
Transport Turnover Rates for Human OCT2 and MATE1 Expressed in Chinese Hamster Ovary Cells

Abstract
1. Introduction
2. Results
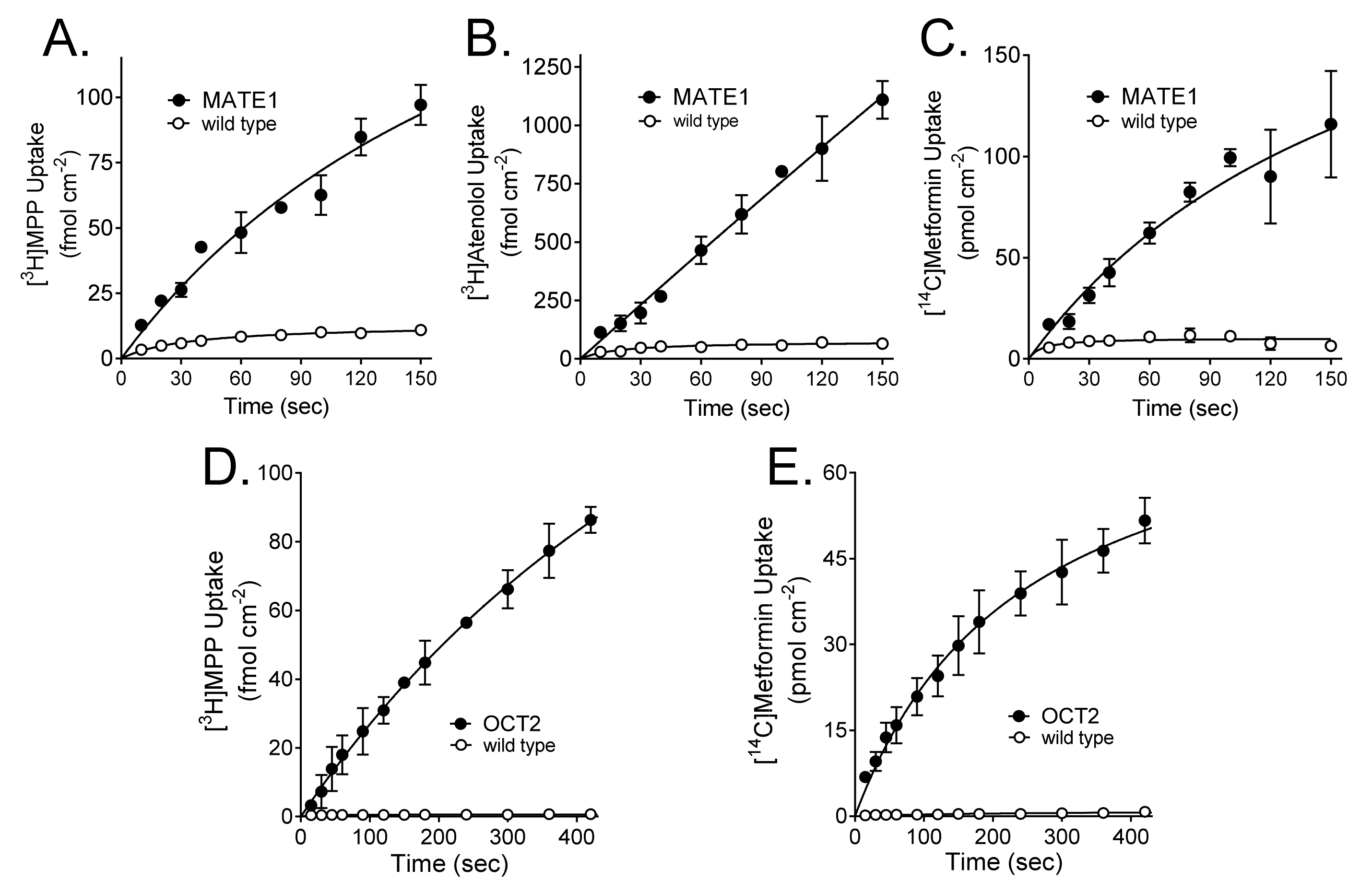
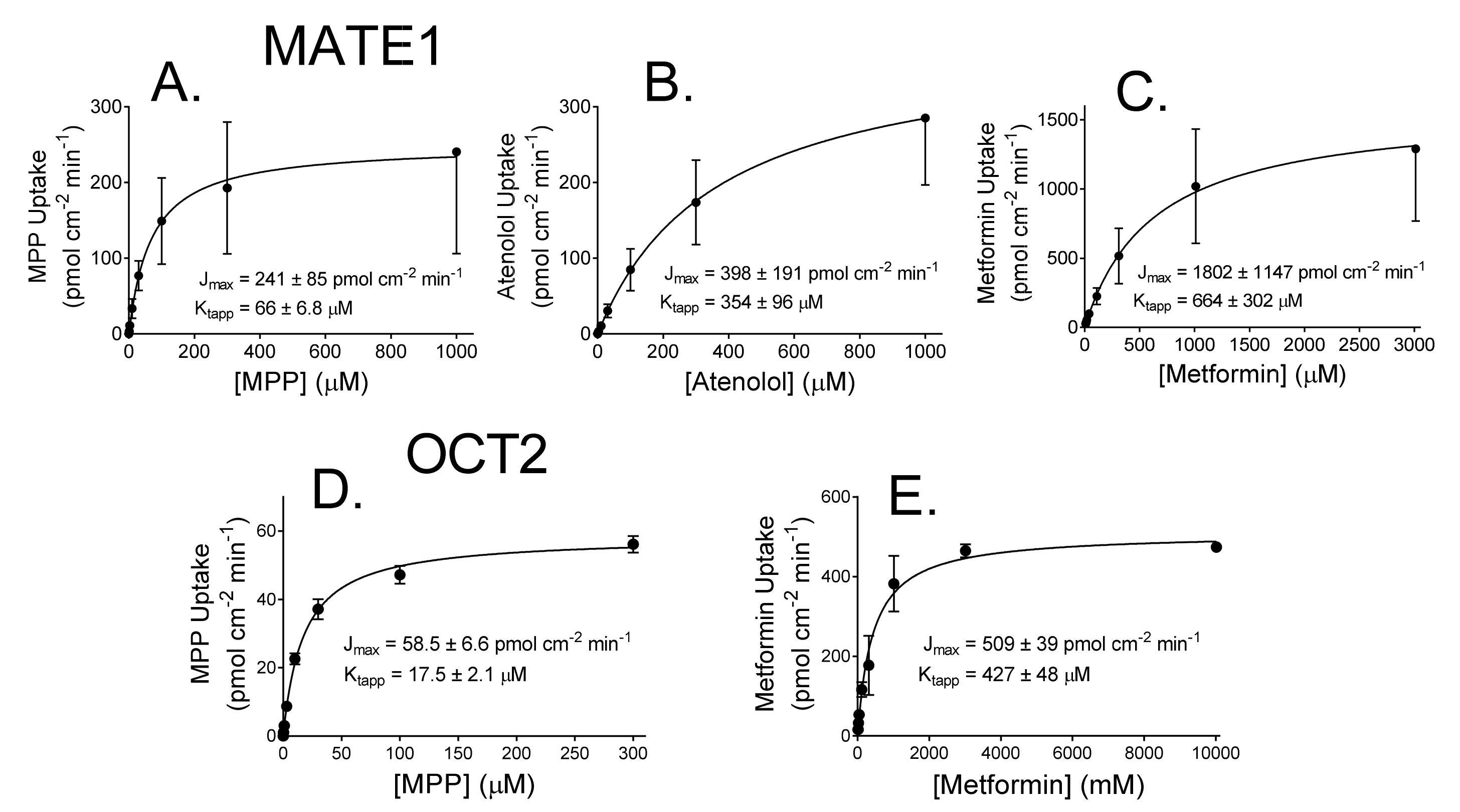
2.1. Kinetics of MATE1 and OCT2-Mediated Transport
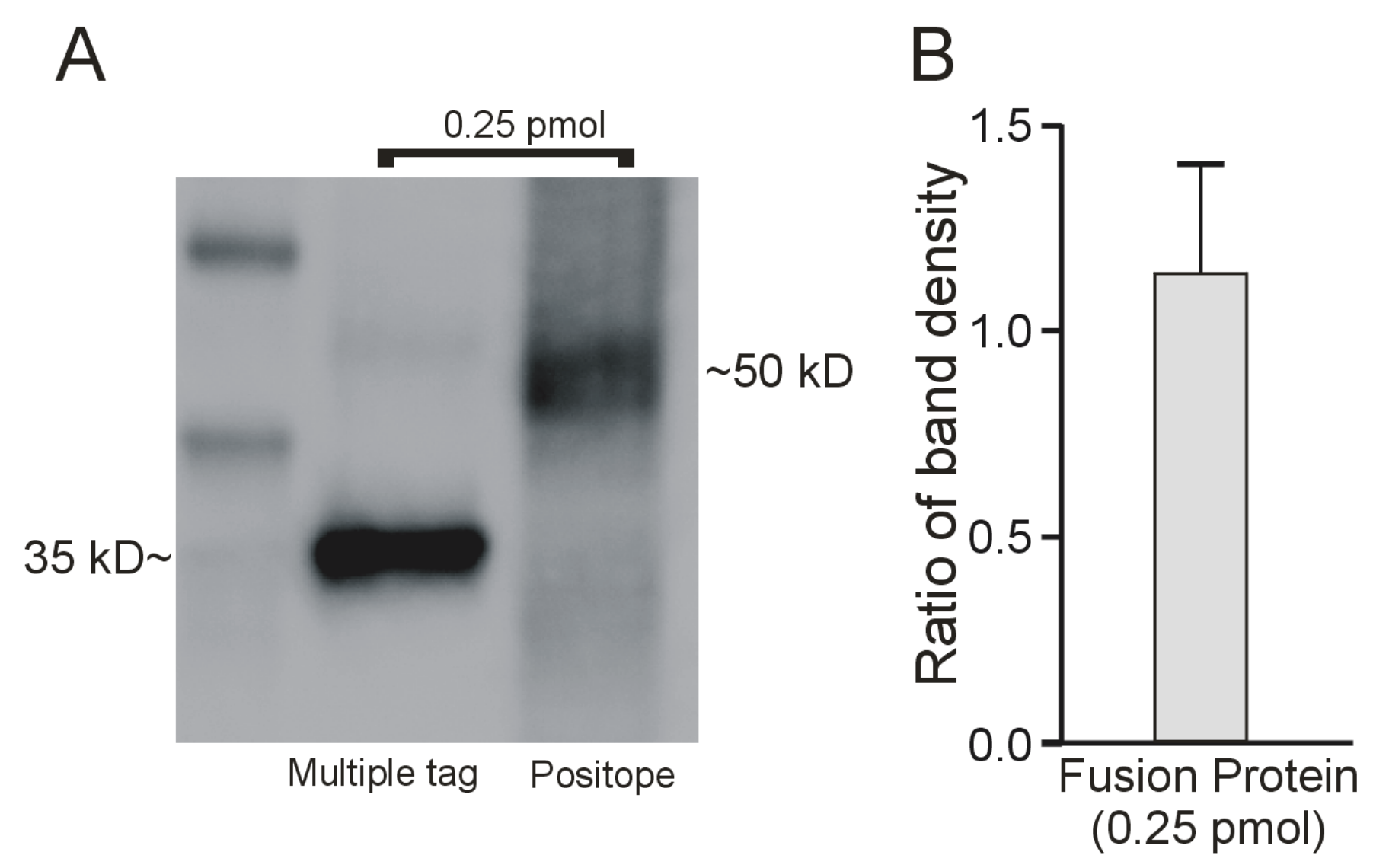
2.2. Interaction of the Monoclonal Anti-V5 Antibody with an Epitope ‘Standard’
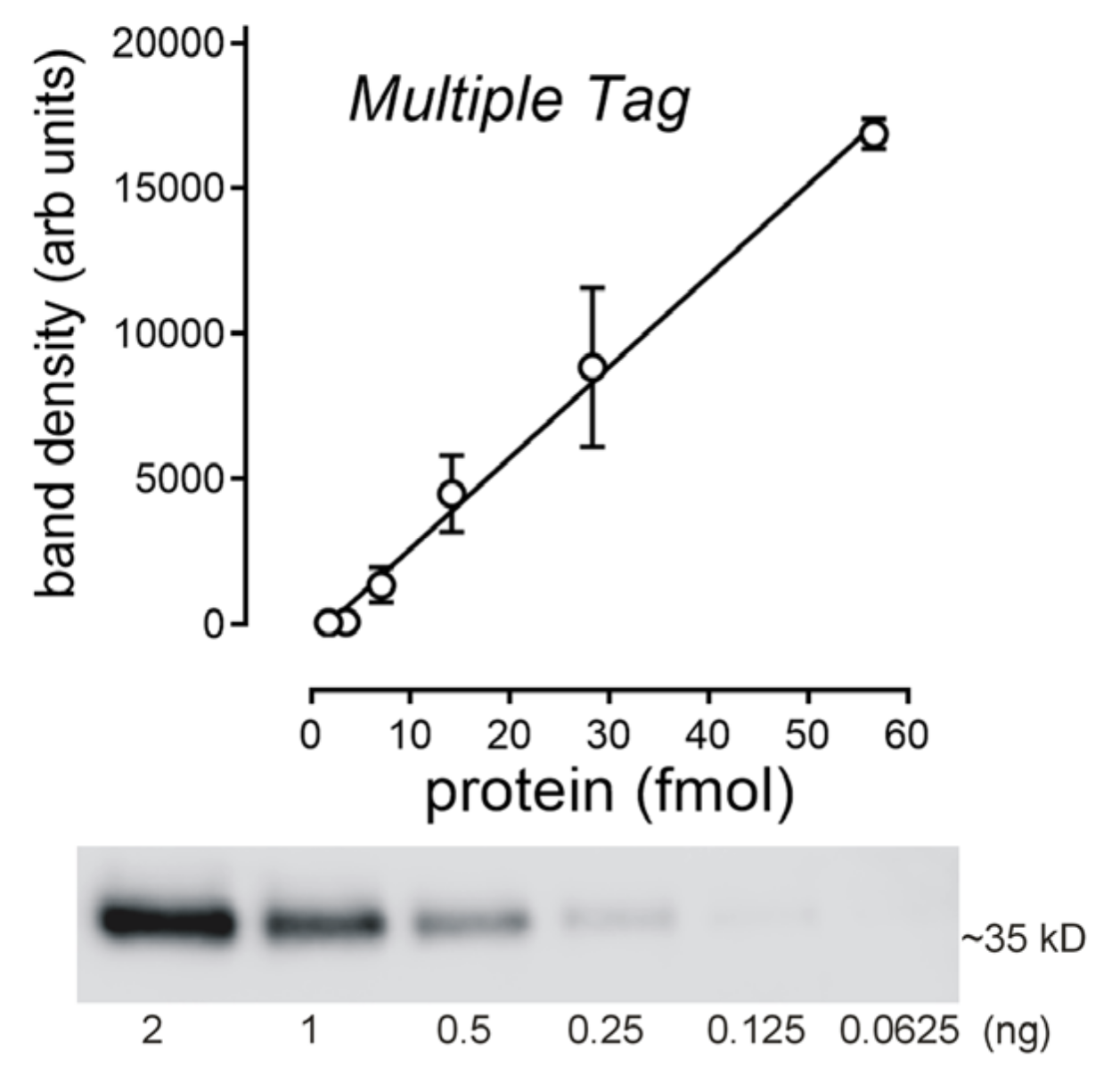
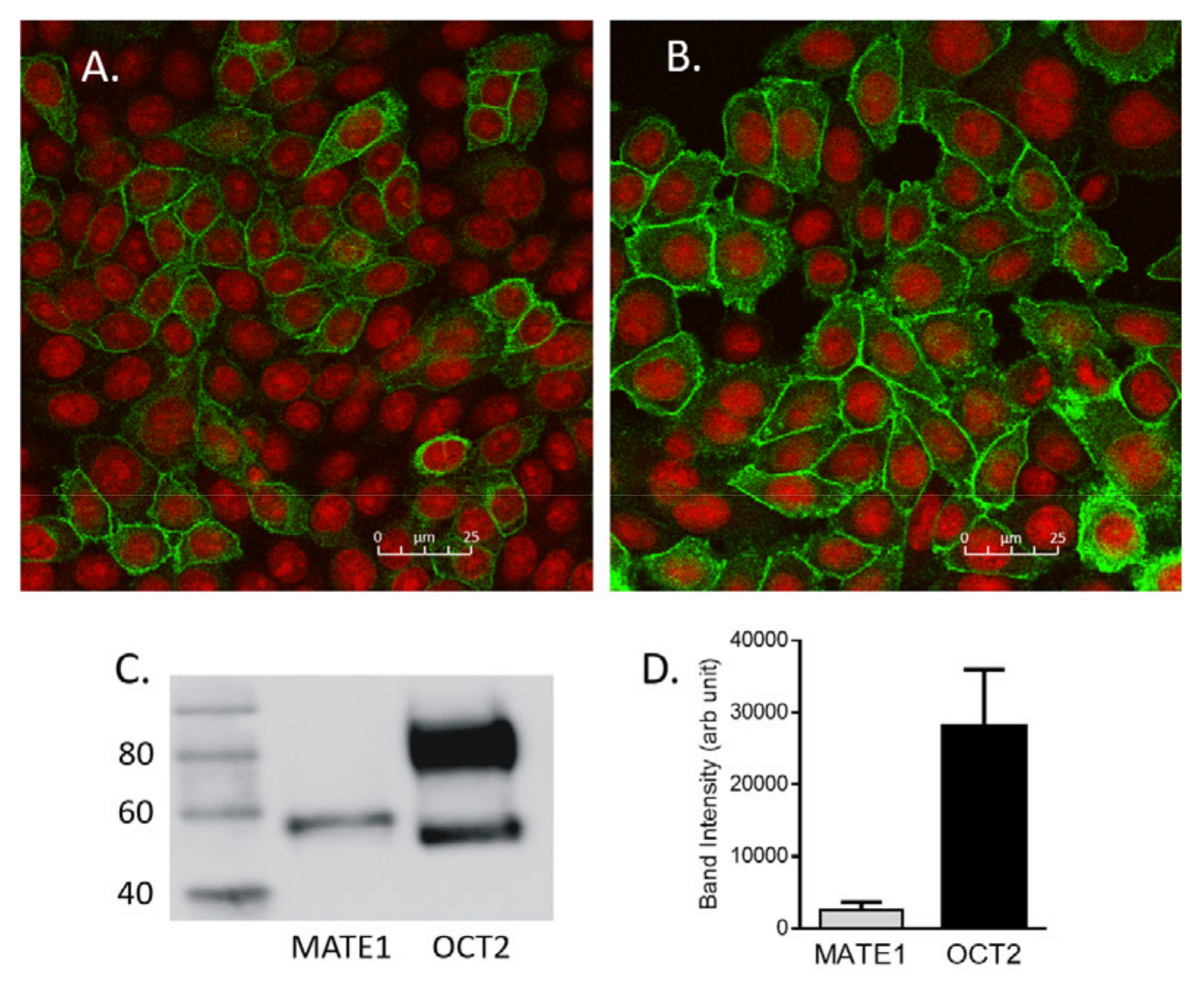
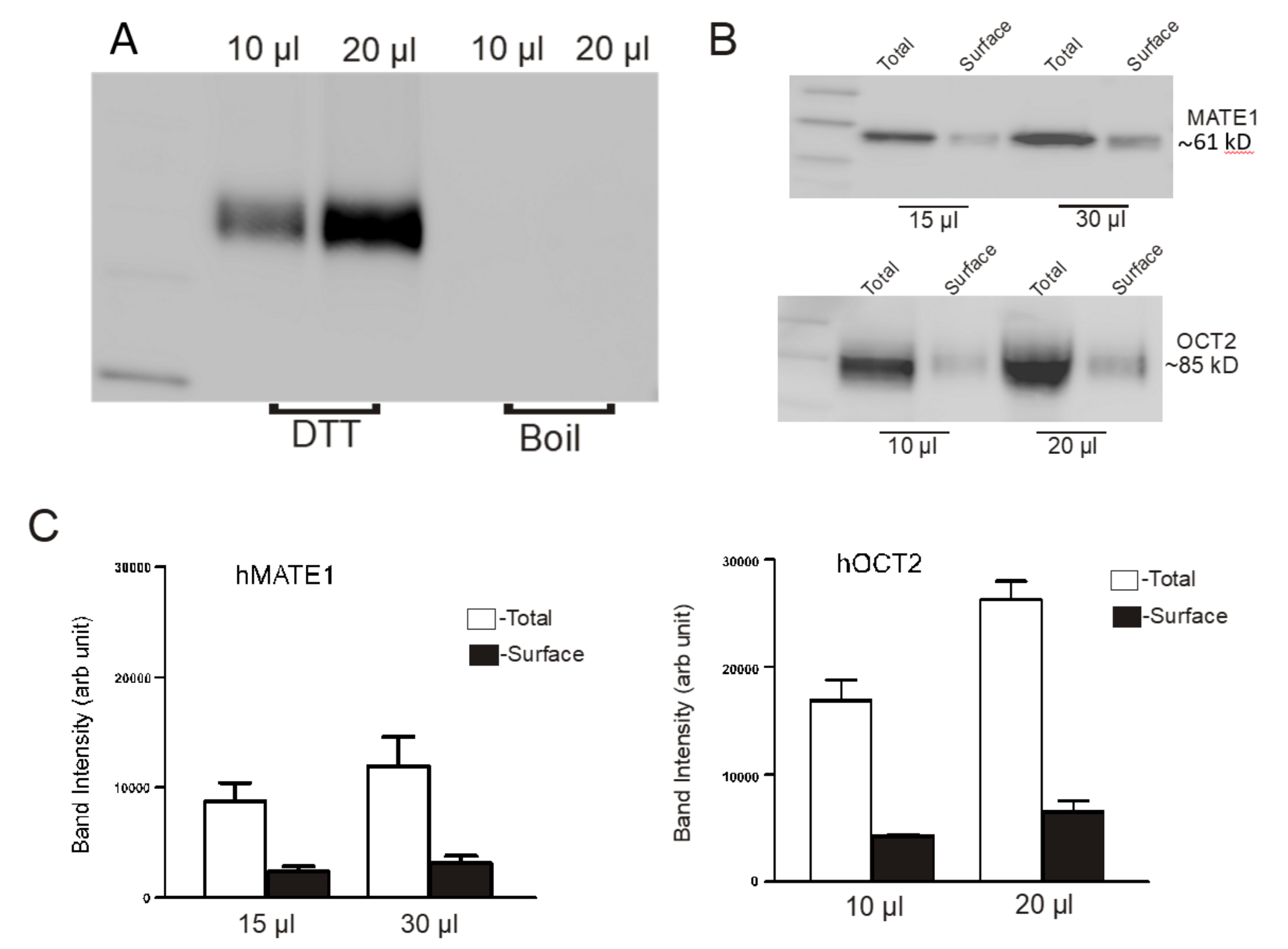
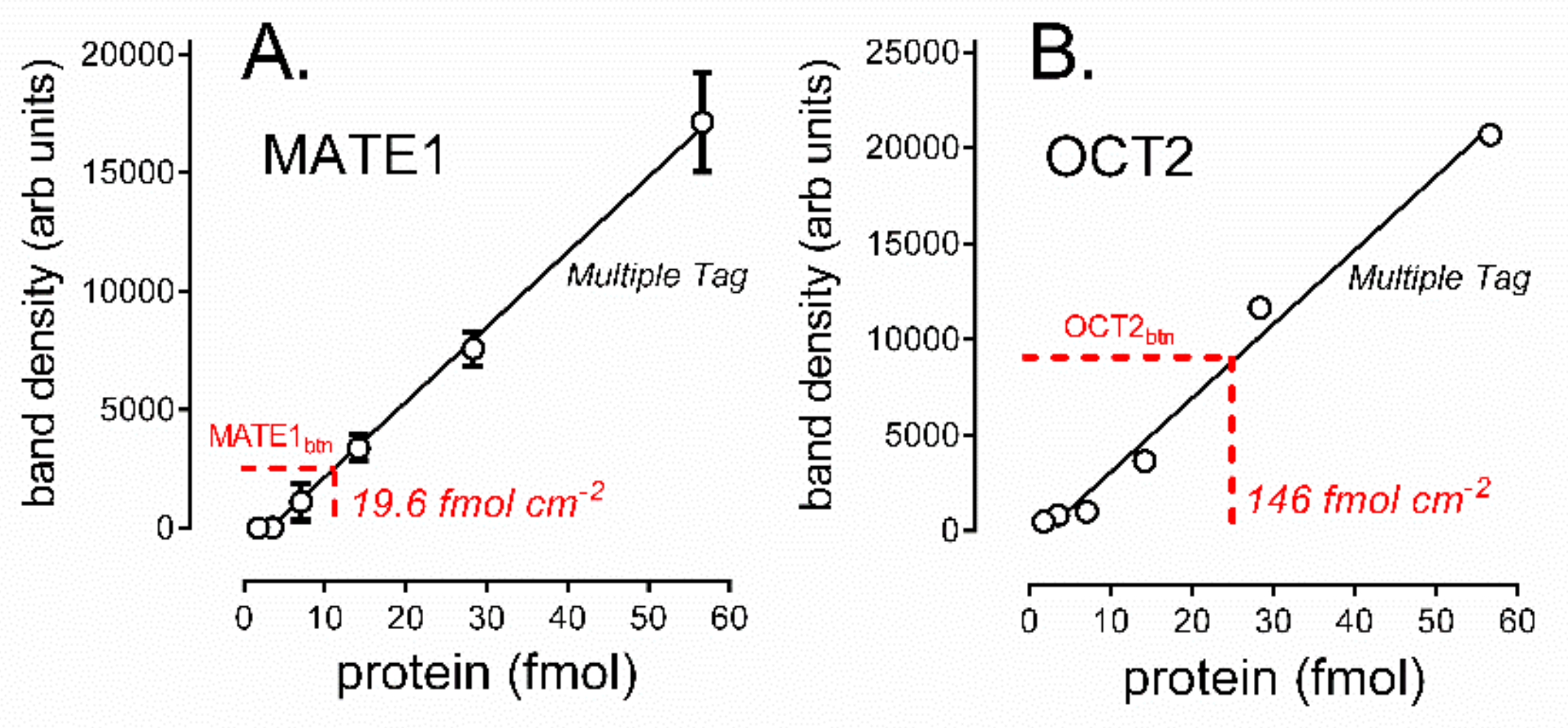
2.3. Determination of the Amount of MATE1 and OCT2 in CHO Cell Plasma Membrane
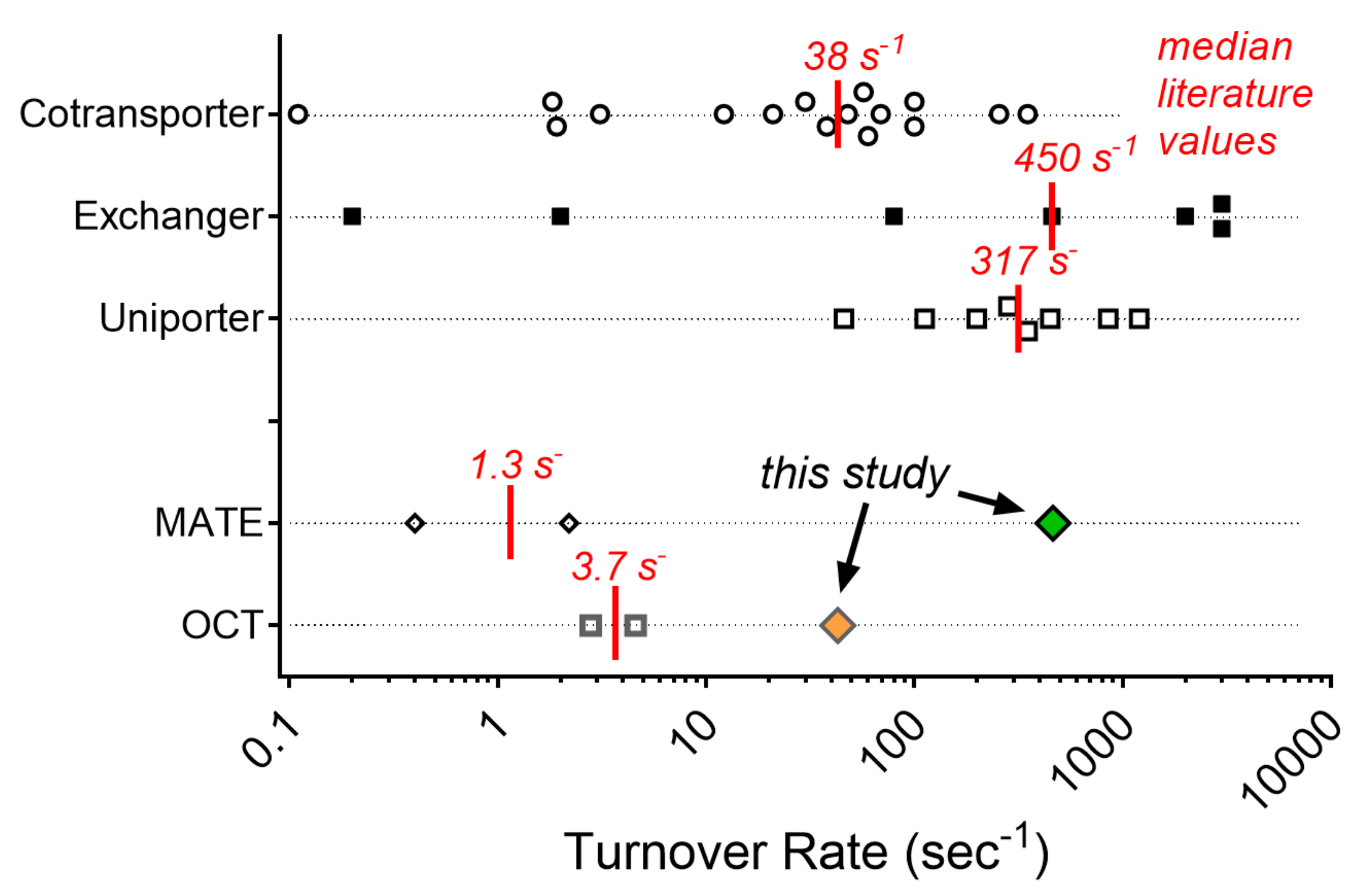
2.4. Turnover Rates (TOR) for MATE1 and OCT2
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture
4.3. Transport Experiments
4.4. Immunocytochemistry
4.5. Cell Surface Biotinylation of MATE1 and OCT2
4.6. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hagenbuch, B. Drug uptake systems in liver and kidney: A historic perspective. Clin. Pharmacol. Ther. 2010, 87, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Ahlin, G.; Karlsson, J.; Pedersen, J.M.; Gustavsson, L.; Larsson, R.; Matsson, P.; Norinder, U.; Bergström, C.A.; Artursson, P. Structural requirements for drug inhibition of the liver specific human Organic Cation Transport protein. J. Med. Chem. 2008, 51, 5932–5942. [Google Scholar] [CrossRef] [PubMed]
- Neuhoff, S.; Ungell, A.L.; Zamora, I.; Artursson, P. pH-dependent bidirectional transport of weakly basic drugs across Caco-2 monolayers: Implications for drug-drug interactions. Pharm. Res. 2003, 20, 1141–1148. [Google Scholar] [CrossRef]
- Kikuchi, R.; Chiou, W.J.; Durbin, K.R.; Savaryn, J.P.; Ma, J.; Emami Riedmaier, A.; de Morais, S.M.; Jenkins, G.J.; Bow, D.A.J. Quantitation of Plasma Membrane Drug Transporters in Kidney Tissue and Cell Lines Using a Novel Proteomic Approach Enabled a Prospective Prediction of Metformin Disposition. Drug Metab. Dispos. 2021, 49, 938–946. [Google Scholar] [CrossRef] [PubMed]
- Pelis, R.M.; Wright, S.H. Renal transport of organic anions and cations. Compr. Physiol. 2011, 1, 1795–1835. [Google Scholar]
