
The Transmembrane Protease TMPRSS2 as a Therapeutic Target for COVID-19 Treatment


Abstract
1. Type II Transmembrane Serine Proteases
2. Basic Features of TMPRSS2
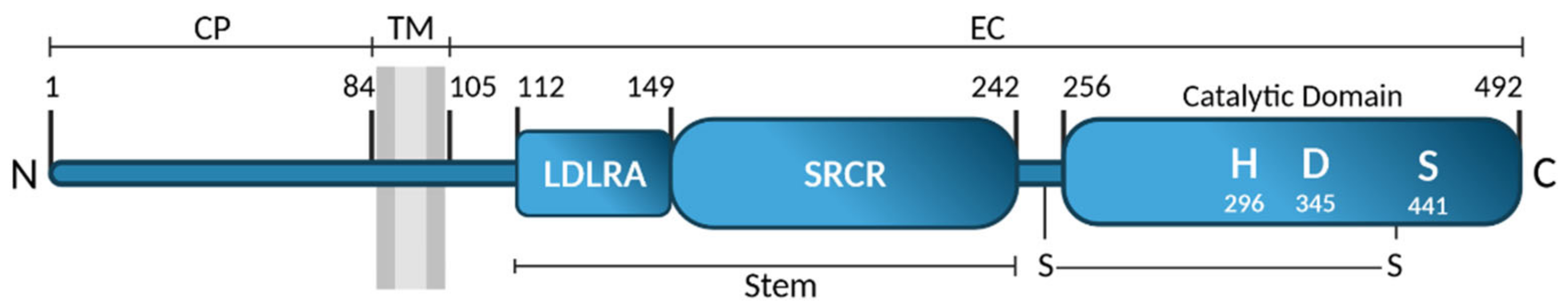
2.1. Identification and Structure
2.2. Expression
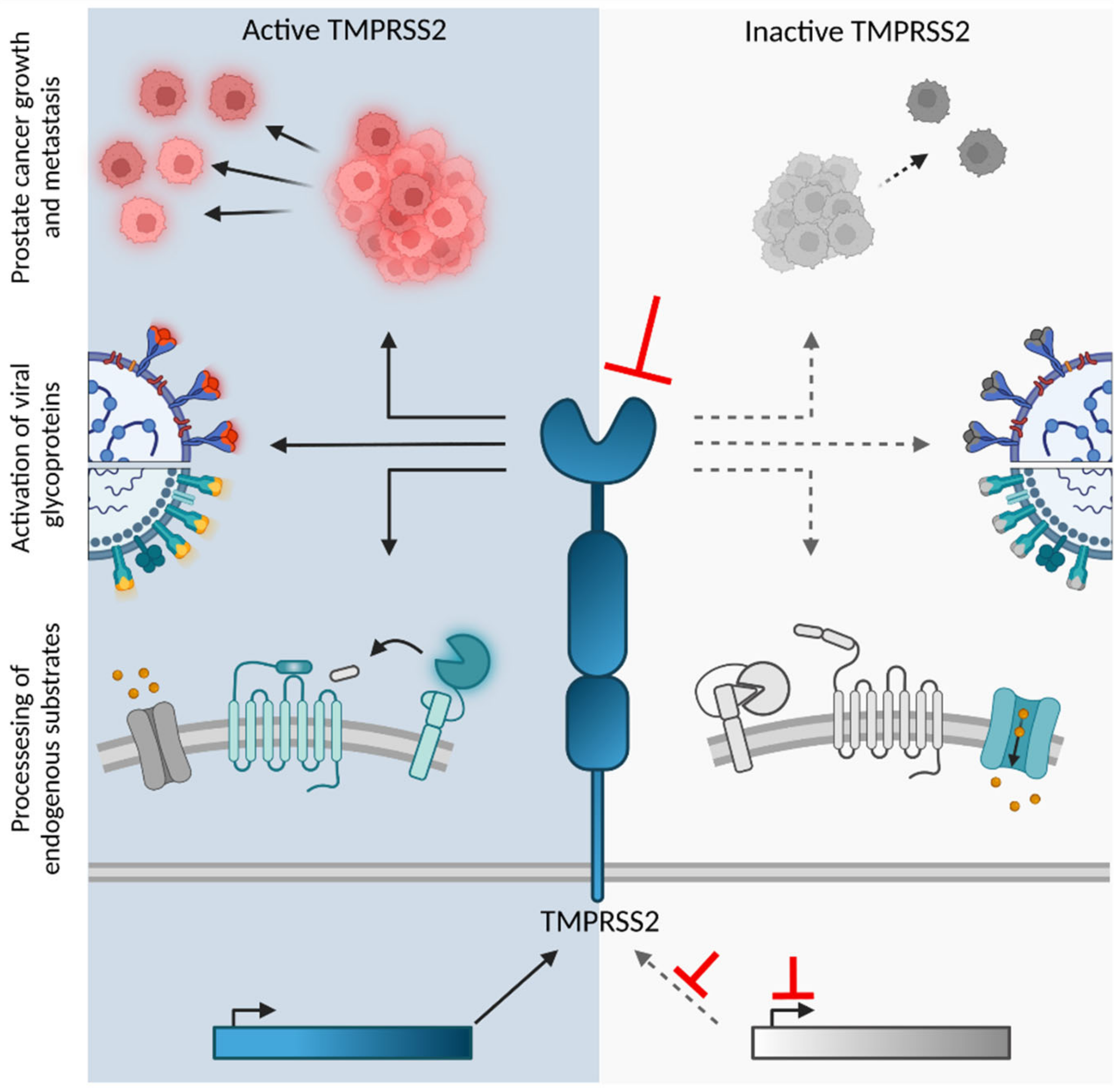
2.3. Endogenous Substrates
3. Role of TMPRSS2 in Viral Entry
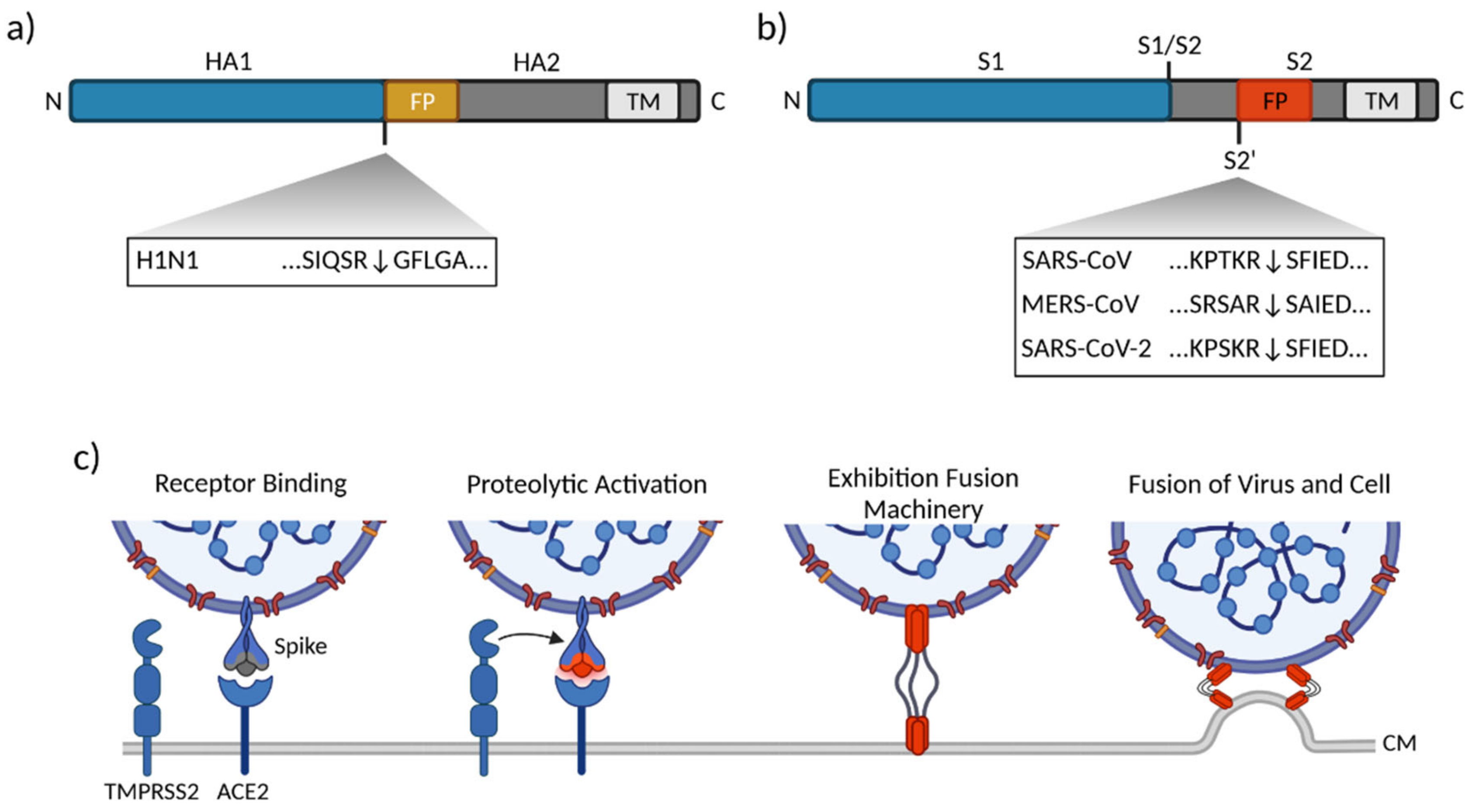
3.1. Influenza Virus
3.2. Human Coronaviruses
3.2.1. SARS-CoV
3.2.2. MERS-CoV
3.2.3. SARS-CoV-2
3.2.4. Common Cold Coronaviruses
3.3. Parainfluenza, Metapneumovirus and Sendaivirus
4. Inhibition of TMPRSS2 Function
4.1. Inhibitors of TMPRSS2 Expression
4.2. Inhibitors of TMPRSS2 Activity
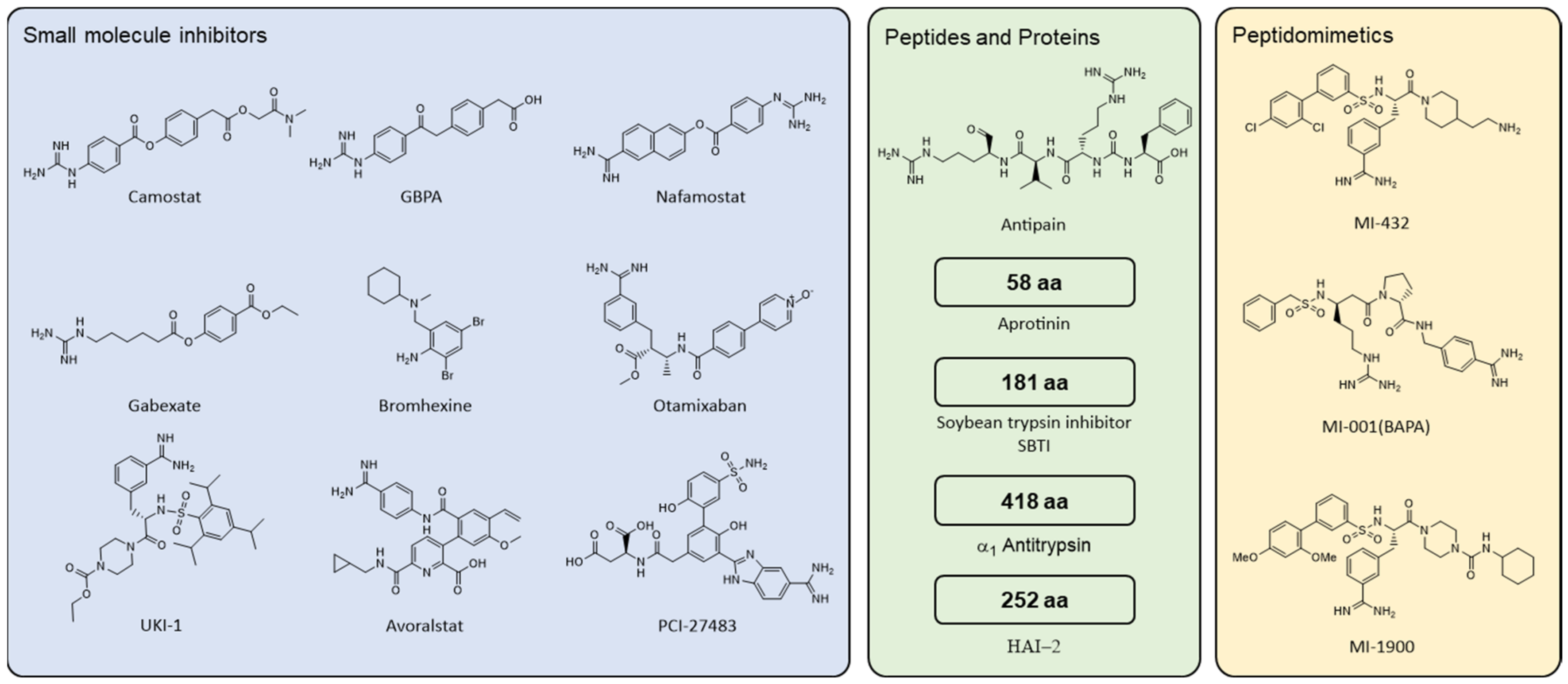
4.2.1. Small Molecule Compounds
4.2.2. Peptides and Proteins
4.2.3. Peptidomimetics

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antiviral Activity | ||||||
|---|---|---|---|---|---|---|
| Compound | Anti-TMPRSS2 | Influenza | SARS-CoV | MERS-CoV | SARS-CoV-2 | |
| Small molecule | Camostat | ✓ [120,123,124,125,126,127] | ✓ [118] | ✓ [74,96,117] | ✓ [76,77,80,100, 113114] | ✓ [74,83,97,117,119,120,121,122,123,124,125] |
| GBPA (FOY251) | ✓ [83,127] | n.a. | n.a. | n.a. | ✓ [83] | |
| Nafamostat | ✓ [125,126,127] | n.a. | ✓ [117] | ✓ [97,116,117] | ✓ [86,100,113,115,116, 118,119] | |
| Gabexate | ✓ [125,127] | ✓/🗴 [118,139] | ✓/🗴 [92,117] | ✓/🗴 [116,117,125] | ~/🗴 [117,125,134] | |
| Bromhexine | ✓/🗴 [14,125] | n.a. | n.a. | n.a. | ✓ [119] | |
| 0591-5323, 4401-0077, 4554-5138, 8008-1235 | ✓ [14] | n.a. | n.a. | n.a. | n.a. | |
| Otamixaban | ✓ [125] | n.a. | n.a. | n.a. | ~ [125] | |
| UKI-1 | ✓ [125] | n.a. | n.a. | n.a. | n.a. | |
| Avoralstat | ✓ [120] | n.a. | n.a. | n.a. | ✓ [120] | |
| PCI-27483 | ✓ [120] | n.a. | n.a. | n.a. | ✓ [120] | |
| Peptide & Protein | Antipain | ✓ [120] | n.a. | n.a. | n.a. | ✓ [120] |
| Aprotinin | n.a. | ✓ [118,143] | n.a. | n.a. | ✓ [13,119,149] | |
| SBTI | ✓ [120] | 🗴 [143] | n.a. | n.a. | ✓ [120] | |
| α1AT | ✓ [123,124] | n.a. | n.a. | n.a. | ✓/🗴 [123,124,149] | |
| HAI-2 | ✓ [135] | ✓ [153] | n.a. | n.a. | n.a. | |
| Peptidomimetic | MI-001 (BAPA) | ✓ [174] | ✓ [57,175] | n.a. | n.a. | n.a. |
| MI-432 | ✓ [174] | ✓ [174,175] | n.a. | n.a. | ✓ [13] | |
| MI-1900 | ✓ [174] | n.a. | n.a. | n.a. | ✓ [13] | |
5. Clinical Evaluation of TMPRSS2 Inhibitors in COVID-19 Patients
| Study Title | Identifier, Status | Inclusion Criteria | Intervention | Primary Outcome |
|---|---|---|---|---|
| A Trial Looking at the Use of Camostat to Reduce Progression of Symptoms of Coronavirus (COVID-19) in People Who Have Tested Positive. (SPIKE-1) | NCT04455815, Active, not recruiting | Confirmed SARS-CoV-2 infection, symptomatic | CM orally, 200 mg q.i.d., 14 d | Safety and efficacy of CM |
| Camostat Mesylate in COVID-19 Outpatients | NCT04353284, Completed | Confirmed SARS-CoV-2 infection, enrolled within 3 d, symptomatic | CM orally, 200 mg q.i.d., 7 d | Change in SARS-CoV-2 viral load |
| Camostat Efficacy vs. Placebo for Outpatient Treatment of COVID-19 (CAMELOT) | NCT04583592, Completed | Confirmed SARS-CoV-2 infection, symptomatic, risk for severe illness | CM orally, 200 mg q.i.d., 14 d | Disease progression (hospital or death within 28 d) |
| Camostat Mesilate Treating Patients With Hospitalized Patients With COVID-19 (RECOVER) | NCT04470544, Recruiting | Confirmed SARS-CoV-2 infection, hospitalized | CM, 200 mg q.i.d. | Proportion of patients alive and free from respiratory failure within 28 d |
| The Utility of Camostat Mesylate in Patients With COVID-19 Associated Coagulopathy (CAC) and Cardiovascular Complications | NCT04435015, Not yet recruiting | Positive SARS-CoV-2 test, COVID-19 associated coagulopathy/cardiac complication | CM orally, 200 mg, t.i.d. | Percent change in plasma D-Dimer within 7 d |
| Evaluation of Efficacy and Safety of Camostat Mesylate for the Treatment of SARS-CoV-2 Infection—COVID-19 in Ambulatory Adult Patients (CAMOVID) | NCT04608266, Recruiting | PCR confirmed SARS-CoV-2 infection, symptomatic, risk of severe COVID-19 | CM orally, 200 mg, t.i.d., 14 d | Hospitalization for COVID-19 deterioration or death |
| Oral Camostat Compared With Standard Supportive Care in Mild-Moderate COVID-19 Patients (COPS-2003) | NCT04524663, Completed | Confirmed SARS-CoV-2 infection, symptomatic, mild to moderate COVID-19 | CM orally, 10 d | Time until cessation of shedding of SARS-CoV-2 |
| The Potential of Oral Camostat in Early COVID-19 Disease in an Ambulatory Setting to Reduce Viral Load and Disease Burden | NCT04625114, Recruiting | PCR confirmed SARS-CoV-2 infection, signs or symptoms of COVID-19, outpatient | CM orally, 100 mg t.i.d., 5–10 d | Reduction of viral load (RT qpCR) |
| The DAWN Camostat Trial for Ambulatory COVID-19 Patients | NCT04730206, Recruiting | Positive SARS-CoV-2 antigen test, symptomatic < 5 d, outpatient | CM orally, 200 mg q.i.d., 7 d | Time to recovery, unplanned hospitalization or death |
| The Impact of Camostat Mesilate on COVID-19 Infection (CamoCO-19) | NCT04321096, [187] | PCR confirmed SARS-CoV-2 infection, hospitalized, symptomatic < 48 h | CM orally, 200 mg t.i.d., 5 d | Time to clinical improvement |
| Multiple-dose Study of FOY-305 in Japanese Healthy Adult Male Subjects | NCT04451083, Completed | Healthy adults | CM orally, multiple doses q.i.d. | Safety and tolerability |
| COVID-19 Outpatient Pragmatic Platform Study (COPPS)-Camostat Sub-Protocol (COPPS) | NCT04662073, Active not recruiting | Confirmed SARS-CoV-2 infection, enrolled within 72 h, mild-moderate COVID-19 | CM orally, 200 mg q.i.d. | Change in SARS-CoV-2 shedding |
| Reconvalescent Plasma/Camostat Mesylate Early in SARS-CoV-2 Q-PCR (COVID-19) Positive High-risk Individuals (RES-Q-HR) | NCT04681430, Recruiting | PCR confirmed SARS-CoV-2 infection, enrolled within 3 d, symptomatic COVID-19 | CM orally, 200 mg t.i.d., 7 d | Clinical status improvement |
| A Study of FOY-305 in Patients With SARS-CoV-2 Infection (COVID-19) | NCT04657497, Completed | Confirmed SARS-CoV-2 infection, symptomatic, enrolled within 5 d after symptom onset | CM orally, 600 mg q.i.d., 14 d | Positivity of SARS-CoV-2 PCR |
| Safety and Pharmacokinetics Evaluation Study According to the Dose of Camostat Mesylate in Healthy Volunteers | NCT04782505, Recruiting | Healthy adults | CM orally, 100–300 mg | Pharmacokinetics |
| COVID-19 Outpatient Pragmatic Platform Study (COPPS)—Master Protocol | NCT04662086, Recruiting | Confirmed SARS-CoV-2 infection, enrolled within <72 h, mild-moderate COVID-19 | CM orally, 200 mg q.i.d. | Change in SARS-CoV-2 shedding Time to sustained symptom resolution |
| Novel Agents for Treatment of High-risk COVID-19 Positive Patients | NCT04374019, Recruiting | Confirmed SARS-CoV-2 infection, high risk feature for clinical deterioration | CM orally, 200 mg t.i.d., 14 d | Clinical deterioration |
| ACTIV-2: A Study for Outpatients With COVID-19 | NCT04518410, Recruiting | Confirmed SARS-CoV-2 infection symptomatic, enrolled within 7 d | CM orally, 200 mg t.i.d., 7 d | COVID-19 symptom duration and SARS-CoV-2 viral load |
| Efficacy and Safety of DWJ1248 in Patients With Mild to Moderate COVID-19 Compared to the Placebo | NCT04521296, Active, not recruiting | Confirmed SARS-CoV-2 infection, mild or moderate COVID-19 | CM orally, 200 mg t.i.d. | SARS-CoV-2 viral load, clinical improvement |
| A Study of DWJ1248 in Prevention of COVID-19 Infection After the Exposure of SARS-CoV-2 | NCT04721535, Not yet recruiting | Contact with SARS-CoV-2 positive patient, asymptomatic and PCR negative subjects | CM orally, 200 mg 1x daily | SARS-CoV-2 positivity (RT-PCR) |
| Efficacy and Safety of DWJ1248 With Remdesivir in Severe COVID-19 Patients | NCT04713176, Recruiting | PCR confirmed SARS-CoV-2 infection, enrolled within 10 d | CM orally, 200 mg 1x daily, up to 14 d Remdesivir | Mortality rate or ECMO patients |
| Clinical Efficacy of Nafamostat Mesylate for COVID-19 Pneumonia | NCT04418128, Not yet recruiting | Confirmed SARS-CoV-2 infection, hospitalized, pneumonia within 72 h, no oxygenation | NM i.v. 0.1–0.2 mg/kg/h over 24 h, up to 14 d | Clinical improvement |
