in the progression of renal fibrosis via NF-kappaB-mediated inflammation. Cell Death Dis. 2021, 12, 44. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.C.; Sung, P.H.; Cheng, B.C.; Li, Y.C.; Chen, Y.L.; Lee, M.S.; Yip, H.K. Safety and efficacy of intrarenal arterial autologous CD34+ cell transfusion in patients with chronic kidney disease: A randomized, open-label, controlled phase II clinical trial. Stem Cells Transl. Med. 2020, 9, 827–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, H.; Ohtake, T.; Tsukiyama, T.; Morota, M.; Ishioka, K.; Moriya, H.; Mochida, Y.; Hidaka, S.; Sato, T.; Asahara, T.; et al. Acute kidney injury successfully treated with autologous granulocyte colony-stimulating factor-mobilized peripheral blood CD34-positive cell transplantation: A first-in-human report. Stem Cells Transl. Med. 2021, 10, 1253–1257. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jang, H.N.; Kim, J.H.; Jung, M.H.; Tak, T.; Jung, J.H.; Lee, S.; Jung, S.; Chang, S.-H.; Kim, H.-J. Human Endothelial Progenitor Cells Protect the Kidney against Ischemia-Reperfusion Injury via the NLRP3 Inflammasome in Mice. Int. J. Mol. Sci. 2022, 23, 1546. https://doi.org/10.3390/ijms23031546
Jang HN, Kim JH, Jung MH, Tak T, Jung JH, Lee S, Jung S, Chang S-H, Kim H-J. Human Endothelial Progenitor Cells Protect the Kidney against Ischemia-Reperfusion Injury via the NLRP3 Inflammasome in Mice. International Journal of Molecular Sciences. 2022; 23(3):1546. https://doi.org/10.3390/ijms23031546
Chicago/Turabian StyleJang, Ha Nee, Jin Hyun Kim, Myeong Hee Jung, Taekil Tak, Jung Hwa Jung, Seunghye Lee, Sehyun Jung, Se-Ho Chang, and Hyun-Jung Kim. 2022. "Human Endothelial Progenitor Cells Protect the Kidney against Ischemia-Reperfusion Injury via the NLRP3 Inflammasome in Mice" International Journal of Molecular Sciences 23, no. 3: 1546. https://doi.org/10.3390/ijms23031546
APA StyleJang, H. N., Kim, J. H., Jung, M. H., Tak, T., Jung, J. H., Lee, S., Jung, S., Chang, S.-H., & Kim, H.-J. (2022). Human Endothelial Progenitor Cells Protect the Kidney against Ischemia-Reperfusion Injury via the NLRP3 Inflammasome in Mice. International Journal of Molecular Sciences, 23(3), 1546. https://doi.org/10.3390/ijms23031546