- Wittwer, M.B.; Zur, A.A.; Khuri, N.; Kido, Y.; Kosaka, A.; Zhang, X.; Morrissey, K.M.; Sali, A.; Huang, Y.; Giacomini, K.M. Discovery of potent, selective MATE1 inhibitors through prescription drug profiling and computational modeling. J. Med. Chem. 2013, 56, 781–795. [Google Scholar] [CrossRef]
- Kido, Y.; Matsson, P.; Giacomini, K.M. Profiling of a prescription drug library for potential renal drug-drug interactions mediated by the organic cation transporter 2. J. Med. Chem. 2011, 54, 4548–4558. [Google Scholar] [CrossRef]
- Sandoval, P.J.; Zorn, K.M.; Clark, A.M.; Ekins, S.; Wright, S.H. Assessment of substrate dependent ligand interactions at the Organic Cation Transporter OCT2 using six model substrates. Mol. Pharmacol. 2018, 94, 1057–1068. [Google Scholar] [CrossRef]
- Martinez-Guerrero, L.J.; Morales, M.H.; Ekins, S.; Wright, S.H. Lack of influence of substrate on ligand interaction with human MATE1. Mol. Pharmacol. 2016, 90, 254–264. [Google Scholar] [CrossRef]
- Koepsell, H. Update on drug-drug interaction at organic cation transporters: Mechanisms, clinical impact, and proposal for advanced in vitro testing. Expert Opin. Drug Metab. Toxicol. 2021, 17, 635–653. [Google Scholar] [CrossRef]
- Koepsell, H. Role of organic cation transporters in drug-drug interaction. Expert Opin. Drug Metab. Toxicol. 2015, 11, 1619–1633. [Google Scholar] [CrossRef]
- Giacomini, K.M.; Huang, S.M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.M.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [PubMed]
- Wright, S.H. Multidrug and Toxin Extrusion Proteins. In Drug Transporters, 2nd ed.; You, G., Morris, M.E., Eds.; Wiley: Hoboken, NJ, USA, 2014; pp. 223–243. [Google Scholar]
- Lepist, E.-I.; Ray, A.S. Renal drug-drug interactions: What we have learned and where we are going. Expert Opin. Drug Metab. Toxicol. 2012, 8, 433–448. [Google Scholar] [CrossRef] [PubMed]
- Burt, H.J.; Neuhoff, S.; Almond, L.; Gaohua, L.; Harwood, M.D.; Jamei, M.; Rostami-Hodjegan, A.; Tucker, G.T.; Rowland-Yeo, K. Metformin and cimetidine: Physiologically based pharmacokinetic modelling to investigate transporter mediated drug-drug interactions. Eur. J. Pharm. Sci. 2016, 88, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Harwood, M.D.; Neuhoff, S.; Carlson, G.L.; Warhurst, G.; Rostami-Hodjegan, A. Absolute abundance and function of intestinal drug transporters: A prerequisite for fully mechanistic in vitro-in vivo extrapolation of oral drug absorption. Biopharm. Drug Dispos. 2013, 34, 2–28. [Google Scholar] [CrossRef]