| Efficacy of Nafamostat in COVID-19 Patients (RACONA Study) (RACONA) | NCT04352400, Recruiting | Confirmed SARS-CoV-2 infection, hospitalized | NM i.v. | Time to clinical improvement |
| Oral Nafamostat in Healthy Volunteers | NCT04406415, Completed | Healthy adults | NM orally, 10–200 mg t.i.d., 5 d | Safety and tolerability |
| Efficacy and Safety Evaluation of Treatment Regimens in Adult COVID-19 Patients in Senegal (SEN-CoV-Fadj) | NCT04390594, Recruiting | Confirmed SARS-CoV-2 infection, pneumonia within 72 h of symptom onset | NM i.v. 0.1–0.2 mg/kg/h, up to 14 d | SARS-CoV-2 viral load |
| Australasian COVID-19 Trial (ASCOT) ADAptive Platform Trial (ASCOT ADAPT) | NCT04483960, Recruiting | Confirmed SARS-CoV-2 infection, symptomatic, hospitalized | NM i.v. 0.2 mg/kg/h, 7 d | Mortality rate or requirement of ICU support |
| DEFINE—Evaluating Therapies for COVID-19 (DEFINE) | NCT04473053, Active, not recruiting | Confirmed SARS-CoV-2 infection, symptomatic | NM 0.2 mg/kg/h, 7 d | Safety and tolerability |
| A Study Evaluating the Efficacy and Safety of CKD-314 (Nafabelltan) in Hospitalized Adult Patients Diagnosed With COVID-19 Pneumonia | NCT04623021, [189] | Confirmed SARS-CoV-2 infection, pneumonia | NM i.v. 4.8 mg/kg/day, 10 d | Time to clinical improvement |
| A Study Evaluating the Efficacy and Safety of CKD-314 in Hospitalized Adult Patients Diagnosed With COVID-19 Pneumonia | NCT04628143, Completed | Confirmed SARS-CoV-2 infection, pneumonia, hospitalized | NM i.v. | Time to clinical improvement |
| Phase 3 Clinical Trial to Evaluate the Efficacy and Safety of CKD-314 | NCT04871646, Recruiting | Confirmed SARS-CoV-2 infection, pneumonia | NM i.v. | Time to recovery |
| Use of Bromhexine and Hydroxychloroquine for Treatment of COVID-19 Pneumonia | NCT04355026, Recruiting | PCR confirmed SARS-CoV-2 infection, hospitalized | BHH orally, 16 mg t.i.d. | Duration of hospitalization and disease |
| BromhexIne And Spironolactone For CoronaVirUs Infection Requiring HospiTalization (BISCUIT) | NCT04424134, Recruiting | Confirmed SARS-CoV-2 infection, pneumonia | BHH, 8 mg q.i.d., 10 d Spironolactone 50 mg, 10 d | Change from baseline in clinical score |
| Clinical Trial With N-acetylcysteine and Bromhexine for COVID-19 | NCT04928495, not yet recruiting | Clinical signs and symptoms of COVID-19 | BHH 32 mg/day 10 d N-acetylcysteine 1800 mg/day, 10 d | Time of recovery |
| Evaluating the Efficacy and Safety of Bromhexine Hydrochloride Tablets Combined with Standard Treatment/Standard Treatment in Patients With Suspected and Mild Novel Coronavirus Pneumonia (COVID-19) | NCT04273763, [97] | Confirmed or suspected mild or moderate COVID-19 | BHH orally, 32 mg t.i.d., 14 d | Time of clinical recovery and deterioration rate |
| Prevention of Infection and Incidence of COVID-19 in Medical Personnel Assisting Patients with New Coronavirus Disease | NCT04405999, [193], preprint | Medical personnel at risk for COVID-19, negative PCR for SARS-CoV-2 infection | BHH | SARS-CoV-2 viral load |
| Study on the Pharmacokinetics of Bromine Hexane Hydrochloride Tablets in Healthy Adults | NCT04672707, not yet recruiting | Healthy adults | BHH orally, 32–80 mg t.i.d., 2 d | Pharmacokinetics |
| Effect of bromhexine on clinical outcomes and mortality in COVID-19 patients: A randomized clinical trial | IRCT202003117046797N4, [190] | Diagnosed COVID-19 pneumonia | BHH orally, 8 mg t.i.d., 14 d | Improvement in rate of ICU admission, intubation and ventilation, 28-day mortality |
| Effect of bromhexine in hospitalized patients with COVID-19 | [191] | PCR confirmed SARS-CoV-2 infection, hospitalized | BHH orally, 8 mg q.i.d., 14 d | Clinical improvement |
| An Open Non-comparative Study of the Efficacy and Safety of Aprotinin in Patients Hospitalized with COVID-19 | NCT04527133, active, not recruiting | PCR confirmed SARS-CoV-2 infection, moderate to severe COVID-19 | Stage 1: Aprotinin i.v., 1,000,000 KIU IV, 3 d Stage 2: Aprotinin inhaled, 625 KIU q.i.d., 5 d or: Aprotinin i.v. 1,000,000 KIU IV 1x daily, 5 d + Favirapir | Time to SARS-CoV-2 negativity, CRP and D-Dimer normalization |
| Study to Evaluate the Safety and Efficacy of Prolastin in Hospitalized Subjects with COVID-19 | NCT04495101, completed | PCR confirmed SARS-CoV-2 infection, symptomatic, hospitalized | α1AT i.v., 120 mg/kg, day 1 and day 8 | Percentage of subjects dying and dependent on ventilation |
| Trial of Alpha One Antitrypsin Inhalation in Treating Patient with Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) | NCT04385836, Recruiting | PCR confirmed SARS-CoV-2 infection, hospitalized | α1AT inhalative, b.i.d., 5 d | Clinical improvement |
| Study to Evaluate the Safety and Efficacy of Liquid Alpha1-Proteinase Inhibitor (Human) in Hospitalized Participants with Coronavirus Disease (COVID-19) | NCT04547140, recruiting | PCR confirmed SARS-CoV-2 infection, symptomatic, hospitalized | α1AT i.v., 120 mg/kg, day 1 and day 8 | Percentage of subjects dying and dependent on ventilation |
| A randomized double-blind placebo-controlled pilot trial of intravenous plasma-purified alpha-1 antitrypsin for severe COVID-19 illness. | EudraCT 2020-001391-15, Completed | Confirmed COVID-19 infection, moderate ARDS | α1AT, i.v. 120 mg/kg weekly, 28 d | Biological activity of A1AT as anti-inflammatory therapy |
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Turk, B. Targeting proteases: Successes, failures and future prospects. Nat. Rev. Drug Discov. 2006, 5, 785–799. [Google Scholar] [CrossRef]
- López-Otín, C.; Bond, J.S. Proteases: Multifunctional enzymes in life and disease. J. Biol. Chem. 2008, 283, 30433–30437. [Google Scholar] [CrossRef] [PubMed]
- Hedstrom, L. Serine protease mechanism and specificity. Chem. Rev. 2002, 102, 4501–4523. [Google Scholar] [CrossRef] [PubMed]
- Bugge, T.H.; Antalis, T.M.; Wu, Q. Type II transmembrane serine proteases. J. Biol. Chem. 2009, 284, 23177–23181. [Google Scholar] [CrossRef]
- Barré, O.; Dufour, A.; Eckhard, U.; Kappelhoff, R.; Béliveau, F.; Leduc, R.; Overall, C.M. Cleavage specificity analysis of six type II transmembrane serine proteases (TTSPs) using PICS with proteome-derived peptide libraries. PLoS ONE 2014, 9, e105984. [Google Scholar] [CrossRef] [PubMed]
- Paoloni-Giacobino, A.; Chen, H.; Peitsch, M.C.; Rossier, C.; Antonarakis, S.E. Cloning of the TMPRSS2 gene, which encodes a novel serine protease with transmembrane, LDLRA, and SRCR domains and maps to 21q22.3. Genomics 1997, 44, 309–320. [Google Scholar] [CrossRef]
- Böttcher-Friebertshauser, E.; Stein, D.A.; Klenk, H.-D.; Garten, W. Inhibition of Influenza Virus Infection in Human Airway Cell Cultures by an Antisense Peptide-Conjugated Morpholino Oligomer Targeting the Hemagglutinin-Activating Protease TMPRSS2. J. Virol. 2011, 85, 1554–1562. [Google Scholar] [CrossRef]
- Inouye, K.; Tomoishi, M.; Yasumoto, M.; Miyake, Y.; Kojima, K.; Tsuzuki, S.; Fushiki, T. Roles of CUB and LDL receptor class A domain repeats of a transmembrane serine protease matriptase in its zymogen activation. J. Biochem. 2013, 153, 51–61. [Google Scholar] [CrossRef]
- Silvestri, L.; Guillem, F.; Pagani, A.; Nai, A.; Oudin, C.; Silva, M.; Toutain, F.; Kannengiesser, C.; Beaumont, C.; Camaschella, C.; et al. Molecular mechanisms of the defective hepcidin inhibition in TMPRSS6 mutations associated with iron-refractory iron deficiency anemia. Blood 2009, 113, 5605–5608. [Google Scholar] [CrossRef] [PubMed]
- Yap, N.V.L.; Whelan, F.J.; Bowdish, D.M.E.; Golding, G.B. The evolution of the scavenger receptor cysteine-rich domain of the class A scavenger receptors. Front. Immunol. 2015, 6, 1–9. [Google Scholar] [CrossRef]
- Hohenester, E.; Sasaki, T.; Timpl, R. Crystal structure of a scavenger receptor cysteine-rich domain sheds light on an ancient superfamily. Nat. Struct. Biol. 1999, 6, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Guipponi, M.; Antonarakis, S.E.; Scott, H.S. TMPRSS3, a type II transmembrane serine protease mutated in non-syndromic autosomal recessive deafness. Front. Biosci. 2008, 13, 1557–1567. [Google Scholar] [CrossRef] [PubMed]
- Bestle, D.; Heindl, M.R.; Limburg, H.; van Lam van, T.; Pilgram, O.; Moulton, H.; Stein, D.A.; Hardes, K.; Eickmann, M.; Dolnik, O.; et al. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci. Alliance 2020, 3, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lucas, J.M.; Heinlein, C.; Kim, T.; Hernandez, S.A.; Malik, M.S.; True, L.D.; Morrissey, C.; Corey, E.; Montgomery, B.; Mostaghel, E.; et al. The androgen-regulated protease TMPRSS2 activates a proteolytic cascade involving components of the tumor microenvironment and promotes prostate cancer metastasis. Cancer Discov. 2014, 4, 1310–1325. [Google Scholar] [CrossRef] [PubMed]
- Afar, D.E.H.; Vivanco, I.; Hubert, R.S.; Kuo, J.; Chen, E.; Saffran, D.C.; Raitano, A.B.; Jakobovits, A. Catalytic cleavage of the androgen-regulated TMPRSS2 protease results in its secretion by prostate and prostate cancer epithelia. Cancer Res. 2001, 61, 1686–1692. [Google Scholar] [PubMed]
- Kim, T.S.; Heinlein, C.; Hackman, R.C.; Nelson, P.S. Phenotypic Analysis of Mice Lacking the Tmprss2 -Encoded Protease. Mol. Cell. Biol. 2006, 26, 965–975. [Google Scholar] [CrossRef]
- Chen, Y.W.; Lee, M.S.; Lucht, A.; Chou, F.P.; Huang, W.; Havighurst, T.C.; Kim, K.; Wang, J.K.; Antalis, T.M.; Johnson, M.D.; et al. TMPRSS2, a serine protease expressed in the prostate on the apical surface of luminal epithelial cells and released into semen in prostasomes, is misregulated in prostate cancer cells. Am. J. Pathol. 2010, 176, 2986–2996. [Google Scholar] [CrossRef]
- Jacquinet, E.; Rao, N.V.; Rao, G.V.; Zhengming, W.; Albertine, K.H.; Hoidal, J.R. Cloning and characterization of the cDNA and gene for human epitheliasin. Eur. J. Biochem. 2001, 268, 2687–2699. [Google Scholar] [CrossRef]
- Lin, B.; Ferguson, C.; White, J.T.; Wang, S.; Vessella, R.; True, L.D.; Hood, L.; Nelson, P.S. Prostate-localized and Androgen-regulated Expression of the Membrane-bound Serine Protease TMPRSS2. Cancer Res. 1999, 59, 4180–4184. [Google Scholar]
- Zmora, P.; Moldenhauer, A.S.; Hofmann-Winkler, H.; Pöhlmann, S. TMPRSS2 isoform 1 activates respiratory viruses and is expressed in viral target cells. PLoS ONE 2015, 10, e0138380. [Google Scholar] [CrossRef]
- Vaarala, M.H.; Porvari, K.S.; Kellokumpu, S.; Kyllönen, A.P.; Vihko, P.T. Expression of transmembrane serine protease TMPRSS2 in mouse and human tissues. J. Pathol. 2001, 193, 134–140. [Google Scholar] [CrossRef]
- Bertram, S.; Heurich, A.; Lavender, H.; Gierer, S.; Danisch, S.; Perin, P.; Lucas, J.M.; Nelson, P.S.; Pöhlmann, S.; Soilleux, E.J. Influenza and SARS-coronavirus activating proteases TMPRSS2 and HAT are expressed at multiple sites in human respiratory and gastrointestinal tracts. PLoS ONE 2012, 7, e0035876. [Google Scholar] [CrossRef] [PubMed]
- Lucas, J.M.; True, L.; Hawley, S.; Morrissey, C.; Vessella, R.; Nelson, P.S. The androgen-regulated type II serine protease TMPRSS2 is differentially expressed and mislocalized in prostate adenocarcinoma. J. Pathol. 2008, 215, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Vaarala, M.H.; Porvari, K.; Kyllönen, A.; Lukkarinen, O.; Vihko, P. The TMPRSS2 gene encoding transmembrane serine protease is overexpressed in a majority of prostate cancer patients: Detection of mutated TMPRSS2 form in a case of aggressive disease. Int. J. Cancer 2001, 94, 705–710. [Google Scholar] [CrossRef]
- Donaldson, S.H.; Hirsh, A.; Li, D.C.; Holloway, G.; Chao, J.; Boucher, R.C.; Gabriel, S.E. Regulation of the epithelial sodium channel by serine proteases in human airways. J. Biol. Chem. 2002, 277, 8338–8345. [Google Scholar] [CrossRef]
- Lukassen, S.; Chua, R.L.; Trefzer, T.; Kahn, N.C.; Schneider, M.A.; Muley, T.; Winter, H.; Meister, M.; Veith, C.; Boots, A.W.; et al. SARS -CoV-2 receptor ACE 2 and TMPRSS 2 are primarily expressed in bronchial transient secretory cells. EMBO J. 2020, 39, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sungnak, W.; Huang, N.; Bécavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020, 26, 681–687. [Google Scholar] [CrossRef]
- Mikkonen, L.; Pihlajamaa, P.; Sahu, B.; Zhang, F.P.; Jänne, O.A. Androgen receptor and androgen-dependent gene expression in lung. Mol. Cell. Endocrinol. 2010, 317, 14–24. [Google Scholar] [CrossRef]
- Li, F.; Han, M.; Dai, P.; Xu, W.; He, J.; Tao, X.; Wu, Y.; Tong, X.; Xia, X.; Guo, W.; et al. Distinct mechanisms for TMPRSS2 expression explain organ-specific inhibition of SARS-CoV-2 infection by enzalutamide. Nat. Commun. 2021, 12, 866. [Google Scholar] [CrossRef]
- Wilson, S.; Greer, B.; Hooper, J.; Zijlstra, A.; Walker, B.; Quigley, J.; Hawthorne, S. The membrane-anchored serine protease, TMPRSS2, activates PAR-2 in prostate cancer cells. Biochem. J. 2005, 388, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Mackie, E.J.; Pagel, C.N.; Smith, R.; De Niese, M.R.; Song, S.J.; Pike, R.N. Protease-activated receptors: A means of converting extracellular proteolysis into intracellular signals. IUBMB Life 2002, 53, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.J.; Huang, C.C.; Lin, H.Y.; Juan, C.P.; Lan, S.W.; Shyu, H.Y.; Wu, S.R.; Hsiao, P.W.; Huang, H.P.; Shun, C.T.; et al. Androgen-induced TMPRSS2 activates matriptase and promotes extracellular matrix degradation, prostate cancer cell invasion, tumor growth, and metastasis. Cancer Res. 2015, 75, 2949–2960. [Google Scholar] [CrossRef]
- Pawar, N.R.; Buzza, M.S.; Antalis, T.M. Membrane-anchored serine proteases and protease-activated receptor-2–mediated signaling: Co-conspirators in cancer progression. Cancer Res. 2019, 79, 301–310. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, M.R.; Derian, C.K.; Leturcq, D.; Baker, S.M.; Brunmark, A.; Ling, P.; Darrow, A.L.; Santulli, R.J.; Brass, L.F.; Andrade-Gordon, P. Characterization of protease-activated receptor-2 immunoreactivity in normal human tissues. J. Histochem. Cytochem. 1998, 46, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, A.L.; Chinni, C.; De Niese, M.R.; Blackhart, B.; Mackie, E.J. Expression of protease-activated receptor-2 during embryonic development. Dev. Dyn. 2000, 218, 465–471. [Google Scholar] [CrossRef]
- Cocks, T.M.; Fong, B.; Chow, J.M.; Anderson, G.P.; Frauman, A.G.; Goldie, R.G.; Henry, P.J.; Carr, M.J.; Hamilton, J.R.; Moffatt, J.D. A protective role for protease-activated receptros in the airways. Nature 1999, 398, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.; Mcconalogue, K.; Khitin, L.M.; Hollenberg, M.D.; Payan, D.G.; Böhm, S.K.; Bunnett, N.W. Luminal trypsin may regulate enterocytes through proteinase-activated receptor 2. Proc. Natl. Acad. Sci. USA 1997, 94, 8884–8889. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.A.; Xu, Y.; De Marzo, A.M.; Isaacs, J.T.; Denmeade, S.R. Prostate-Specific Antigen (PSA) Is Activated by KLK2 in Prostate Cancer Ex Vivo Models and in Prostate-Targeted PSA/KLK2 Double Transgenic Mice. Prostate 2010, 70, 788–796. [Google Scholar] [CrossRef]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2020, 19, 155–170. [Google Scholar] [CrossRef]
- Laporte, M.; Naesens, L. Airway proteases: An emerging drug target for influenza and other respiratory virus infections. Curr. Opin. Virol. 2017, 24, 16–24. [Google Scholar] [CrossRef]
- Bertram, S.; Glowacka, I.; Steffen, I.; Kühl, A.; Pöhlmann, S. Novel insights into proteolytic cleavage of influenza virus hemagglutinin. Rev. Med. Virol. 2010, 20, 298–310. [Google Scholar] [CrossRef]
- Bestle, D.; Limburg, H.; Kruhl, D.; Harbig, A.; Stein, D.A.; Moulton, H.; Matrosovich, M.; Abdelwhab, E.M.; Stech, J.; Böttcher-Friebertshäuser, E. Hemagglutinins of Avian Influenza Viruses Are Proteolytically Activated by TMPRSS2 in Human and Murine Airway Cells. J. Virol. 2021, 95, e0090621. [Google Scholar] [CrossRef]
- Böttcher, E.; Matrosovich, T.; Beyerle, M.; Klenk, H.-D.; Garten, W.; Matrosovich, M. Proteolytic Activation of Influenza Viruses by Serine Proteases TMPRSS2 and HAT from Human Airway Epithelium. J. Virol. 2006, 80, 9896–9898. [Google Scholar] [CrossRef]
- Chaipan, C.; Kobasa, D.; Bertram, S.; Glowacka, I.; Steffen, I.; Solomon Tsegaye, T.; Takeda, M.; Bugge, T.H.; Kim, S.; Park, Y.; et al. Proteolytic Activation of the 1918 Influenza Virus Hemagglutinin. J. Virol. 2009, 83, 3200–3211. [Google Scholar] [CrossRef]
- Galloway, S.E.; Reed, M.L.; Russell, C.J.; Steinhauer, D.A. Influenza HA Subtypes Demonstrate Divergent Phenotypes for Cleavage Activation and pH of Fusion: Implications for Host Range and Adaptation. PLoS Pathog. 2013, 9, e1003151. [Google Scholar] [CrossRef]