- Ball, K.; Bouzom, F.; Scherrmann, J.-M.; Walther, B.; Declèves, X. Development of a physiologically based pharmacokinetic model for the rat central nervous system and determination of an in vitro-in vivo scaling methodology for the blood-brain barrier permeability of two transporter substrates, morphine and oxycodone. J. Pharm. Sci. 2012, 101, 4277–4292. [Google Scholar] [CrossRef]
- Kumar, A.R.; Prasad, B.; Bhatt, D.K.; Mathialagan, S.; Varma, M.V.S.; Unadkat, J.D. In Vivo-to-In Vitro Extrapolation of Transporter-Mediated Renal Clearance: Relative Expression Factor Versus Relative Activity Factor Approach. Drug Metab. Dispos. 2020, 49, 470–478. [Google Scholar] [CrossRef]
- Stein, W.D. Transport and Diffusion across Cell Membranes; Academic Press: New York, NY, USA, 1986; 685p. [Google Scholar]
- Qian, C.Q.; Zhao, K.J.; Chen, Y.; Liu, L.; Liu, X.D. Simultaneously predict pharmacokinetic interaction of rifampicin with oral versus intravenous substrates of cytochrome P450 3A/P-glycoprotein to healthy human using a semi-physiologically based pharmacokinetic model involving both enzyme and transporter turnover. Eur. J. Pharm. Sci. 2019, 134, 194–204. [Google Scholar]
- Yin, J.; Duan, H.; Shirasaka, Y.; Prasad, B.; Wang, J. Atenolol renal secretion is mediated by human Organic Cation Transporter 2 and Multidrug and Toxin Extrusion proteins. Drug Metab. Dispos. 2015, 43, 1872–1881. [Google Scholar] [CrossRef]
- Severance, A.C.; Sandoval, P.J.; Wright, S.H. Correlation between apparent substrate affinity and OCT2 transporter turnover. J. Pharmacol. Exp. Ther. 2017, 362, 405–412. [Google Scholar] [CrossRef]
- Belzer, M.; Morales, M.; Jagadish, B.; Mash, E.A.; Wright, S.H. Substrate-dependent ligand inhibition of the human Organic Cation Transporter, OCT2. J. Pharmacol. Exp. Ther. 2013, 346, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, P.J.; Morales, M.N.; Secomb, T.W.; Wright, S.H. Kinetic basis of metformin-MPP interactions with the Organic Cation Transporter OCT2. Am. J. Physiol. Renal Physiol. 2019, 317, F720–F734. [Google Scholar] [CrossRef] [PubMed]
- Pelis, R.M.; Suhre, W.M.; Wright, S.H. Functional influence of N-glycosylation in OCT2-mediated tetraethylammonium transport. Am. J. Physiol. Renal Physiol. 2006, 290, F1118–F1126. [Google Scholar] [CrossRef] [PubMed]
- Prasad, B.; Unadkat, J.D. Optimized approaches for quantification of drug transporters in tissues and cells by MRM proteomics. AAPS J. 2014, 16, 634–648. [Google Scholar] [CrossRef]
- Achour, B.; Al-Majdoub, Z.M.; Rostami-Hodjegan, A.; Barber, J. Mass Spectrometry of Human Transporters. Annu. Rev. Anal. Chem. 2020, 13, 223–247. [Google Scholar] [CrossRef]
- Goodyear, L.J.; Hirshman, M.F.; Valyou, P.M.; Horton, E.S. Glucose transporter number, function, and subcellular distribution in rat skeletal muscle after exercise training. Diabetes 1992, 41, 1091–1099. [Google Scholar] [CrossRef] [PubMed]
- Wright, S.H.; Pajor, A.M.; Moon, D.A.; Wunz, T.M. High affinity phlorizin binding in Mytilus gill. Biochim. Biophys. Acta Biomembr. 1992, 1103, 212–218. [Google Scholar] [CrossRef]
- Ward, J.L.; Sherali, A.; Mo, Z.-P.; Tse, C.-M. Kinetic and pharmacological properties of cloned human equilibrative nucleoside transporters, ENT1 and ENT2, stably expressed in nucleoside transporter-deficient PK15 cells. Ent2 exhibits a low affinity for guanosine and cytidine but a high affinity for inosine. J. Biol. Chem. 2000, 275, 8375–8381. [Google Scholar]
- Mackenzie, B.; Loo, D.D.; Panayotova-Heiermann, M.; Wright, E.M. Biophysical characteristics of the pig kidney Na+/glucose cotransporter SGLT2 reveal a common mechanism for SGLT1 and SGLT2. J. Biol. Chem. 1996, 271, 32678–32683. [Google Scholar] [CrossRef]
- Loo, D.D.; Hazama, A.; Supplisson, S.; Turk, E.; Wright, E.M. Relaxation kinetics of the Na+/glucose cotransporter. Proc. Natl. Acad. Sci. USA 1993, 90, 5767–5771. [Google Scholar] [CrossRef]
- Boorer, K.J.; Frommer, W.B.; Bush, D.R.; Kreman, M.; Loo, D.D.; Wright, E.M. Kinetics and specificity of a H+/amino acid transporter from Arabidopsis thaliana. J. Biol. Chem. 1996, 271, 2213–2220. [Google Scholar] [CrossRef] [PubMed]
- Omote, H.; Al-Shawi, M.K. A novel electron paramagnetic resonance approach to determine the mechanism of drug transport by P-glycoprotein. J. Biol. Chem. 2002, 277, 45688–45694. [Google Scholar] [CrossRef] [PubMed]