- Bertram, S.; Glowacka, I.; Blazejewska, P.; Soilleux, E.; Allen, P.; Danisch, S.; Steffen, I.; Choi, S.-Y.; Park, Y.; Schneider, H.; et al. TMPRSS2 and TMPRSS4 Facilitate Trypsin-Independent Spread of Influenza Virus in Caco-2 Cells. J. Virol. 2010, 84, 10016–10025. [Google Scholar] [CrossRef]
- Baron, J.; Tarnow, C.; Mayoli-Nüssle, D.; Schilling, E.; Meyer, D.; Hammami, M.; Schwalm, F.; Steinmetzer, T.; Guan, Y.; Garten, W.; et al. Matriptase, HAT, and TMPRSS2 Activate the Hemagglutinin of H9N2 Influenza A Viruses. J. Virol. 2013, 87, 1811–1820. [Google Scholar] [CrossRef]
- Kühn, N.; Bergmann, S.; Kösterke, N.; Lambertz, R.L.O.; Keppner, A.; van den Brand, J.M.A.; Pöhlmann, S.; Weiß, S.; Hummler, E.; Hatesuer, B.; et al. The Proteolytic Activation of (H3N2) Influenza A Virus Hemagglutinin Is Facilitated by Different Type II Transmembrane Serine Proteases. J. Virol. 2016, 90, 4298–4307. [Google Scholar] [CrossRef] [PubMed]
- Böttcher-Friebertshäuser, E.; Freuer, C.; Sielaff, F.; Schmidt, S.; Eickmann, M.; Uhlendorff, J.; Steinmetzer, T.; Klenk, H.-D.; Garten, W. Cleavage of Influenza Virus Hemagglutinin by Airway Proteases TMPRSS2 and HAT Differs in Subcellular Localization and Susceptibility to Protease Inhibitors. J. Virol. 2010, 84, 5605–5614. [Google Scholar] [CrossRef] [PubMed]
- Hatesuer, B.; Bertram, S.; Mehnert, N.; Bahgat, M.M.; Nelson, P.S.; Pöhlman, S.; Schughart, K. Tmprss2 Is Essential for Influenza H1N1 Virus Pathogenesis in Mice. PLoS Pathog. 2013, 9, e1003774. [Google Scholar] [CrossRef] [PubMed]
- Tarnow, C.; Engels, G.; Arendt, A.; Schwalm, F.; Sediri, H.; Preuss, A.; Nelson, P.S.; Garten, W.; Klenk, H.-D.; Gabriel, G.; et al. TMPRSS2 Is a Host Factor That Is Essential for Pneumotropism and Pathogenicity of H7N9 Influenza A Virus in Mice. J. Virol. 2014, 88, 4744–4751. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Ami, Y.; Nakajima, N.; Nakajima, K.; Kitazawa, M.; Anraku, M.; Takayama, I.; Sangsriratanakul, N.; Komura, M.; Sato, Y.; et al. TMPRSS2 Independency for Haemagglutinin Cleavage in Vivo Differentiates Influenza B Virus from Influenza A Virus. Sci. Rep. 2016, 6, 29430. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Ami, Y.; Tahara, M.; Kubota, T.; Anraku, M.; Abe, M.; Nakajima, N.; Sekizuka, T.; Shirato, K.; Suzaki, Y.; et al. The Host Protease TMPRSS2 Plays a Major Role in In Vivo Replication of Emerging H7N9 and Seasonal Influenza Viruses. J. Virol. 2014, 88, 5608–5616. [Google Scholar] [CrossRef] [PubMed]
- Lambertz, R.L.O.; Gerhauser, I.; Nehlmeier, I.; Leist, S.R.; Kollmus, H.; Pöhlmann, S.; Schughart, K. Tmprss2 knock-out mice are resistant to H10 influenza a virus pathogenesis. J. Gen. Virol. 2019, 100, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Sekizuka, T.; Ami, Y.; Nakajima, N.; Kitazawa, M.; Sato, Y.; Nakajima, K.; Anraku, M.; Kubota, T.; Komase, K.; et al. A Mutant H3N2 Influenza Virus Uses an Alternative Activation Mechanism in TMPRSS2 Knockout Mice by Loss of an Oligosaccharide in the Hemagglutinin Stalk Region. J. Virol. 2015, 89, 5154–5158. [Google Scholar] [CrossRef] [PubMed]
- Limburg, H.; Harbig, A.; Bestle, D.; Stein, D.A.; Moulton, H.M.; Jaeger, J.; Janga, H.; Hardes, K.; Koepke, J.; Schulte, L.; et al. TMPRSS2 Is the Major Activating Protease of Influenza A Virus in Primary Human Airway Cells and Influenza B Virus in Human Type II Pneumocytes. J. Virol. 2019, 93, 649–668. [Google Scholar] [CrossRef] [PubMed]
- Böttcher-Friebertshäuser, E.; Lu, Y.; Meyer, D.; Sielaff, F.; Steinmetzer, T.; Klenk, H.D.; Garten, W. Hemagglutinin activating host cell proteases provide promising drug targets for the treatment of influenza A and B virus infections. Vaccine 2012, 30, 7374–7380. [Google Scholar] [CrossRef]
- Braun, E.; Sauter, D. Furin-mediated protein processing in infectious diseases and cancer. Clin. Transl. Immunol. 2019, 8, e1073. [Google Scholar] [CrossRef]
- Belouzard, S.; Chu, V.C.; Whittaker, G.R. Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc. Natl. Acad. Sci. USA 2009, 106, 5871–5876. [Google Scholar] [CrossRef]
- Millet, J.K.; Whittaker, G.R. Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc. Natl. Acad. Sci. USA 2014, 111, 15214–15219. [Google Scholar] [CrossRef]
- Millet, J.K.; Whittaker, G.R. Physiological and molecular triggers for SARS-CoV membrane fusion and entry into host cells. Virology 2018, 517, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Simmons, G.; Reeves, J.D.; Rennekamp, A.J.; Amberg, S.M.; Piefer, A.J.; Bates, P. Characterization of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) spike glycoprotein-mediated viral entry. Proc. Natl. Acad. Sci. USA 2004, 101, 4240–4245. [Google Scholar] [CrossRef] [PubMed]
- Simmons, G.; Gosalia, D.N.; Rennekamp, A.J.; Reeves, J.D.; Diamond, S.L.; Bates, P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. USA 2005, 102, 11876–11881. [Google Scholar] [CrossRef] [PubMed]
- Glowacka, I.; Bertram, S.; Muller, M.A.; Allen, P.; Soilleux, E.; Pfefferle, S.; Steffen, I.; Tsegaye, T.S.; He, Y.; Gnirss, K.; et al. Evidence that TMPRSS2 Activates the Severe Acute Respiratory Syndrome Coronavirus Spike Protein for Membrane Fusion and Reduces Viral Control by the Humoral Immune Response. J. Virol. 2011, 85, 4122–4134. [Google Scholar] [CrossRef]
- Matsuyama, S.; Nagata, N.; Shirato, K.; Kawase, M.; Takeda, M.; Taguchi, F. Efficient Activation of the Severe Acute Respiratory Syndrome Coronavirus Spike Protein by the Transmembrane Protease TMPRSS2. J. Virol. 2010, 84, 12658–12664. [Google Scholar] [CrossRef]
- Shulla, A.; Heald-Sargent, T.; Subramanya, G.; Zhao, J.; Perlman, S.; Gallagher, T. A Transmembrane Serine Protease Is Linked to the Severe Acute Respiratory Syndrome Coronavirus Receptor and Activates Virus Entry. J. Virol. 2011, 85, 873–882. [Google Scholar] [CrossRef]
- Hoffmann, M.; Pöhlmann, S. How SARS-CoV-2 makes the cut. Nat. Microbiol. 2021, 6, 828–829. [Google Scholar] [CrossRef]
- Kleine-Weber, H.; Elzayat, M.T.; Hoffmann, M.; Pöhlmann, S. Functional analysis of potential cleavage sites in the MERS-coronavirus spike protein. Sci. Rep. 2018, 8, 16597. [Google Scholar] [CrossRef]
- Gierer, S.; Müller, M.A.; Heurich, A.; Ritz, D.; Springstein, B.L.; Karsten, C.B.; Schendzielorz, A.; Gnirß, K.; Drosten, C.; Pöhlmann, S. Inhibition of proprotein convertases abrogates processing of the middle eastern respiratory syndrome coronavirus spike protein in infected cells but does not reduce viral infectivity. J. Infect. Dis. 2015, 211, 889–897. [Google Scholar] [CrossRef]
- Gierer, S.; Bertram, S.; Kaup, F.; Wrensch, F.; Heurich, A.; Kramer-Kuhl, A.; Welsch, K.; Winkler, M.; Meyer, B.; Drosten, C.; et al. The Spike Protein of the Emerging Betacoronavirus EMC Uses a Novel Coronavirus Receptor for Entry, Can Be Activated by TMPRSS2, and Is Targeted by Neutralizing Antibodies. J. Virol. 2013, 87, 5502–5511. [Google Scholar] [CrossRef]
- Shirato, K.; Kawase, M.; Matsuyama, S. Middle East Respiratory Syndrome Coronavirus Infection Mediated by the Transmembrane Serine Protease TMPRSS2. J. Virol. 2013, 87, 12552–12561. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Li, K.; Barlan, A.; Fehr, A.R.; Perlman, S.; McCray, P.B.; Gallagher, T. Proteolytic processing of middle east respiratory syndrome coronavirus spikes expands virus tropism. Proc. Natl. Acad. Sci. USA 2016, 113, 12262–12267. [Google Scholar] [CrossRef]
- Matsuyama, S.; Shirato, K.; Kawase, M.; Terada, Y.; Kawachi, K.; Fukushi, S.; Kamitani, W. Middle East Respiratory Syndrome Coronavirus Spike Protein Is Not Activated Directly by Cellular Furin during Viral Entry into Target Cells. J. Virol. 2018, 92, e00683-18. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Coutard, B.; Valle, C.; de Lamballerie, X.; Canard, B.; Seidah, N.G.; Decroly, E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Res. 2020, 176, 104742. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784.e5. [Google Scholar] [CrossRef]
- Tang, T.; Jaimes, J.A.; Bidon, M.K.; Straus, M.R.; Daniel, S.; Whittaker, G.R. Proteolytic Activation of SARS-CoV-2 Spike at the S1/S2 Boundary: Potential Role of Proteases beyond Furin. ACS Infect. Dis. 2021, 7, 264–272. [Google Scholar] [CrossRef]