- Siczkowski, M.; Davies, J.E.; Ng, L.L. Activity and density of the Na+/H+ antiporter in normal and transformed human lymphocytes and fibroblasts. Am. J. Physiol. 1994, 267, C745–C752. [Google Scholar] [CrossRef] [PubMed]
- Siczkowski, M.; Ng, L.L. Culture density and the activity, abundance and phosphorylation of the Na+/H+ exchanger isoform 1 in human fibroblasts. Biochem. Biophys. Res. Commun. 1995, 209, 191–197. [Google Scholar] [CrossRef]
- Cavet, M.E.; Akhter, S.; de Medina, F.S.; Donowitz, M.; Tse, C.M. Na+/H+ exchangers (NHE1-3) have similar turnover numbers but different percentages on the cell surface. Am. J. Physiol. 1999, 277, C1111–C1121. [Google Scholar] [CrossRef]
- Taylor, S.C.; Posch, A. The design of a quantitative western blot experiment. BioMed Res. Int. 2014, 2014, 361590. [Google Scholar] [CrossRef]
- Taylor, S.C.; Berkelman, T.; Yadav, G.; Hammond, M. A defined methodology for reliable quantification of Western blot data. Mol. Biotechnol. 2013, 55, 217–226. [Google Scholar] [CrossRef]
- Bünger, S.; Roblick, U.J.; Habermann, J.K. Comparison of five commercial extraction kits for subsequent membrane protein profiling. Cytotechnology 2009, 61, 153–159. [Google Scholar] [CrossRef]
- Hensley, C.B.; Bradley, M.E.; Mircheff, A.K. Subcellular distribution of Na+/H+ antiport activity in rat renal cortex. Kidney Int. 1990, 37, 707–716. [Google Scholar] [CrossRef]
- Hensley, C.B.; Mircheff, A.K. Complex subcellular distribution of sodium-dependent amino acid transport systems in kidney cortex and LLC-PK1/Cl4 cells. Kidney Int. 1994, 45, 110–122. [Google Scholar] [CrossRef][Green Version]
- Keller, T.; Egenberger, B.; Gorboulev, V.; Bernhard, F.; Uzelac, Z.; Gorbunov, D.; Wirth, C.; Koppatz, S.; Dotsch, V.; Hunte, C.; et al. The large extracellular loop of organic cation transporter 1 influences substrate affinity and is pivotal for oligomerization. J. Biol. Chem. 2011, 286, 37874–37886. [Google Scholar] [CrossRef] [PubMed]
- Brast, S.; Grabner, A.; Sucic, S.; Sitte, H.H.; Hermann, E.; Pavenstädt, H.; Schlatter, E.; Ciarimboli, G. The cysteines of the extracellular loop are crucial for trafficking of human organic cation transporter 2 to the plasma membrane and are involved in oligomerization. FASEB J. 2011, 26, 976–986. [Google Scholar] [CrossRef]