- Peacock, T.P.; Goldhill, D.H.; Zhou, J.; Baillon, L.; Frise, R.; Swann, O.C.; Kugathasan, R.; Penn, R.; Brown, J.C.; Sanchez-David, R.Y.; et al. The furin cleavage site in the SARS-CoV-2 spike protein is required for transmission in ferrets. Nat. Microbiol. 2021, 6, 899–909. [Google Scholar] [CrossRef]
- Tsatsakis, A.; Calina, D.; Falzone, L.; Petrakis, D.; Mitrut, R.; Siokas, V.; Pennisi, M.; Lanza, G.; Libra, M.; Doukas, S.G.; et al. SARS-CoV-2 pathophysiology and its clinical implications: An integrative overview of the pharmacotherapeutic management of COVID-19. Food Chem. Toxicol. 2020, 146, 111769. [Google Scholar] [CrossRef] [PubMed]
- Katopodis, P.; Randeva, H.S.; Spandidos, D.A.; Saravi, S.; Kyrou, I.; Karteris, E. Host cell entry mediators implicated in the cellular tropism of SARS-CoV-2, the pathophysiology of COVID-19 and the identification of microRNAs that can modulate the expression of these mediators (Review). Int. J. Mol. Med. 2021, 49, 20. [Google Scholar] [CrossRef] [PubMed]
- Katopodis, P.; Kerslake, R.; Davies, J.; Randeva, H.; Chatha, K.; Hall, M.; Spandidos, D.; Anikin, V.; Polychronis, A.; Robertus, J.; et al. COVID-19 and SARS-CoV-2 host cell entry mediators: Expression profiling of TMRSS4 in health and disease. Int. J. Mol. Med. 2021, 47, 64. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Hofmann-Winkler, H.; Smith, J.C.; Krüger, N.; Arora, P.; Sørensen, L.K.; Søgaard, O.S.; Hasselstrøm, J.B.; Winkler, M.; Hempel, T.; et al. Camostat mesylate inhibits SARS-CoV-2 activation by TMPRSS2-related proteases and its metabolite GBPA exerts antiviral activity. EBioMedicine 2021, 65, 103255. [Google Scholar] [CrossRef] [PubMed]
- Appleyard, G.; Tisdale, M. Inhibition of the growth of human coronavirus 229E by leupeptin. J. Gen. Virol. 1985, 66, 363–366. [Google Scholar] [CrossRef]
- Kawase, M.; Shirato, K.; Matsuyama, S.; Taguchi, F. Protease-Mediated Entry via the Endosome of Human Coronavirus 229E. J. Virol. 2009, 83, 712–721. [Google Scholar] [CrossRef]
- Bertram, S.; Dijkman, R.; Habjan, M.; Heurich, A.; Gierer, S.; Glowacka, I.; Welsch, K.; Winkler, M.; Schneider, H.; Hofmann-Winkler, H.; et al. TMPRSS2 Activates the Human Coronavirus 229E for Cathepsin-Independent Host Cell Entry and Is Expressed in Viral Target Cells in the Respiratory Epithelium. J. Virol. 2013, 87, 6150–6160. [Google Scholar] [CrossRef]
- Shirato, K.; Kawase, M.; Matsuyama, S. Wild-type human coronaviruses prefer cell-surface TMPRSS2 to endosomal cathepsins for cell entry. Virology 2018, 517, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Shirato, K.; Kanou, K.; Kawase, M.; Matsuyama, S. Clinical Isolates of Human Coronavirus 229E Bypass the Endosome for Cell Entry. J. Virol. 2017, 91, e01387-16. [Google Scholar] [CrossRef]
- Hofmann, H.; Pyrc, K.; Van Der Hoek, L.; Geier, M.; Berkhout, B.; Pöhlmann, S. Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc. Natl. Acad. Sci. USA 2005, 102, 7988–7993. [Google Scholar] [CrossRef]
- Huang, I.C.; Bosch, B.J.; Li, F.; Li, W.; Kyoung, H.L.; Ghiran, S.; Vasilieva, N.; Dermody, T.S.; Harrison, S.C.; Dormitzer, P.R.; et al. SARS coronavirus, but not human coronavirus NL63, utilizes cathepsin L to infect ACE2-expressing cells. J. Biol. Chem. 2006, 281, 3198–3203. [Google Scholar] [CrossRef]
- Milewska, A.; Nowak, P.; Owczarek, K.; Szczepanski, A.; Zarebski, M.; Hoang, A.; Berniak, K.; Wojarski, J.; Zeglen, S.; Baster, Z.; et al. Entry of Human Coronavirus NL63 into the Cell. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Kawase, M.; Shirato, K.; van der Hoek, L.; Taguchi, F.; Matsuyama, S. Simultaneous Treatment of Human Bronchial Epithelial Cells with Serine and Cysteine Protease Inhibitors Prevents Severe Acute Respiratory Syndrome Coronavirus Entry. J. Virol. 2012, 86, 6537–6545. [Google Scholar] [CrossRef] [PubMed]
- Millet, J.K.; Whittaker, G.R. Host cell proteases: Critical determinants of coronavirus tropism and pathogenesis. Virus Res. 2015, 202, 120–134. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Hofmann-Winkler, H.; Pöhlmann, S. Priming Time: How Cellular Proteases Arm Coronavirus Spike Proteins. In Activation of Viruses by Host Proteases; Springer: Cham, Switzerland, 2018; pp. 1–335. [Google Scholar] [CrossRef]
- Iwata-Yoshikawa, N.; Okamura, T.; Shimizu, Y.; Hasegawa, H.; Takeda, M.; Nagata, N. TMPRSS2 Contributes to Virus Spread and Immunopathology in the Airways of Murine Models after Coronavirus Infection. J. Virol. 2019, 93, e01933-17. [Google Scholar] [CrossRef]
- Zhou, Y.; Vedantham, P.; Lu, K.; Agudelo, J.; Carrion, R.; Nunneley, J.W.; Barnard, D.; Pöhlmann, S.; McKerrow, J.H.; Renslo, A.R.; et al. Protease inhibitors targeting coronavirus and filovirus entry. Antiviral Res. 2015, 116, 76–84. [Google Scholar] [CrossRef]
- Li, K.; Meyerholz, D.K.; Bartlett, J.A.; McCray, P.B. The Tmprss2 Inhibitor Nafamostat Reduces SARS-CoV-2 Pulmonary Infection in Mouse Models of covid-19. MBio 2021, 12, e00970-21. [Google Scholar] [CrossRef]
- Abe, M.; Tahara, M.; Sakai, K.; Yamaguchi, H.; Kanou, K.; Shirato, K.; Kawase, M.; Noda, M.; Kimura, H.; Matsuyama, S.; et al. TMPRSS2 Is an Activating Protease for Respiratory Parainfluenza Viruses. J. Virol. 2013, 87, 11930–11935. [Google Scholar] [CrossRef] [PubMed]
- Shirogane, Y.; Takeda, M.; Iwasaki, M.; Ishiguro, N.; Takeuchi, H.; Nakatsu, Y.; Tahara, M.; Kikuta, H.; Yanagi, Y. Efficient Multiplication of Human Metapneumovirus in Vero Cells Expressing the Transmembrane Serine Protease TMPRSS2. J. Virol. 2008, 82, 8942–8946. [Google Scholar] [CrossRef]
- Schmidt, A.C.; Schaap-Nutt, A.; Bartlett, E.J.; Schomacker, H.; Boonyaratanakornkit, J.; Karron, R.A.; Collins, P.L. Progress in the development of human parainfluenza virus vaccines. Expert Rev. Respir. Med. 2011, 5, 515–526. [Google Scholar] [CrossRef]
- Paulsson-Habegger, L.; Snabaitis, A.K.; Wren, S.P. Enzyme inhibition as a potential therapeutic strategy to treat COVID-19 infection. Bioorganic Med. Chem. 2021, 48, 116389. [Google Scholar] [CrossRef]
- Shen, L.W.; Mao, H.J.; Wu, Y.L.; Tanaka, Y.; Zhang, W. TMPRSS2: A potential target for treatment of influenza virus and coronavirus infections. Biochimie 2017, 142, 1–10. [Google Scholar] [CrossRef]
- Shaw, G.L.; Whitaker, H.; Corcoran, M.; Dunning, M.J.; Luxton, H.; Kay, J.; Massie, C.E.; Miller, J.L.; Lamb, A.D.; Ross-Adams, H.; et al. The Early Effects of Rapid Androgen Deprivation on Human Prostate Cancer. Eur. Urol. 2016, 70, 214–218. [Google Scholar] [CrossRef]
- Schröder, F.; Crawford, E.D.; Axcrona, K.; Payne, H.; Keane, T.E. Androgen deprivation therapy: Past, present and future. BJU Int. 2012, 109, 1–12. [Google Scholar] [CrossRef]
- Feldman, B.J.; Feldman, D. The development of androgen-independent prostate cancer. Nat. Rev. Cancer 2001, 1, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Pienta, K.J.; Bradley, D. Mechanisms underlying the development of androgen-independent prostate cancer. Clin. Cancer Res. 2006, 12, 1665–1671. [Google Scholar] [CrossRef] [PubMed]
- Summerton, J.; Weller, D. Morpholino antisense oligomers: Design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 1997, 7, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lear, T.B.; Evankovich, J.W.; Larsen, M.B.; Lin, B.; Alfaras, I.; Kennerdell, J.R.; Salminen, L.; Camarco, D.P.; Lockwood, K.C.; et al. A high-throughput screen for TMPRSS2 expression identifies FDA-approved compounds that can limit SARS-CoV-2 entry. Nat. Commun. 2021, 12, 3907. [Google Scholar] [CrossRef]
- Keller, T.L.; Zocco, D.; Sundrud, M.S.; Hendrick, M.; Edenius, M.; Yum, J.; Kim, Y.J.; Lee, H.K.; Cortese, J.F.; Wirth, D.F.; et al. Halofuginone and other febrifugine derivatives inhibit prolyl-tRNA synthetase. Nat. Chem. Biol. 2012, 8, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Song, Y.J. Anti-varicella-zoster virus activity of cephalotaxine esters in vitro. J. Microbiol. 2019, 57, 74–79. [Google Scholar] [CrossRef]
- Kaur, P.; Thiruchelvan, M.; Lee, R.C.H.; Chen, H.; Chen, K.C.; Ng, M.L.; Chu, J.J.H. Inhibition of Chikungunya virus replication by harringtonine, a novel antiviral that suppresses viral protein expression. Antimicrob. Agents Chemother. 2013, 57, 155–167. [Google Scholar] [CrossRef]
- Cao, J.; Forrest, J.C.; Zhang, X. A screen of the NIH Clinical Collection small molecule library identifies potential anti-coronavirus drugs. Antiviral Res. 2015, 114, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tamura, Y.; Hirado, M.; Okamura, K.; Minato, Y.; Fujii, S. Synthetic inhibitors of trypsin, plasmin, kallikrein, thrombin, C1r, and C1 esterase. BBA - Enzymol. 1977, 484, 417–422. [Google Scholar] [CrossRef]
- Kono, K.; Takahashi, A.; Sugai, H.; Umekawa, T.; Yano, T.; Kamiyasu, K.; Teramatsu, M.; Fujii, H. Oral trypsin inhibitor can improve reflux esophagitis after distal gastrectomy concomitant with decreased trypsin activity. Am. J. Surg. 2005, 190, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Ohshio, G.; Saluja, A.K.; Leli, U.; Sengupta, A.; Steer, M.L. Esterase inhibitors prevent lysosomal enzyme redistribution in two noninvasive models of experimental pancreatitis. Gastroenterology 1989, 96, 853–859. [Google Scholar] [CrossRef]
- Yamamoto, M.; Matsuyama, S.; Li, X.; Takeda, M.; Kawaguchi, Y.; Inoue, J.I.; Matsuda, Z. Identification of nafamostat as a potent inhibitor of middle east respiratory syndrome Coronavirus s protein-mediated membrane fusion using the split-protein-based cell-cell fusion assay. Antimicrob. Agents Chemother. 2016, 60, 6532–6539. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Schroeder, S.; Kleine-Weber, H.; Müller, M.A.; Drosten, C.; Pöhlmann, S. Nafamostat mesylate blocks activation of SARS-CoV-2: New treatment option for COVID-19. Antimicrob. Agents Chemother. 2020, 64, 19–21. [Google Scholar] [CrossRef] [PubMed]
- Yamaya, M.; Shimotai, Y.; Hatachi, Y.; Lusamba Kalonji, N.; Tando, Y.; Kitajima, Y.; Matsuo, K.; Kubo, H.; Nagatomi, R.; Hongo, S.; et al. The serine protease inhibitor camostat inhibits influenza virus replication and cytokine production in primary cultures of human tracheal epithelial cells. Pulm. Pharmacol. Ther. 2015, 33, 66–74. [Google Scholar] [CrossRef]
- Lee, J.; Lee, J.; Kim, H.J.; Ko, M.; Jee, Y.; Kim, S. TMPRSS2 and RNA-Dependent RNA Polymerase Are Effective Targets of Therapeutic Intervention for Treatment of COVID-19 Caused by SARS-CoV-2 Variants (B.1.1.7 and B.1.351). Microbiol. Spectr. 2021, 9, 2–8. [Google Scholar] [CrossRef]
- Sun, Y.J.; Velez, G.; Parsons, D.E.; Li, K.; Ortiz, M.E.; Sharma, S.; McCray, P.B.; Bassuk, A.G.; Mahajan, V.B. Structure-based phylogeny identifies avoralstat as a TMPRSS2 inhibitor that prevents SARS-CoV-2 infection in mice. J. Clin. Invest. 2021, 131. [Google Scholar] [CrossRef]
- Hoffmann, M.; Arora, P.; Groß, R.; Seidel, A.; Hörnich, B.F.; Hahn, A.S.; Krüger, N.; Graichen, L.; Hofmann-Winkler, H.; Kempf, A.; et al. SARS-CoV-2 variants B.1.351 and P.1 escape from neutralizing antibodies. Cell 2021, 184, 2384–2393. [Google Scholar] [CrossRef]
- Hoffmann, M.; Hofmann-Winkler, H.; Krüger, N.; Kempf, A.; Nehlmeier, I.; Graichen, L.; Arora, P.; Sidarovich, A.; Moldenhauer, A.S.; Winkler, M.S.; et al. SARS-CoV-2 variant B.1.617 is resistant to bamlanivimab and evades antibodies induced by infection and vaccination. Cell Rep. 2021, 36, 109415. [Google Scholar] [CrossRef] [PubMed]
- Azouz, N.P.; Klingler, A.M.; Callahan, V.; Akhrymuk, I.V.; Elez, K.; Raich, L.; Henry, B.M.; Benoit, J.L.; Benoit, S.W.; Noé, F.; et al. Alpha 1 antitrypsin is an inhibitor of the sars-cov-2–priming protease tmprss2. Pathog. Immun. 2021, 6, 55–74. [Google Scholar] [CrossRef] [PubMed]
- Wettstein, L.; Weil, T.; Conzelmann, C.; Müller, J.A.; Groß, R.; Hirschenberger, M.; Seidel, A.; Klute, S.; Zech, F.; Bozzo, C.P.; et al. Alpha-1 antitrypsin inhibits TMPRSS2 protease activity and SARS-CoV-2 infection. Nat. Commun. 2021, 12, 1726. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Shrimp, J.H.; Guo, H.; Xu, M.; Chen, C.Z.; Zhu, W.; Zakharov, A.V.; Jain, S.; Shinn, P.; Simeonov, A.; et al. Discovery of TMPRSS2 Inhibitors from Virtual Screening as a Potential Treatment of COVID-19. ACS Pharmacol. Transl. Sci. 2021, 4, 1124–1135. [Google Scholar] [CrossRef] [PubMed]
- Hempel, T.; Raich, L.; Olsson, S.; Azouz, N.P.; Klingler, A.M.; Hoffmann, M.; Pöhlmann, S.; Rothenberg, M.E.; Noé, F. Molecular mechanism of inhibiting the SARS-CoV-2 cell entry facilitator TMPRSS2 with camostat and nafamostat. Chem. Sci. 2021, 12, 983–992. [Google Scholar] [CrossRef]
- Shrimp, J.H.; Kales, S.C.; Sanderson, P.E.; Simeonov, A.; Shen, M.; Hall, M.D. An Enzymatic TMPRSS2 Assay for Assessment of Clinical Candidates and Discovery of Inhibitors as Potential Treatment of COVID-19. ACS Pharmacol. Transl. Sci. 2020, 3, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Beckh, K.; Weidenbach, H.; Weidenbach, F.; Müller, R.; Adler, G. Hepatic and pancreatic metabolism and biliary excretion of the protease inhibitor camostat mesilate. Int. J. Pancreatol. 1991, 10, 197–205. [Google Scholar] [CrossRef]
- Midgley, I.; Hood, A.J.; Proctor, P.; Chasseaud, L.F.; Irons, S.R.; Cheng, K.N.; Brindley, C.J.; Bonn, R. Metabolic fate of 14c-camostat mesylate in man, rat and dog after intravenous administration. Xenobiotica 1994, 24, 79–92. [Google Scholar] [CrossRef]
- Fujii, S.; Hitomi, Y. New Synthetic Inhibitors of Clr, Clesterase, Thrombin, Plasmin, Kallikrein and Trypsin. Japanese J. Clin. Chem. 1981, 10, 248–252. [Google Scholar] [CrossRef]
- Akizawa, T.; Koshikawa, S.; Ota, K.; Kazama, M.; Mimura, N.; Hirasawa, Y. Nafamostat Mesilate: A Regional Anticoagulant for Hemodialysis in Patients at High Risk for Bleeding. Nephron 1993, 64, 376–381. [Google Scholar] [CrossRef]
- Han, S.J.; Kim, H.S.; Kim, K.; Whang, S.M.; Hong, K.S.; Lee, W.K.; Lee, S.H. Use of nafamostat mesilate as an anticoagulant during extracorporeal membrane oxygenation. J. Korean Med. Sci. 2011, 26, 945–950. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.W.; Han, B.G.; Kim, B.R.; Lee, Y.H.; Kim, Y.S.; Yu, J.M.; Choi, S.O. Superior outcome of nafamostat mesilate as an anticoagulant in patients undergoing maintenance hemodialysis with intracerebral hemorrhage. Ren. Fail. 2009, 31, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Kiso, M.; Sakai-Tagawa, Y.; Iwatsuki-Horimoto, K.; Imai, M.; Takeda, M.; Kinoshita, N.; Ohmagari, N.; Gohda, J.; Semba, K.; et al. The anticoagulant nafamostat potently inhibits SARS-CoV-2 S protein-mediated fusion in a cell fusion assay system and viral infection in vitro in a cell-type-dependent manner. Viruses 2020, 12, 629. [Google Scholar] [CrossRef]
- Ko, C.J.; Hsu, T.W.; Wu, S.R.; Lan, S.W.; Hsiao, T.F.; Lin, H.Y.; Lin, H.H.; Tu, H.F.; Lee, C.F.; Huang, C.C.; et al. Inhibition of TMPRSS2 by HAI-2 reduces prostate cancer cell invasion and metastasis. Oncogene 2020, 39, 5950–5963. [Google Scholar] [CrossRef] [PubMed]
- Ramjee, M.K.; Henderson, I.M.J.; McLoughlin, S.B.; Padova, A. The kinetic and structural characterization of the reaction of nafamostat with bovine pancreatic trypsin. Thromb. Res. 2000, 98, 559–569. [Google Scholar] [CrossRef]
- Zerner, B.; Bond, R.P.M.; Bender, M.L. Kinetic Evidence for the Formation of Acyl-Enzyme Intermediates in the α-Chymotrypsin-Catalyzed Hydrolyses of Specific Substrates12. J. Am. Chem. Soc. 1964, 86, 3674–3679. [Google Scholar] [CrossRef]
- Fraser, B.J.; Beldar, S.; Seitova, A.; Hutchinson, A.; Mannar, D.; Arrowsmith, C.H.; Bénard, F. Structure, activity and inhibition of human TMPRSS2, a protease implicated in SARS-CoV-2 activation. BioRxiv 2021. [Google Scholar]
- Kosai, K.; Seki, M.; Yanagihara, K.; Nakamura, S.; Kurihara, S.; Izumikawa, K.; Kakeya, H.; Yamamoto, Y.; Tashiro, T.; Kohno, S. Gabexate mesilate suppresses influenza pneumonia in mice through inhibition of cytokines. J. Int. Med. Res. 2008, 36, 322–328. [Google Scholar] [CrossRef]
- Chang, C.C.; Cheng, A.C.; Chang, A.B. Over-the-counter (OTC) medications to reduce cough as an adjunct to antibiotics for acute pneumonia in children and adults. Cochrane Database Syst. Rev. 2014, 2014. [Google Scholar] [CrossRef]
- Mannucci, P.M. Hemostatic Drugs. N. Engl. J. Med. 1998, 339, 245–253. [Google Scholar] [CrossRef]
- Fritz, H.; Wunderer, G. Biochemistry and applications of aprotinin, the kallikrein inhibitor from bovine organs. Arzneimittel-Forschung/Drug Res. 1983, 33, 479–494. [Google Scholar] [CrossRef]