- Zamek-Gliszczynski, M.J.; Lee, C.A.; Poirier, A.; Bentz, J.; Chu, X.; Ellens, H.; Ishikawa, T.; Jamei, M.; Kalvass, J.C.; Nagar, S.; et al. ITC recommendations for transporter kinetic parameter estimation and translational modeling of transport-mediated PK and DDIs in humans. Clin. Pharmacol. Ther. 2013, 94, 64–79. [Google Scholar] [CrossRef]
- Poirier, A.; Lavé, T.; Portmann, R.; Brun, M.-E.; Senner, F.; Kansy, M.; Grimm, H.-P.; Funk, C. Design, data analysis, and simulation of in vitro drug transport kinetic experiments using a mechanistic in vitro model. Drug Metab. Dispos. 2008, 36, 2434–2444. [Google Scholar] [CrossRef] [PubMed]
- Budiman, T.; Bamberg, E.; Koepsell, H.; Nagel, G. Mechanism of electrogenic cation transport by the cloned organic cation transporter 2 from rat. J. Biol. Chem. 2000, 275, 29413–29420. [Google Scholar] [CrossRef]
- Kumar, V.; Yin, J.; Billington, S.; Prasad, B.; Brown, C.D.A.; Wang, J.; Unadkat, J.D. The importance of incorporating OCT2 plasma membrane expression and membrane potential in IVIVE of metformin renal secretory clearance. Drug Metab. Dispos. 2018, 46, 1441–1445. [Google Scholar] [CrossRef]
- Yin, J.; Duan, H.; Wang, J. Impact of substrate-dependent inhibition on renal organic cation transporters hOCT2 and hMATE1/2-K-mediated drug transport and intracellular accumulation. J. Pharmacol. Exp. Ther. 2016, 359, 401–410. [Google Scholar] [CrossRef]
- Martinez-Guerrero, L.J.; Evans, K.K.; Dantzler, W.H.; Wright, S.H. The multidrug transporter MATE1 sequesters OCs within an intracellular compartment that has no influence on OC secretion in renal proximal tubules. Am. J. Physiol. Renal Physiol. 2016, 310, F57–F67. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dangprapai, Y.; Wright, S.H. Interaction of H+ with the extracellular and intracellular aspects of hMATE1. Am. J. Physiol. Renal Physiol. 2011, 301, F520–F528. [Google Scholar] [CrossRef]
- Wright, S.H.; Wunz, T.M. Mechanism of cis- and trans-substrate interactions at the tetraethylammonium/H+ exchanger of rabbit renal brush-border membrane vesicles. J. Biol. Chem. 1988, 263, 19494–19497. [Google Scholar] [CrossRef]