- Böttcher, E.; Freuer, C.; Steinmetzer, T.; Klenk, H.D.; Garten, W. MDCK cells that express proteases TMPRSS2 and HAT provide a cell system to propagate influenza viruses in the absence of trypsin and to study cleavage of HA and its inhibition. Vaccine 2009, 27, 6324–6329. [Google Scholar] [CrossRef]
- Zhirnov, O.P.; Klenk, H.D.; Wright, P.F. Aprotinin and similar protease inhibitors as drugs against influenza. Antiviral Res. 2011, 92, 27–36. [Google Scholar] [CrossRef]
- Zhirnov, O.P.; Matrosovich, T.Y.; Matrosovich, M.N.; Klenk, H.D. Aprotinin, a protease inhibitor, suppresses proteolytic activation of pandemic H1N1v influenza virus. Antivir. Chem. Chemother. 2011, 21, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Zhirnov, O.P.; Ovcharenko, A.V.; Bukrinskaya, A.G. Myxovirus replication in chicken embryos can be suppressed by aprotinin due to the blockage of viral glycoprotein cleavage. J. Gen. Virol. 1985, 66, 1633–1638. [Google Scholar] [CrossRef] [PubMed]
- Zhirnov, O.P.; Ovcharenko, A.V.; Bukrinskaya, A.G. Suppression of influenza virus replication in infected mice by protease inhibitors. J. Gen. Virol. 1984, 65, 191–196. [Google Scholar] [CrossRef]
- Zhirnov, O.P. High protection of animals lethally infected with influenza virus by aprotinin-rimantadine combination. J. Med. Virol. 1987, 21, 161–167. [Google Scholar] [CrossRef]
- Bojkova, D.; Bechtel, M.; McLaughlin, K.M.; McGreig, J.E.; Klann, K.; Bellinghausen, C.; Rohde, G.; Jonigk, D.; Braubach, P.; Ciesek, S.; et al. Aprotinin Inhibits SARS-CoV-2 Replication. Cells 2020, 9, 2377. [Google Scholar] [CrossRef]
- Ovcharenko, A.V.; Zhirnov, O.P. Aprotinin aerosol treatment of influenza and paramyxovirus bronchopneumonia of mice. Antiviral Res. 1994, 23, 107–118. [Google Scholar] [CrossRef]
- Szabo, R.; Hobson, J.P.; List, K.; Molinolo, A.; Lin, C.Y.; Bugge, T.H. Potent inhibition and global co-localization implicate the transmembrane Kunitz-type serine protease inhibitor hepatocyte growth factor activator inhibitor-2 in the regulation of epithelial matriptase activity. J. Biol. Chem. 2008, 283, 29495–29504. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Qin, L.; Shimomura, T.; Kondo, J.; Matsumoto, K.; Denda, K.; Kitamura, N. Purification and cloning of hepatocyte growth factor activator inhibitor type 2, a Kunitz-type serine protease inhibitor. J. Biol. Chem. 1997, 272, 27558–27564. [Google Scholar] [CrossRef]
- Straus, M.R.; Kinder, J.T.; Segall, M.; Dutch, R.E.; Whittaker, G.R. SPINT2 inhibits proteases involved in activation of both influenza viruses and metapneumoviruses. Virology 2020, 543, 43–53. [Google Scholar] [CrossRef]
- Hamilton, B.S.; Chung, C.; Cyphers, S.Y.; Rinaldi, V.D.; Marcano, V.C.; Whittaker, G.R. Inhibition of influenza virus infection and hemagglutinin cleavage by the protease inhibitor HAI-2. Biochem. Biophys. Res. Commun. 2014, 450, 1070–1075. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Janciauskiene, S.; Welte, T. Well-known and less well-known functions of Alpha-1 antitrypsin: Its role in chronic obstructive pulmonary disease and other disease developments. Ann. Am. Thorac. Soc. 2016, 13, S280–S288. [Google Scholar] [CrossRef]
- Jonigk, D.; Al-Omari, M.; Maegel, L.; Muller, M.; Izykowski, N.; Hong, J.; Hong, K.; Kim, S.-H.; Dorsch, M.; Mahadeva, R.; et al. Anti-inflammatory and immunomodulatory properties of 1-antitrypsin without inhibition of elastase. Proc. Natl. Acad. Sci. USA 2013, 110, 15007–15012. [Google Scholar] [CrossRef] [PubMed]
- McElvaney, O.J.; McEvoy, N.L.; McElvaney, O.F.; Carroll, T.P.; Murphy, M.P.; Dunlea, D.M.; Ní Choileáin, O.; Clarke, J.; O’Connor, E.; Hogan, G.; et al. Characterization of the Inflammatory Response to Severe COVID-19 Illness. Am. J. Respir. Crit. Care Med. 2020, 202, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Shapira, G.; Shomron, N.; Gurwitz, D. Ethnic differences in alpha-1 antitrypsin deficiency allele frequencies may partially explain national differences in COVID-19 fatality rates. FASEB J. 2020, 34, 14160–14165. [Google Scholar] [CrossRef]
- Yang, C.; Keshavjee, S.; Liu, M. Alpha-1 Antitrypsin for COVID-19 Treatment: Dual Role in Antiviral Infection and Anti-Inflammation. Front. Pharmacol. 2020, 11, 2113. [Google Scholar] [CrossRef]
- Yang, C.; Chapman, K.R.; Wong, A.; Mingyao, L. α1-Antitrypsin deficiency and the risk of COVID-19: An urgent call to action. Lancet Respir Med 2021, 9, 337–339. [Google Scholar] [CrossRef]
- McElvaney, O.J.; O’Connor, E.; McEvoy, N.L.; Fraughan, D.D.; Clarke, J.; McElvaney, O.F.; Gunaratnam, C.; O’Rourke, J.; Curley, G.F.; McElvaney, N.G. Alpha-1 antitrypsin for cystic fibrosis complicated by severe cytokinemic COVID-19. J. Cyst. Fibros. 2021, 20, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Campos, M.A.; Kueppers, F.; Stocks, J.M.; Strange, C.; Chen, J.; Griffin, R.; Wang-Smith, L.; Brantly, M.L. Safety and Pharmacokinetics of 120 mg/kg versus 60 mg/kg Weekly Intravenous Infusions of Alpha-1 Proteinase Inhibitor in Alpha-1 Antitrypsin Deficiency: A Multicenter, Randomized, Double-Blind, Crossover Study (SPARK). COPD J. Chronic Obstr. Pulm. Dis. 2013, 10, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Campos, M.A.; Geraghty, P.; Holt, G.; Mendes, E.; Newby, P.R.; Ma, S.; Luna-Diaz, L.V.; Turino, G.M.; Stockley, R.A. The biological effects of double-dose alpha-1 antitrypsin augmentation therapy a pilot clinical trial. Am. J. Respir. Crit. Care Med. 2019, 200, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, R.C.; Sellers, S.; Czerski, D.; Stephens, L.; Crystal, R.G. Biochemical Efficacy and Safety of Monthly Augmentation Therapy for α1-Antitrypsin Deficiency. JAMA J. Am. Med. Assoc. 1988, 260, 1259–1264. [Google Scholar] [CrossRef]
- Franciosi, A.N.; McCarthy, C.; McElvaney, N.G. The efficacy and safety of inhaled human α-1 antitrypsin in people with α-1 antitrypsin deficiency-related emphysema. Expert Rev. Respir. Med. 2015, 9, 143–151. [Google Scholar] [CrossRef]
- Brand, P.; Schulte, M.; Wencker, M.; Herpich, C.H.; Klein, G.; Hanna, K.; Meyer, T. Lung deposition of inhaled α1-proteinase inhibitor in cystic fibrosis and α1-antitrypsin deficiency. Eur. Respir. J. 2008, 34, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Sielaff, F.; Böttcher-Friebertshäuser, E.; Meyer, D.; Saupe, S.M.; Volk, I.M.; Garten, W.; Steinmetzer, T. Development of substrate analogue inhibitors for the human airway trypsin-like protease HAT. Bioorganic Med. Chem. Lett. 2011, 21, 4860–4864. [Google Scholar] [CrossRef]
- Hammami, M.; Rühmann, E.; Maurer, E.; Heine, A.; Gütschow, M.; Klebe, G.; Steinmetzer, T. New 3-amidinophenylalanine-derived inhibitors of matriptase. Medchemcomm 2012, 3, 807–813. [Google Scholar] [CrossRef]
- Sisay, M.T.; Steinmetzer, T.; Stirnberg, M.; Maurer, E.; Hammami, M.; Bajorath, J.; Gütschow, M. Identification of the first low-molecular-weight inhibitors of matriptase-2. J. Med. Chem. 2010, 53, 5523–5535. [Google Scholar] [CrossRef]
- Steinmetzer, T.; Schweinitz, A.; Stürzebecher, A.; Dönnecke, D.; Uhland, K.; Schuster, O.; Steinmetzer, P.; Müller, F.; Friedrich, R.; Than, M.E.; et al. Secondary amides of sulfonylated 3-amidinophenylalanine. New potent and selective inhibitors of matriptase. J. Med. Chem. 2006, 49, 4116–4126. [Google Scholar] [CrossRef] [PubMed]
- Schweinitz, A.; Steinmetzer, T.; Banke, I.J.; Arlt, M.J.E.; Stürzebecher, A.; Schuster, O.; Geissler, A.; Giersiefen, H.; Zeslawska, E.; Jacob, U.; et al. Design of novel and selective inhibitors of urokinase-type plasminogen activator with improved pharmacokinetic properties for use as antimetastatic agents. J. Biol. Chem. 2004, 279, 33613–33622. [Google Scholar] [CrossRef] [PubMed]
- Stürzebecher, J.; Prasa, D.; Hauptmann, J.; Vieweg, H.; Wikström, P. Synthesis and structure-activity relationships of potent thrombin inhibitors: Piperazides of 3-amidinophenylalanine. J. Med. Chem. 1997, 40, 3091–3099. [Google Scholar] [CrossRef]
- Stürzebecher, J.; Vieweg, H.; Steinmetzer, T.; Schweinitz, A.; Stubbs, M.T.; Renatus, M.; Wikström, P. 3-Amidinophenylalanine-based inhibitors of urokinase. Bioorganic Med. Chem. Lett. 1999, 9, 3147–3152. [Google Scholar] [CrossRef]