- Astorga, B.; Ekins, S.; Morales, M.; Wright, S.H. Molecular determinants of ligand selectivity for the human Multidrug And Toxin Extrusion proteins, MATE1 and MATE2-K. J. Pharmacol. Exp. Ther. 2012, 341, 743–755. [Google Scholar] [CrossRef]
- Meyer zu Schwabedissen, H.E.; Verstuyft, C.; Kroemer, H.K.; Becquemont, L.; Kim, R.B. Human multidrug and toxin extrusion 1 (MATE1/SLC47A1) transporter: Functional characterization, interaction with OCT2 (SLC22A2), and single nucleotide polymorphisms. Am. J. Physiol. Renal Physiol. 2010, 298, F997–F1005. [Google Scholar] [CrossRef]
- Wright, S.H.; Wunz, T.M. Transport of tetraethylammonium by rabbit renal brush-border and basolateral membrane vesicles. Am. J. Physiol. Renal Physiol. 1987, 253, F1040–F1050. [Google Scholar] [CrossRef]
- Masuda, S.; Terada, T.; Yonezawa, A.; Tanihara, Y.; Kishimoto, K.; Katsura, T.; Ogawa, O.; Inui, K.I. Identification and functional characterization of a new human kidney-specific H+/organic cation antiporter, Kidney-Specific Multidrug and Toxin Extrusion 2. J. Am. Soc. Nephrol. 2006, 17, 2127–2135. [Google Scholar] [CrossRef]
- Terada, T.; Masuda, S.; Asaka, J.I.; Tsuda, M.; Katsura, T.; Inui, K.I. Molecular cloning, functional characterization and tissue distribution of rat H(+)/organic cation antiporter MATE1. Pharm. Res. 2006, 23, 1696–1701. [Google Scholar] [CrossRef]
- Dantzler, W.H.; Brokl, O.; Wright, S.H. Brush-border TEA transport in intact proximal tubules and isolated membrane vesicles. Am. J. Physiol. Renal Physiol. 1989, 256, F290–F297. [Google Scholar] [CrossRef]
- Brasen, J.C.; Burford, J.L.; McDonough, A.A.; Holstein-Rathlou, N.-H.; Peti-Peterdi, J. Local pH domains regulate NHE3-mediated Na+ reabsorption in the renal proximal tubule. Am. J. Physiol. Renal Physiol. 2014, 307, F1249–F1262. [Google Scholar] [CrossRef]
- Roos, A.; Boron, W.F. Intracellular pH. Physiol. Rev. 1981, 61, 296–434. [Google Scholar] [CrossRef]
- Martinez Guerrero, L.J.; Zhang, X.; Zorn, K.M.; Ekins, S.; Wright, S.H. Cationic compounds with SARS-CoV-2 antiviral activity and their interaction with OCT/MATE secretory transporters. J. Pharmacol. Exp. Ther. 2021, 379, 96–107. [Google Scholar] [CrossRef]
- He, X.; Szewczyk, P.; Karyakin, A.; Evin, M.; Chang, G. Structure of a cation-bound Multidrug and Toxin Compound Extrusion (MATE) transporter. Nature 2010, 467, 991–994. [Google Scholar] [CrossRef]