- Meyer, D.; Sielaff, F.; Hammami, M.; Böttcher-Friebertshäuser, E.; Garten, W.; Steinmetzer, T. Identification of the first synthetic inhibitors of the type II transmembrane serine protease TMPRSS2 suitable for inhibition of influenza virus activation. Biochem. J. 2013, 452, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Garten, W.; Braden, C.; Arendt, A.; Peitsch, C.; Baron, J.; Lu, Y.; Pawletko, K.; Hardes, K.; Steinmetzer, T.; Böttcher-Friebertshäuser, E. Influenza virus activating host proteases: Identification, localization and inhibitors as potential therapeutics. Eur. J. Cell Biol. 2015, 94, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Hellstern, P.; Stürzebecher, U.; Wuchold, B.; Haubelt, H.; Seyfert, U.T.; Bauer, M.; Vogt, A.; Stürzebecher, J. Preservation of in vitro function of platelets stored in the presence of a synthetic dual inhibitor of factor Xa and thrombin. J. Thromb. Haemost. 2007, 5, 2119–2126. [Google Scholar] [CrossRef] [PubMed]
- Montopoli, M.; Zumerle, S.; Vettor, R.; Rugge, M.; Zorzi, M.; Catapano, C.V.; Carbone, G.M.; Cavalli, A.; Pagano, F.; Ragazzi, E.; et al. Androgen-deprivation therapies for prostate cancer and risk of infection by SARS-CoV-2: A population-based study (N = 4532). Ann. Oncol. 2020, 31, 1040–1045. [Google Scholar] [CrossRef]
- Klein, E.A.; Li, J.; Milinovich, A.; Schold, J.D.; Sharifi, N.; Kattan, M.W.; Jehi, L. Androgen Deprivation Therapy in Men with Prostate Cancer Does Not Affect Risk of Infection with SARS-CoV-2. J. Urol. 2021, 295, 441–443. [Google Scholar] [CrossRef]
- Gedeborg, R.; Styrke, J.; Loeb, S.; Garmo, H.; Stattin, P. Androgen deprivation therapy and excess mortality in men with prostate cancer during the initial phase of the COVID-19 pandemic. PLoS ONE 2021, 16, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.G.; Zhong, X.; Shah, N.J.; Pina Martina, L.; Hawley, J.; Lin, E.; Gartrell, B.A.; Adorno Febles, V.R.; Wise, D.R.; Qin, Q.; et al. The role of androgen deprivation therapy on the clinical course of COVID-19 infection in men with prostate cancer. J. Clin. Oncol. 2021, 39, 41. [Google Scholar] [CrossRef]
- Jiménez-Alcaide, E.; García-Fuentes, C.; Hernández, V.; De la Peña, E.; Pérez-Fernández, E.; Castro, A.; Caballero-Perea, B.; Guijarro, A.; Llorente, C. Influence of androgen deprivation therapy on the severity of COVID-19 in prostate cancer patients. Prostate 2021, 81, 1349–1354. [Google Scholar] [CrossRef]
- Schmidt, A.L.; Tucker, M.D.; Bakouny, Z.; Labaki, C.; Hsu, C.-Y.; Shyr, Y.; Armstrong, A.J.; Beer, T.M.; Bijjula, R.R.; Bilen, M.A.; et al. Association Between Androgen Deprivation Therapy and Mortality Among Patients With Prostate Cancer and COVID-19. JAMA Netw. Open 2021, 4, e2134330. [Google Scholar] [CrossRef] [PubMed]
- Hofmann-Winkler, H.; Moerer, O.; Alt-Epping, S.; Bräuer, A.; Büttner, B.; Müller, M.; Fricke, T.; Grundmann, J.; Harnisch, L.-O.; Heise, D.; et al. Camostat Mesylate May Reduce Severity of Coronavirus Disease 2019 Sepsis: A First Observation. Crit. Care Explor. 2020, 2, e0284. [Google Scholar] [CrossRef] [PubMed]
- Doi, K.; Ikeda, M.; Hayase, N.; Moriya, K.; Morimura, N.; Maehara, H.; Tagami, S.; Fukushima, K.; Misawa, N.; Inoue, Y.; et al. Nafamostat mesylate treatment in combination with favipiravir for patients critically ill with Covid-19: A case series. Crit. Care 2020, 24, 20–23. [Google Scholar] [CrossRef]
- Jang, S.; Rhee, J. Three cases of treatment with nafamostat in elderly patients with COVID-19 pneumonia who need oxygen therapy. Int. J. Infect. Dis. 2020, 96, 500–502. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, W.; Yoneda, T.; Koba, H.; Ueda, T.; Tsuji, N.; Ogawa, H.; Asakura, H. Potential mechanisms of nafamostat therapy for severe COVID-19 pneumonia with disseminated intravascular coagulation. Int. J. Infect. Dis. 2021, 102, 529–531. [Google Scholar] [CrossRef]
- Gunst, J.D.; Staerke, N.B.; Pahus, M.H.; Kristensen, L.H.; Bodilsen, J.; Lohse, N.; Dalgaard, L.S.; Brønnum, D.; Fröbert, O.; Hønge, B.; et al. Efficacy of the TMPRSS2 inhibitor camostat mesilate in patients hospitalized with Covid-19-a double-blind randomized controlled trial. EClinicalMedicine 2021, 35, 100849. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, J.; Arai, H.; Iida, H.; Mukai, J.; Furukawa, K.; Ohtsu, S.; Nakade, S.; Hikima, T.; Haranaka, M.; Uemura, N. A phase I study of high dose camostat mesylate in healthy adults provides a rationale to repurpose the TMPRSS2 inhibitor for the treatment of COVID-19. Clin. Transl. Sci. 2021, 14, 1967–1976. [Google Scholar] [CrossRef]
- Zhuravel, S.V.; Khmelnitskiy, O.K.; Burlaka, O.O.; Gritsan, A.I.; Goloshchekin, B.M.; Kim, S.; Hong, K.Y. Nafamostat in hospitalized patients with moderate to severe COVID-19 pneumonia: A randomised Phase II clinical trial. EClinicalMedicine 2021, 41, 101169. [Google Scholar] [CrossRef] [PubMed]
- Ansarin, K.; Tolouian, R.; Ardalan, M.; Taghizadieh, A.; Varshochi, M.; Teimouri, S.; Vaezi, T.; Valizadeh, H.; Saleh, P.; Safiri, S.; et al. Effect of bromhexine on clinical outcomes and mortality in COVID-19 patients: A randomized clinical trial. BioImpacts 2020, 10, 209–215. [Google Scholar] [CrossRef]
- Tolouian, R.; Mulla, Z.D.; Jamaati, H.; Babamahmoodi, A.; Marjani, M.; Eskandari, R.; Dastan, F. Effect of bromhexine in hospitalized patients with COVID-19. J. Investig. Med. 2021, 1–6. [Google Scholar] [CrossRef]
- Li, T.; Sun, L.; Zhang, W.; Zheng, C.; Jiang, C.; Chen, M.; Chen, D.; Dai, Z.; Bao, S.; Shen, X. Bromhexine Hydrochloride Tablets for the Treatment of Moderate COVID-19: An Open-Label Randomized Controlled Pilot Study. Clin. Transl. Sci. 2020, 13, 1096–1102. [Google Scholar] [CrossRef] [PubMed]
- Mikhaylov, E.N.; Lyubimtseva, T.A.; Vakhrushev, A.D.; Stepanov, D.; Lebedev, D.S.; Vasilieva, E.Y.; Konradi, A.O.; Shlyakhto, E.V. Bromhexine Hydrochloride Prophylaxis of COVID-19 for Medical Personnel: A Randomized Open-Label Study. medRxiv 2021, 007. [Google Scholar] [CrossRef]
- He, X.; Lau, E.H.Y.; Wu, P.; Deng, X.; Wang, J.; Hao, X.; Lau, Y.C.; Wong, J.Y.; Guan, Y.; Tan, X.; et al. Temporal dynamics in viral shedding and transmissibility of COVID-19. Nat. Med. 2020, 26, 672–675. [Google Scholar] [CrossRef] [PubMed]
- To, K.K.W.; Tsang, O.T.Y.; Leung, W.S.; Tam, A.R.; Wu, T.C.; Lung, D.C.; Yip, C.C.Y.; Cai, J.P.; Chan, J.M.C.; Chik, T.S.H.; et al. Temporal profiles of viral load in posterior oropharyngeal saliva samples and serum antibody responses during infection by SARS-CoV-2: An observational cohort study. Lancet Infect. Dis. 2020, 20, 565–574. [Google Scholar] [CrossRef]
- Bai, X.; Hippensteel, J.; Leavitt, A.; Maloney, J.P.; Beckham, D.; Garcia, C.; Li, Q.; Freed, B.M.; Ordway, D.; Sandhaus, R.A.; et al. Hypothesis: Alpha-1-antitrypsin is a promising treatment option for COVID-19. Med. Hypotheses 2020, 146, 110394. [Google Scholar] [CrossRef]
- Torres Acosta, M.A.; Singer, B.D. Pathogenesis of COVID-19-induced ARDS: Implications for an ageing population. Eur. Respir. J. 2020, 56. [Google Scholar] [CrossRef]
- Gupta, A.; Madhavan, M.V.; Sehgal, K.; Nair, N.; Mahajan, S.; Sehrawat, T.S.; Bikdeli, B.; Ahluwalia, N.; Ausiello, J.C.; Wan, E.Y.; et al. Extrapulmonary manifestations of COVID-19. Nat. Med. 2020, 26, 1017–1032. [Google Scholar] [CrossRef]
- Jamilloux, Y.; Henry, T.; Belot, A.; Viel, S.; Fauter, M.; El, T.; Walzer, T.; François, B.; Sève, P. Should we suppress or stimulate immune responses for Covid-19? Cytokine and anti-cytokine interventions. Autoimmun. Rev. 2020, 19, 102567. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wettstein, L.; Kirchhoff, F.; Münch, J. The Transmembrane Protease TMPRSS2 as a Therapeutic Target for COVID-19 Treatment. Int. J. Mol. Sci. 2022, 23, 1351. https://doi.org/10.3390/ijms23031351
Wettstein L, Kirchhoff F, Münch J. The Transmembrane Protease TMPRSS2 as a Therapeutic Target for COVID-19 Treatment. International Journal of Molecular Sciences. 2022; 23(3):1351. https://doi.org/10.3390/ijms23031351
Chicago/Turabian StyleWettstein, Lukas, Frank Kirchhoff, and Jan Münch. 2022. "The Transmembrane Protease TMPRSS2 as a Therapeutic Target for COVID-19 Treatment" International Journal of Molecular Sciences 23, no. 3: 1351. https://doi.org/10.3390/ijms23031351
APA StyleWettstein, L., Kirchhoff, F., & Münch, J. (2022). The Transmembrane Protease TMPRSS2 as a Therapeutic Target for COVID-19 Treatment. International Journal of Molecular Sciences, 23(3), 1351. https://doi.org/10.3390/ijms23031351