- Reddy, V.S.; Shlykov, M.A.; Castillo, R.; Sun, E.I.; Saier, M.H., Jr. The major facilitator superfamily (MFS) revisited. FEBS J. 2012, 279, 2022–2035. [Google Scholar] [CrossRef]
- Pelis, R.M.; Zhang, X.; Dangprapai, Y.; Wright, S.H. Cysteine accessibility in the hydrophilic cleft of human organic cation transporter 2. J. Biol. Chem. 2006, 281, 35272–35280. [Google Scholar] [CrossRef]
- Zhang, X.; He, X.; Baker, J.; Tama, F.; Chang, G.; Wright, S.H. Twelve transmembrane helices form the functional core of mammalian MATE1 (Multidrug and Toxin Extruder 1) protein. J. Biol. Chem. 2012, 287, 27971–27982. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | MATE1 | OCT2 | ||
|---|---|---|---|---|
| Jmax (pmol cm−2 min−1) | Kt (µM) | Jmax (pmol cm−2 min−1) | Kt (µM) | |
| MPP | 241 ± 84.9 (n = 4) | 66.0 ± 6.8 | 58.5 ± 6.6 (n = 3) | 17.5 ± 2.1 |
| Atenolol | 398 ± 191 (n = 4) | 354 ± 95.9 | ND | ND |
| Metformin | 1802 ± 1147 (n = 4) | 664 ± 302 | 509 ± 39.0 (n = 2) | 427 ± 48.0 |
| Substrate | MATE1 | OCT2 | ||
|---|---|---|---|---|
| Abundance (fmol cm−2) | TOR (s−1) | Abundance (fmol cm−2) | TOR (s−1) | |
| 21.9 ± 18.1 | 154 ± 70.7 | |||
| MPP | 297 ± 201 | 7.1 ± 2.4 | ||
| Atenolol | 463 ± 313 | ND | ||
| Metformin | 1894 ± 1282 | 62.2 ± 20.5 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Wright, S.H. Transport Turnover Rates for Human OCT2 and MATE1 Expressed in Chinese Hamster Ovary Cells. Int. J. Mol. Sci. 2022, 23, 1472. https://doi.org/10.3390/ijms23031472
Zhang X, Wright SH. Transport Turnover Rates for Human OCT2 and MATE1 Expressed in Chinese Hamster Ovary Cells. International Journal of Molecular Sciences. 2022; 23(3):1472. https://doi.org/10.3390/ijms23031472
Chicago/Turabian StyleZhang, Xiaohong, and Stephen H. Wright. 2022. "Transport Turnover Rates for Human OCT2 and MATE1 Expressed in Chinese Hamster Ovary Cells" International Journal of Molecular Sciences 23, no. 3: 1472. https://doi.org/10.3390/ijms23031472
APA StyleZhang, X., & Wright, S. H. (2022). Transport Turnover Rates for Human OCT2 and MATE1 Expressed in Chinese Hamster Ovary Cells. International Journal of Molecular Sciences, 23(3), 1472. https://doi.org/10.3390/ijms23031472